

Summary
The presence of supernumerary centrosomes in cells infected with Chlamydia trachomatis may provide a mechanism to explain the association of C. trachomatis genital infection with cervical cancer. We show that the amplified centrosomal foci induced during a chlamydial infection contain both centriolar and pericentriolar matrix markers, demonstrating that they are bona fide centrosomes. As there were multiple immature centrioles but approximately one mature centriole per cell, aborted cytokinesis alone cannot account for centrosome amplification during a chlamydial infection. Production of supernumerary centrosomes required the kinase activities of Cdk2 and Plk4, which are known regulators of centrosome duplication, and progression through S-phase, which is the stage in the cell cycle when duplication of the centrosome occurs. These requirements indicate that centrosome amplification during a chlamydial infection depends on the host centrosome duplication pathway, which normally produces a single procentriole from each template centriole. However, C. trachomatis induces a loss of numerical control so that multiple procentrioles are formed per template.
Introduction
Chlamydia trachomatis is a leading cause of sexually transmitted infection with a potential role in the development of cancer (Zenilman, 2001). More than a million cases of C. trachomatis genital infection are reported to the Centers for Disease Control and Prevention (CDC) each year, making it by far the most common notifiable infection (CDC, 2006). C. trachomatis genital infections have been epidemiologically linked with cervical cancer (Koskela et al., 2000; Naucler et al., 2007), one of the three most prevalent malignancies in women worldwide (Zenilman, 2001). In addition to being an independent risk factor, C. trachomatis has been implicated as a cofactor for human papilloma virus (HPV) in the development of cervical cancer (Samoff et al., 2005; Silins et al., 2005). The possibility of a more general link between Chlamydia and cancer has been raised by a recent report showing an association between another chlamydial species, Chlamydia psittaci, and lymphoma (Ponzoni et al., 2008).
Chlamydia trachomatis infection may contribute to oncogenesis through effects on the centrosome of the infected host cell that can lead to genetic instability. Grieshaber et al. (2006) have shown that this obligate intra-cellular bacterium induces centrosome abnormalities in an infected cell. They noted the presence of supernumerary centrosomes by staining with antibodies to γ-tubulin, a marker protein of the pericentriolar matrix (PCM). This phenotype was more pronounced at later time points in the chlamydial developmental cycle. Centrosomes were also mislocalized away from their normal perinuclear position and appeared to be recruited to the chlamydial inclusion, which is the membrane-bound compartment in which chlamydiae grow and replicate. Infected cells displayed chromosome segregation errors consistent with defects of centrosome function in nucleating spindle microtubules during mitosis. Interestingly, these centrosome abnormalities persisted even after cells were cured of Chlamydia with antibiotics.
In uninfected cells, centrosome number is tightly regulated by controlling centrosome duplication during S-phase. The centrosome, which is the major microtubule organizing centre of the cell, is composed of two centrioles surrounded by an electron-dense cloud of about 100 proteins called the PCM (Andersen et al., 2003; Bettencourt-Dias and Glover, 2007). In S-phase, the single centrosome is duplicated through a semi-conservative mechanism, in which each centriole is a template for the formation of a procentriole that ultimately elongates into a daughter centriole (Nigg, 2007; Strnad and Gönczy, 2008). Thus by G2, the centrosome consists of four centrioles organized as two pairs. At the onset of mitosis these centriole pairs migrate to opposite ends of the cell to form the spindle poles. Each daughter cell will inherit one spindle pole, composed of two centrioles, which will eventually become its centrosome. Known regulators of centrosome duplication include the protein kinases, Plk4, Cdk2 and Mps1, and centrosome structural proteins, such as SAS-4 and SAS-6 (Strnad and Gönczy, 2008). Dysregulation of Plk4, SAS-4 or SAS-6 through knockdown or overexpression has been shown to induce alterations in the number of centrioles (Leidel and Gönczy, 2003; Habedanck et al., 2005; Leidel et al., 2005).
Centrosome abnormalities that affect centrosome organization and number are a hallmark of many human cancers including solid tumours of the breast and lung, and haematological malignancies such as leukaemia (Pihan et al., 1998; Giehl et al., 2005; Nigg, 2006). Abnormal centrosomes affect the fidelity of chromosome segregation leading to genetic instability (Pihan et al., 1998; Lingle et al., 2002). Several studies in breast and cervical cancer have detected centrosome abnormalities as early lesions (Lingle et al., 2002; Pihan et al., 2003). Centrosome abnormalities have also been shown to be caused by HPV, a sexually transmitted virus strongly associated with cervical and anogenital cancers (zur Hausen, 2002; Duensing and Munger, 2003; Psyrri and DiMaio, 2008).
In this study, we investigate how supernumerary centrosomes are generated during an infection with C. trachomatis. To distinguish between different mechanisms of formation, we examined the composition of the amplified centrosomes and whether there was an increase in the number of mature centrioles. To determine if the centrosome duplication machinery of the infected cell is involved, we tested if known host factors, such as specific regulatory kinases and cell cycle progression, are necessary for production of supernumerary centrosomes. Our findings indicate that centrosome amplification during a chlamydial infection depends on the host centrosome duplication pathway, although the numerical control of procentriole formation is dysregulated.
Results
The supernumerary centrosomes of Chlamydia-infected cells contain components of the centriole and the pericentriolar matrix
We examined the supernumerary centrosomes generated during a C. trachomatis infection for the presence of additional centrosomal components besides γ-tubulin. We first performed immunofluorescence analysis with antibodies against centrin2, a well-established component of the centriole (Salisbury et al., 2002). Ninety-five per cent of uninfected HeLa cells either had two centrin2 foci, representing the two centrioles of an unduplicated centrosome, or four centrin2 foci found in a duplicated centrosome in G2 or early mitosis (Fig. 1A and B). In contrast, 54% of HeLa cells infected with C. trachomatis serovar L2 contained more than four centrin2 foci (P < 0.0001). As HeLa cells are transformed cells, we repeated the experiment with a primary cell line. We observed a similar increase in the percentage of cells with supernumerary centrin2 foci in primary human foreskin fibroblasts (HFF) (Fig. 1A and B). These results validate the findings of Grieshaber et al. (2006), who demonstrated the presence of supernumerary centrosomes in 60% of Chlamydia-infected HeLa cells by staining for the PCM marker, γ-tubulin.
Fig. 1. Cells infected with Chlamydia trachomatis contain extra centrioles.

A. Epifluorescence images of HeLa and HFF cells stained with antibodies against Chlamydia (red) and the centriolar marker protein, centrin2 (white). The inset in the bottom right hand of each image is a magnified view of the centrioles in a single cell. Cells were also stained with the DNA dye Hoechst 33342 (blue). The scale bar corresponds to 10 μm.
B. The percentage of cells with greater than four centrin2 foci was determined in HeLa and HFF cells that were mock infected (uninfected control) or infected with Chlamydia trachomatis serovar L2 for 36 h (infected). A minimum of 100 cells was quantified for each of three independent experiments. A statistically significant difference between uninfected and infected cells was observed for both HeLa and HFF cells (*P < 0.0001, Z-test).
We then examined additional markers of the centriole and PCM in uninfected and Chlamydia-infected cells. γ-Tubulin colocalized at each of the centrosomal foci with centrin2 and the centriolar marker, C-Nap1, which functions as an intercentriolar linker (Fig. 2, upper panels on the left) (Mayor et al., 2000). Centrin2 also colocalized with HsSAS-6, a marker of the daughter centriole (Strnad et al., 2007). Each centrin2 focus was also associated with the PCM components, kendrin, which is permanently found at the centrosome (Flory and Davis, 2003), and GCP2, a component of the γ-TuRC complex, which is transiently recruited to the centrosome from the cytosol (Fig. 2, upper panels on the right) (Murphy et al., 1998). The supernumerary centrosomes in HFF cells showed a similar staining pattern (Fig. 2, lower panels). These results lead us to conclude that the supernumerary centrosomal foci in Chlamydia-infected cells are bona fide centrosomes consisting of centrioles that are surrounded by the PCM.
Fig. 2.

The supernumerary centrosomes induced by a chlamydial infection contain centriole and pericentriolar matrix markers. The centrosome morphology of uninfected or C. trachomatis-infected HeLa (top two panels) and HFF cells (bottom two panels) was examined by immunofluorescence. In the first two columns from the left, centrin2 and C-Nap1, which are markers of the centriole, are shown in green, and the pericentriolar marker, γ-tubulin, is in red. In the remaining columns, the centriole marker, HsSAS-6, and the pericentriolar matrix markers, kendrin and GCP2, are shown in green, and costained with centrin2 in red. The scale bar corresponds to 2 μm.
Supernumerary centrioles in Chlamydia-infected cells are mostly immature
We next investigated whether multiple centrioles are the result of defective cytokinesis, which has been described in a Chlamydia infection (Greene and Zhong, 2003; Alzhanov et al., 2009). As centrosome duplication occurs before cell division, aborted cytokinesis produces a centrosome amplification phenotype characterized by the presence of multiple mature centrioles (Meraldi et al., 2002). These mature centrioles can be distinguished from immature centrioles by the presence of subdistal appendages that specifically stain with antibodies to Cep170 (Guarguaglini et al., 2005). Ninety-two per cent of uninfected cells (Fig. 3A, top panel) and 71% of Chlamydia-infected cells contained a single Cep170-positive centriole (Fig. 3A, middle panel). In contrast, when we blocked cytokinesis by treating uninfected cells with cytochalasin D, only 3% of cells had a single Cep170-positive focus and 97% had two, three, four or more mature centrioles (Fig. 3A, bottom panel showing a cell with two Cep170-staining foci). Thus, the phenotype of multiple immature centrioles in Chlamydia-infected cells is different from the presence of multiple mature centrioles when cytokinesis is blocked. We conclude that the majority of Chlamydia-infected cells have multiple immature procentrioles that have formed from a single maternal centriole.
Fig. 3. Amplified centrosomes in Chlamydia-infected cells contain predominantly immature centrioles.
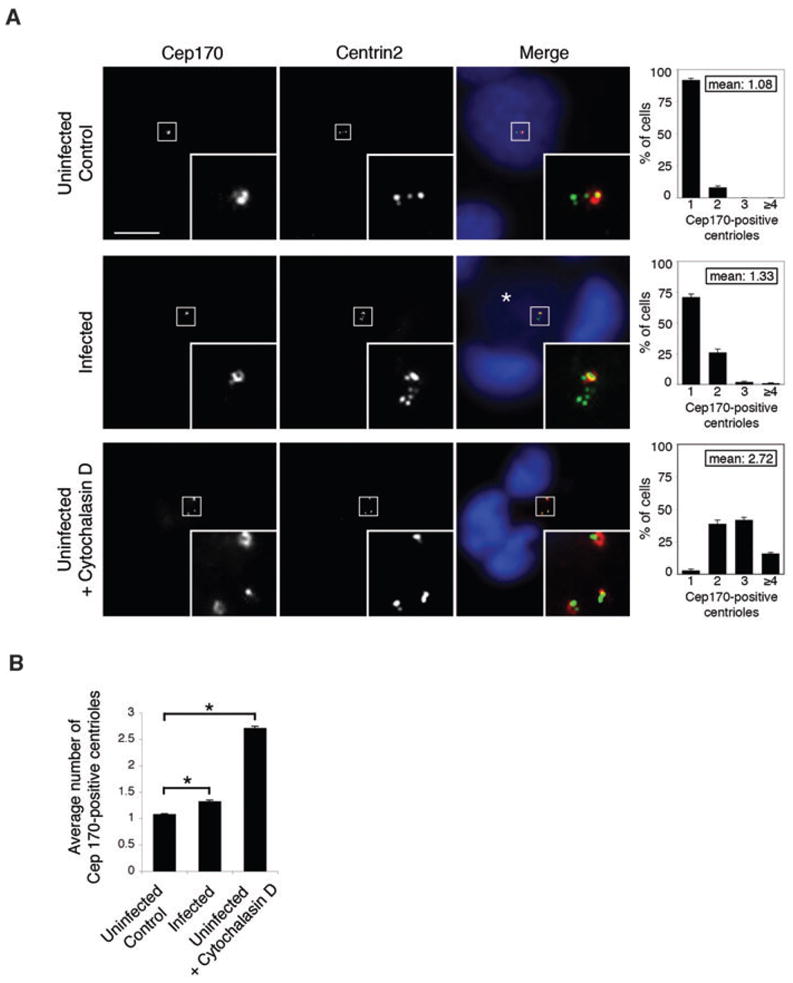
A. Centrioles of uninfected control (upper panel), Chlamydia-infected (middle panel) and cytochalasin D-treated (lower panel) HeLa cells were stained with antibodies to Cep170 and centrin2, as indicated. In the merged images, Cep170 is shown in red, centrin2 in green, and host cell nuclei and chlamydial nucleoids (labelled with an asterisk) are stained with the DNA dye Hoechst 33342 in blue. Each inset, which corresponds to the boxed area in each image, shows a magnified view of the centrioles of a single cell. The scale bar corresponds to 10 μm. The graphs on the right show the percentage of Cep170-positive centrioles for the respective cells. A minimum of 100 cells was analysed per experiment and three independent experiments were performed. The mean number of Cep170-positive centrioles is also shown for each experimental condition.
B. A comparison of the mean number of Cep170-positive centrioles. Statistical analyses using the Student’s t-test and Matlab revealed significant differences between uninfected and infected cells (*P < 0.0001) and between uninfected and cytochalasin D-treated cells (*P < 0.0001).
Our results indicate that cytokinesis was blocked in a small proportion of Chlamydia-infected cells. We detected this effect by measuring a statistically significant difference in the average number of mature centrioles between infected and uninfected cells (P < 0.0001), even though the absolute difference was small (1.3 mature centrioles in infected cells versus 1.1 mature centrioles in uninfected cells, Fig. 3B). For reference, the average number of mature centrioles in cytochalasin D-treated cells was 2.7 (P < 0.0001 compared with untreated, uninfected control cells).
Parameters of the chlamydial infection affect the proportion of cells with increased centrosomes
We next investigated if the time course of infection or the infectious inoculum had an effect on the number of cells with supernumerary centrosomes. In these and subsequent experiments, we defined the presence of supernumerary centrosomes as greater than four centrin2-staining foci. In HeLa cells infected with C. trachomatis at an multiplicity of infection (moi) of 3, we observed little effect on centrosome number at 18 or 24 hours post infection (hpi) (Fig. 4). However, we measured a dramatic increase in the proportion of infected cells with supernumerary centrosomes at 30 and 36 hpi (48% and 60% respectively), which is consistent with published studies in HFF cells (Grieshaber et al., 2006). At an moi of 0.3, we also observed a large increase in cells with supernumerary centrosomes at 30 and 36 hpi but not at 18 or 24 hpi. However, the proportion of infected cells with supernumerary centrosomes was significantly reduced at this lower moi compared with an moi of 3 (33% versus 48% at 30 hpi, *P < 0.0001, and 44% versus 60% at 36 hpi, *P < 0.0001). We obtained similar results in HeLa and HFF cells when we stained with antibodies against ninein, γ-tubulin and kendrin (data not shown). Thus, the prevalence of this centrosome phenotype in infected cells was greater at later time points during a chlamydial infection and when a higher inoculum was used for the infection. Regardless of the moi, however, we did not detect obvious differences in the actual centrosome amplification phenotype at the level of an individual infected cell.
Fig. 4.
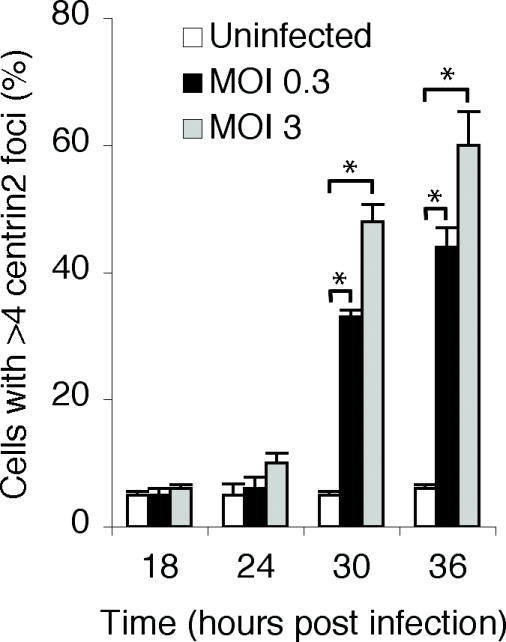
The proportion of cells with supernumerary centrioles increased with the length of chlamydial infection and the infectious inoculum. HeLa cells were infected with C. trachomatis serovar L2 at an moi of 0.3 or 3 and fixed at different times after infection, as indicated, prior to staining with antibodies to centrin2. For each time point, a minimum of 100 cells was counted and the percentage of cells with greater than four centrin2 foci was determined. Three independent experiments were performed for each moi. At both 30 and 36 hpi, there was a statistically significant difference between uninfected and infected cells, and also between cells infected at the higher and lower moi (*P < 0.0001, Z-test).
Centrosome amplification during a chlamydial infection involves the canonical centrosome duplication pathway
To determine if the production of supernumerary centrosomes during a chlamydial infection involves the normal centrosome duplication machinery of the host cell, we examined two known regulators of centrosome duplication. Cdk2 and Plk4 are protein kinases required for formation of a daughter centriole from a template centriole during S-phase (Lacey et al., 1999; Habedanck et al., 2005). Expression of an inactive form of either kinase acts as a dominant negative and blocks centrosome duplication so that cells only contain two centrioles (Fig. 5A). When we expressed GFP-tagged kinase-inactive forms of Cdk2 or Plk4 in Chlamydia-infected cells, only 2–3% of the cells had more than four centrin2 foci, demonstrating that formation of supernumerary centrosomes was abrogated (Fig. 5A and B). In control experiments, 56% of infected but untransfected cells, and 58% of infected cells transfected with an empty GFP vector, contained supernumerary centrosomes (Fig. 5A and B). In contrast, a dominant negative form of the mitotic kinase Cdk1 did not prevent centrosome amplification in Chlamydia-infected cells (Fig. 5A, bottom panel, and Fig. 5B). Cdk1 is required for entry of cells into mitosis, but not for centrosome duplication, demonstrating that the effects of dominant negative Cdk2 and Plk4 are specific. These results indicate that Chlamydia-induced centrosome amplification involves the normal centrosome duplication machinery of the host cell.
Fig. 5. Centrosome amplification during a Chlamydia infection involves the canonical centrosome duplication pathway.

A. Uninfected or Chlamydia-infected HeLa cells were left untreated as a control (top), or transfected with an empty GFP vector (second panel) or with a GFP-tagged dominant negative form of Cdk2, Plk4 or Cdk1 (bottom three panels). Centrosome morphology was visualized by staining cells with antibodies to centrin2 (in red in the merged image, and in white in the magnified images in the insets). The DNA dye Hoechst 33342 was used to stain DNA (blue), and antibodies to Chlamydia were used to detect the chlamydial inclusions (cyan), which are marked with an asterisk. The scale bar corresponds to 5 μm.
B. The percentage of cells with greater than four centrin2 foci for each treatment condition. Data are from three independent experiments with a minimum of 100 cells counted per experiment.
C. The effect of these treatments on the production of infectious progeny. For comparison, the infectious yield was determined for an untreated control infection. Each assay was performed in triplicate. The results are reported as a mean with standard deviations marked by error bars.
The ability to block the production of supernumerary centrosomes with dominant negative Cdk2 or Plk4 provided us with a means to test if these adverse effects on the host centrosome are important for the chlamydial infection itself. Interestingly, the number of infectious progeny recovered from GFP-expressing control cells and cells expressing dominant negative Cdk2 or Plk4 were indistinguishable (Fig. 5C). Thus, it appears that the effects of a C. trachomatis infection on the centrosome are not necessary for efficient replication and propagation of this bacterium in cell culture, although they may have detrimental consequences for the infected host cell.
Centrosome amplification during a chlamydial infection requires progression through S-phase
We performed cell cycle arrest studies to determine if production of supernumerary centrosomes in Chlamydia-infected cells required progression through S-phase, which is the stage in the cell cycle when centrosome duplication occurs under normal circumstances. When we induced an S-phase arrest by blocking DNA synthesis at the G1/S transition with thymidine treatment, the number of infected cells with supernumerary centrosomes was only 12% compared with 62% in asynchronous cells (P < 0.0001) (Fig. 6). In contrast, we did not prevent the formation of supernumerary centrosomes in infected cells when we induced a cell cycle arrest in G2 by expressing a kinase inactive form of Cdk1 to block cells from entering mitosis (Fig. 6). Thus, the increase in centrosome number during a chlamydial infection requires progression through the stage in the host cell cycle when centrosomes are normally duplicated.
Fig. 6.
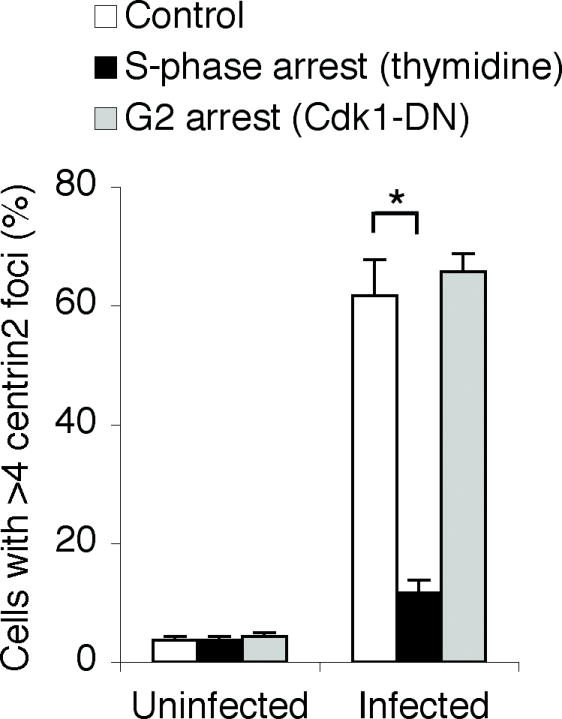
Centrosome amplification during a chlamydial infection requires progression through S-phase. Uninfected or Chlamydia-infected HeLa cells were arrested in S-phase by treatment with thymidine, or in G2, by transfection with GFP-Cdk1-DN, prior to fixation and immunofluorescence analysis with centrin2-specific antibodies. The graph shows the percentage of cells with greater than four centrin2 foci for each treatment condition. Data are from three independent experiments with a minimum of 100 cells counted per experiment (*P < 0.0001, Z-test).
Discussion
In this study, we demonstrate that centrosomal abnormalities observed during a Chlamydia infection result from dysregulation of centrosome duplication. Production of supernumerary centrosomes required the kinase activities of Cdk2 and Plk4, which are known regulators of centrosome duplication, and progression through S-phase, which is the stage in the cell cycle when duplication of the centrosome occurs. Together, these requirements indicate that the centrosome duplication machinery of the host cell is involved, although there is loss of numerical control. The production of multiple immature centrioles leads us to conclude that Chlamydia infection induces the uncontrolled initiation of procentriole formation from the maternal template centriole.
Although blocked cytokinesis and centrosome fragmentation can lead to supernumerary centrosomes, these processes do not appear to be the major cause of the centrosome phenotype in C. trachomatis-infected cells. Chlamydia infection is known to block cytokinesis of the host cell (Greene and Zhong, 2003), and expression of the C. trachomatis gene CT223 in the absence of an infection blocked cytokinesis in 22% of cells (Alzhanov et al., 2009). However, our Cep170 staining experiments showed that Chlamydia-infected cells contain mostly immature centrioles with only a slight increase in the average number of mature centrioles, which differs from the phenotype of multiple mature centrioles seen with aborted cytokinesis. Our results suggest that at most 29% of cells infected with C. trachomatis serovar L2 have a block in cytokinesis, as determined by the presence of more than one mature centriole in a cell. The actual number is likely to be lower, as 8% of uninfected cells had more than one mature centriole, which is indicative of interphase cells that have undergone centrosome duplication but have not yet divided. Thus, while defective cytokinesis may contribute to centrosome amplification during a chlamydial infection, it cannot account for supernumerary centrosomes in 60% of Chlamydia-infected cells, and an additional mechanism to produce multiple immature centrioles must be involved. Multiple centrosomal foci can also result from centrosome fragmentation induced by the depletion of specific structural components of the centrosome (Mikule et al., 2007). However, the supernumerary centrosomes in Chlamydia-infected cells appear to be homogeneous in size and protein composition, and thus do not resemble the centrosome fragmentation phenotype.
It is unclear if centrosome amplification has beneficial consequences for chlamydiae or whether it is purely collateral damage for the infected host cell that may lead to genetic instability. Centrosome amplification did not appear to be necessary for survival and replication of chlamydiae in cell culture, as measured by a progeny assay. However, we do not know if centrosome amplification may have effects on an in vivo infection.
Centrosome amplification has been observed when the levels of specific regulatory or structural components of the centrosome have been experimentally altered. For example, overexpression of the protein kinase, Plk4, or the structural component, HsSAS-6, leads to the formation of multiple immature procentrioles formed at a single template centriole (Habedanck et al., 2005; Leidel et al., 2005; Strnad et al., 2007). These phenotypes are consistent with the centrosome phenotype we have observed in Chlamydia-infected cells, although we do not know whether a Chlamydia infection targets a specific component of the centrosome duplication machinery.
The induction of centrosome abnormalities during infection by an intracellular pathogen has been linked with cancer. The best studied example is the oncogenic virus, HPV, which induces centrosome amplification and chromosomal instability (Duensing and Munger, 2002). Two HPV oncoproteins, E6 and E7, have been shown to cause centriole overduplication characterized by a single mother centriole template and multiple daughter centrioles in a Cdk2-dependent process (Duensing et al., 2006; Duensing et al., 2007). This phenotype appears to be strikingly similar to the centrosome phenotype we have described in Chlamydia-infected cells. Centrosome amplification has also been associated with adult T-cell leukaemia and its causative virus, human T-cell lymphotropic virus (HTLV-1). The Tax protein of HTLV-1 induces centrosome amplification by increasing Cdk2 activity (Nitta et al., 2006), and, by targeting Tax1BP2, a centrosomal protein of the host cell implicated in the control of centrosome duplication (Ching et al., 2006). In contrast to these oncogenic viruses, Chlamydia is the only bacterium to date that has been shown to cause centrosome amplification.
The dysregulation of centrosome duplication by Chlamydia and HPV is intriguing, especially as Chlamydia has been associated with both HPV and cervical cancer. These two pathogens are the main causes of sexually transmitted infection in the USA. While HPV has been firmly established as the causative agent for cervical and anogenital cancers (zur Hausen, 2002; Psyrri and DiMaio, 2008), only a small minority of HPV-infected women develop invasive carcinoma, which suggests that other factors are involved. Both the persistence of HPV infection (Silins et al., 2005) and the prevalence of high-risk HPV types (Samoff et al., 2005) have been associated with C. trachomatis co-infection. In addition to a possible role as a cofactor for HPV, C. trachomatis has been shown to be an independent risk factor for cervical cancer in prospective seroepidemiologic studies (Koskela et al., 2000; Naucler et al., 2007).
In summary, our studies indicate that the supernumerary centrosomes observed during a Chlamydia infection result from dysregulation of the canonical centrosome duplication pathway of the host cell at the stage of procentriole formation. Further exploration of this intriguing host–pathogen interaction may have important implications for the potential role of Chlamydia in cancer development.
Experimental procedures
Antibodies and reagents
Primary antibodies used in this study: mouse anti-centrin2 20H5 (generous gift of Dr Jeffrey L. Salisbury, Mayo Clinic, Rochester, MN, USA), rabbit anti-γ-tubulin and rabbit anti-ninein (Abcam, Cambridge, MA, USA), rabbit anti-kendrin and rabbit anti-GCP2 (kindly provided by Dr Mikiko Takahasi, Kobe University, Japan), rabbit anti-HsSAS6 (generous gift of Dr Karen Oegema, UC San Diego), rabbit anti-Cep170 (kindly provided by Dr Giulia Guarguaglini, Institute of Molecular Biology and Pathology, CNR, Rome, Italy), rabbit anti-Chlamydia muridarum EBs and mouse antibodies to the VD4 epitope of the major outer membrane protein (MOMP) from C. trachomatis serovar E (BOUR) (both kindly provided by Dr Ellena Peterson, UC Irvine), mouse anti-BrdU (kind gift of Dr Vivek Malhotra, Center for Genomic Regulation, Barcelona, Spain), and mouse anti-phospho-Histone H3 (Millipore, Norcross, GA, USA). Secondary antibodies conjugated to Alexa 488, Alexa 594, or Alexa 647 were from Molecular Probes/Invitrogen, Eugene, OR.
Additional reagents used in this study: Hoechst 33342 DNA dye (Molecular Probes/Invitrogen, Eugene, OR, USA), Triton-X 100 (Sigma Aldrich, St Louis, MO, USA), thymidine (Acros Organics/Thermo Fisher Scientific, NJ, USA), cytochalasin D (BIOMOL International, L.P., Plymouth Meeting, PA, USA), and BrdU (provided by Dr Vivek Malhotra, Center for Genomic Regulation).
Cell culture
HeLa cells (ATCC, Manassas, VA, USA) were grown in Advanced DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 2% fetal bovine serum (FBS) (HyClone/Thermo Fisher, Logan, UT, USA) and 2 mM GlutaMAX-I (Invitrogen). HFF cells (ATCC) were cultured in DMEM (Invitrogen) with 10% FBS and 2 mM GlutaMAX-1. Cells were seeded onto glass coverslips in 24-well plates and grown at 37°C in an incubator supplied with 5% CO2. All cell lines used in this study were tested for the absence of Mycoplasma using the MycoTect kit (Gibco/Invitrogen).
Expression constructs
The mammalian expression constructs, pCS399 (pEGFP-Cdk1-DN) and pCS341 (pEGFP-Cdk2-DN), were constructed by cloning the cDNAs for kinase inactive forms of Cdk2 or Cdk1 from either pGEX-Cdk2-DN or pRSET-Cdk1-DN (both gifts from Dr John Newport, UC San Diego) into pEGFP-C3 or pEGFP-C1 (Clonetech, Mountain View). GFP-Plk4-DN was kindly provided by Dr Erich Nigg (Max-Plank Institute of Biochemistry, Martinsried, Germany).
Chlamydia infection and transfection
Monolayers of HeLa or HFF cells were infected with C. trachomatis EBs, serovar L2 (L2/434/Bu), LGV Biovar, at an moi of 3 in sucrose-phosphate-glutamic acid (SPG). Uninfected control experiments were performed as mock infections with SPG alone. Infections were carried out by centrifugation at 700 g in a Beckman Allegra 6 centrifuge for 1 h at room temperature. After centrifugation, the inoculum was replaced with 500 μl fresh cell culture medium and the monolayers were incubated at 37°C and 5% CO2 for 36 h. In experiments to determine the effect of moi and the time in the chlamydial developmental cycle on centrosome morphology, the infections were performed at an moi of 0.3 or 3, and the infected cells were fixed at 18, 24, 30 and 36 hpi.
In experiments in which a protein was exogenously expressed, the expression construct was transfected at 4 hpi with FuGENE 6 (Roche, Basel, Switzerland) according to the manufacturer’s protocol.
Chlamydia trachomatis EBs were tested for Mycoplasma contaminations using Mycoplasma group-specific PCR as described (Ossewaarde et al., 1996).
Immunofluorescence microscopy and quantification
Cells grown on glass coverslips were fixed in −20°C methanol on ice for 7 min, permeabilized and blocked in 10% blocking buffer (0.1% Triton X-100, 10% FBS in PBS) for 30 min. Cells were incubated for 1 h at room temperature with primary antibodies, washed with PBS, incubated for 30 min with secondary antibodies, washed again with PBS and mounted on glass slides using gelvatol. Primary and secondary antibodies were diluted in 2.5% staining buffer (0.1% Triton X-100, 2.5% FBS in PBS). Host and chlamydial DNA were stained with Hoechst 33342 (Molecular Probes/Invitrogen) during incubation with secondary antibodies. Cells were imaged on a Zeiss Axiovert 200 M microscope (Carl Zeiss MicroImaging, Thornwood, NY, USA) and analysed with linear adjustments with the Zeiss Axiovision software. Images were processed using Adobe Photoshop CS3.
For quantifications, the percentage of cells with more than four centrin2 foci were scored in a minimum of 100 cells present in a total of 10 different fields of view. For experiments with a Chlamydia infection, we only counted infected cells. Each experiment was repeated three independent times and a mean and standard deviation were calculated. The statistical significance of our results was determined with the Student’s two-tailed t-test or the Z-test using Matlab software.
Progeny assay
HeLa cells infected at an moi of 3 were transfected with the expression constructs for Plk4-DN, Cdk2-DN and Cdk1-DN at 4 hpi. Cells were lysed at 36 hpi by incubation at −80°C for 15 min, followed by incubation at 37°C for 15 min. Cell lysates and debris were harvested by scraping and washed with SPG. Serial dilutions of the lysates were applied to HeLa cell monolayers in 48-well plates and centrifuged at 700 g in a Beckman Allegra 6 centrifuge for 1 h at room temperature. After 36 h, the cells were fixed and chlamydial inclusions were stained with anti-MOMP and goat anti-mouse secondary antibodies conjugated to horseradish peroxidase. The average number of cells containing inclusions within 10 optical fields, as viewed through the 40× objective of a Zeiss Axiovert 40 CFL microscope (Carl Zeiss MicroImaging, Inc, Thornwood, NY, USA) was used to calculate the number of infectious units per millilitre for each condition.
Cell cycle synchronization
Mock- or C. trachomatis-infected HeLa cells on coverslips were arrested in S-phase at 8 hpi by adding 2 mM thymidine for 24 h. The cells were then either fixed or washed three times and placed in fresh culture medium for 9 h to allow entry into mitosis. S-phase arrest and release were verified by determining the percentage of cells in mitosis as visualized by staining with phospho-Histone H3-specific antibodies.
HeLa cells were synchronized in G2 by transfection with pEGFP-Cdk1-DN. Arrest in G2 was confirmed by comparing the mitotic index of cells transfected with pEGFP-Cdk1-DN and pEGFP-C1 that were released for 9 h from an S-phase arrest induced by treatment with 2 mM thymidine for 20 h.
To block cytokinesis, HeLa cells were treated with 0.6 μg ml−1 cytochalasin D for 48 h as described (Guarguaglini et al., 2005).
BrdU incorporation assay
S-phase-arrested HeLa cells were pulse-labelled with 10 μM BrdU for 30 min. Cells were either fixed or released from the S-phase arrest by washing the cells with PBS and placing them in fresh cell culture medium for 9 h. Fixed cells were denatured with 4 N HCl for 20 min, quenched in 0.1 M sodium borate for 2 min, blocked for 30 min with 10% blocking buffer and stained with anti-BrdU antibody.
Acknowledgments
We thank Dr Giulia Guarguaglini, Dr Vivek Malhotra, Dr John Newport, Dr Erich Nigg, Dr Karen Oegema, Dr Ellena Peterson, Dr Jeffrey Salisbury and Dr Mikiko Takahasi for generously providing us with reagents. We are grateful to Elizabeth DiRusso Case, Johnny Akers, Allan Chen, Eric Cheng and Dr Chris Rosario for critical reading of the manuscript and to Dr Jeffrey Johnson for help with statistical methods. M.T. is supported by an NIH Independent Scientist Award (AI057563).
References
- Alzhanov DT, Weeks SK, Burnett JR, Rockey DD. Cytokinesis is blocked in mammalian cells transfected with Chlamydia trachomatis gene CT223. BMC Microbiol. 2009;9:2. doi: 10.1186/1471-2180-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–574. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- Bettencourt-Dias M, Glover DM. Centrosome biogenesis and function: centrosomics brings new understanding. Nat Rev Mol Cell Biol. 2007;8:451–463. doi: 10.1038/nrm2180. [DOI] [PubMed] [Google Scholar]
- CDC. Summary of notifiable diseases – United States, 2006. MMWR. 2006;55:2–94. [PubMed] [Google Scholar]
- Ching YP, Chan SF, Jeang KT, Jin DY. The retroviral oncoprotein Tax targets the coiled-coil centrosomal protein TAX1BP2 to induce centrosome over-duplication. Nat Cell Biol. 2006;8:717–724. doi: 10.1038/ncb1432. [DOI] [PubMed] [Google Scholar]
- Duensing A, Liu Y, Perdreau SA, Kleylein-Sohn J, Nigg EA, Duensing S. Centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal templates. Oncogene. 2007;26:6280–6288. doi: 10.1038/sj.onc.1210456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing A, Liu Y, Tseng M, Malumbres M, Barbacid M, Duensing S. Cyclin-dependent kinase 2 is dispensable for normal centrosome duplication but required for oncogene-induced centrosome overduplication. Oncogene. 2006;25:2943–2949. doi: 10.1038/sj.onc.1209310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S, Munger K. The human papilloma-virus type 16, E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002;62:7075–7082. [PubMed] [Google Scholar]
- Duensing S, Munger K. Centrosome abnormalities and genomic instability induced by human papilloma-virus oncoproteins. Prog Cell Cycle Res. 2003;5:383–391. [PubMed] [Google Scholar]
- Flory MR, Davis TN. The centrosomal proteins pericentrin and kendrin are encoded by alternatively spliced products of one gene. Genomics. 2003;82:401–405. doi: 10.1016/s0888-7543(03)00119-8. [DOI] [PubMed] [Google Scholar]
- Giehl M, Fabarius A, Frank O, Hochhaus A, Hafner M, Hehlmann R, Seifarth W. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia. 2005;19:1192–1197. doi: 10.1038/sj.leu.2403779. [DOI] [PubMed] [Google Scholar]
- Greene W, Zhong G. Inhibition of host cell cytokinesis by Chlamydia trachomatis infection. J Infect. 2003;47:45–51. doi: 10.1016/s0163-4453(03)00039-2. [DOI] [PubMed] [Google Scholar]
- Grieshaber SS, Grieshaber NA, Miller N, Hackstadt T. Chlamydia trachomatis causes centrosomal defects resulting in chromosomal segregation abnormalities. Traffic. 2006;7:940–949. doi: 10.1111/j.1600-0854.2006.00439.x. [DOI] [PubMed] [Google Scholar]
- Guarguaglini G, Duncan PI, Stierhof YD, Holmstrom T, Duensing S, Nigg EA. The forkhead-associated domain protein Cep170 interacts with Polo-like kinase 1 and serves as a marker for mature centrioles. Mol Biol Cell. 2005;16:1095–1107. doi: 10.1091/mbc.E04-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol. 2005;7:1140–1146. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- Koskela P, Anttila T, Bjorge T, Brunsvig A, Dillner J, Hakama M, et al. Chlamydia trachomatis infection as a risk factor for invasive cervical cancer. Int J Cancer. 2000;85:35–39. doi: 10.1002/(sici)1097-0215(20000101)85:1<35::aid-ijc6>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Lacey KR, Jackson PK, Stearns T. Cyclin-dependent kinase control of centrosome duplication. Proc Natl Acad Sci USA. 1999;96:2817–2822. doi: 10.1073/pnas.96.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidel S, Gönczy P. SAS-4 is essential for centrosome duplication in C. elegans and is recruited to daughter centrioles once per cell cycle. Dev Cell. 2003;4:431–439. doi: 10.1016/s1534-5807(03)00062-5. [DOI] [PubMed] [Google Scholar]
- Leidel S, Delattre M, Cerutti L, Baumer K, Gönczy P. SAS-6 defines a protein family required for centrosome duplication in C. elegans and in human cells. Nat Cell Biol. 2005;7:115–125. doi: 10.1038/ncb1220. [DOI] [PubMed] [Google Scholar]
- Lingle WL, Barrett SL, Negron VC, D’Assoro AB, Boeneman K, Liu W, et al. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci USA. 2002;99:1978–1983. doi: 10.1073/pnas.032479999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor T, Stierhof YD, Tanaka K, Fry AM, Nigg EA. The centrosomal protein C-Nap1 is required for cell cycle-regulated centrosome cohesion. J Cell Biol. 2000;151:837–846. doi: 10.1083/jcb.151.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg EA. Aurora-A over-expression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. Embo J. 2002;21:483–492. doi: 10.1093/emboj/21.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikule K, Delaval B, Kaldis P, Jurcyzk A, Hergert P, Doxsey S. Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat Cell Biol. 2007;9:160–170. doi: 10.1038/ncb1529. [DOI] [PubMed] [Google Scholar]
- Murphy SM, Urbani L, Stearns T. The mammalian gamma-tubulin complex contains homologues of the yeast spindle pole body components spc97p and spc98p. J Cell Biol. 1998;141:663–674. doi: 10.1083/jcb.141.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naucler P, Chen HC, Persson K, You SL, Hsieh CY, Sun CA, et al. Seroprevalence of human papillomaviruses and Chlamydia trachomatis and cervical cancer risk: nested case-control study. J General Virol. 2007;88:814–822. doi: 10.1099/vir.0.82503-0. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Origins and consequences of centrosome aberrations in human cancers. Int J Cancer. 2006;119:2717–2723. doi: 10.1002/ijc.22245. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Centrosome duplication: of rules and licenses. Trends Cell Biol. 2007;17:215–221. doi: 10.1016/j.tcb.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Nitta T, Kanai M, Sugihara E, Tanaka M, Sun B, Nagasawa T, et al. Centrosome amplification in adult T-cell leukemia and human T-cell leukemia virus type 1 Tax-induced human T cells. Cancer Sci. 2006;97:836–841. doi: 10.1111/j.1349-7006.2006.00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossewaarde JM, de Vries A, Bestebroer T, Angulo AF. Application of a Mycoplasma group-specific PCR for monitoring decontamination of Mycoplasma-infected Chlamydia sp. strains. Appl Environ Microbiol. 1996;62:328–331. doi: 10.1128/aem.62.2.328-331.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihan GA, Purohit A, Wallace J, Knecht H, Woda B, Quesenberry P, Doxsey SJ. Centrosome defects and genetic instability in malignant tumors. Cancer Res. 1998;58:3974–3985. [PubMed] [Google Scholar]
- Pihan GA, Wallace J, Zhou Y, Doxsey SJ. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res. 2003;63:1398–1404. [PubMed] [Google Scholar]
- Ponzoni M, Ferreri AJ, Guidoboni M, Lettini AA, Cangi MG, Pasini E, et al. Chlamydia infection and lymphomas: association beyond ocular adnexal lymphomas highlighted by multiple detection methods. Clin Cancer Res. 2008;14:5794–5800. doi: 10.1158/1078-0432.CCR-08-0676. [DOI] [PubMed] [Google Scholar]
- Psyrri A, DiMaio D. Human papillomavirus in cervical and head-and-neck cancer. Nat Clin Pract Oncol. 2008;5:24–31. doi: 10.1038/ncponc0984. [DOI] [PubMed] [Google Scholar]
- Salisbury JL, Suino KM, Busby R, Springett M. Centrin-2 is required for centriole duplication in mammalian cells. Curr Biol. 2002;12:1287–1292. doi: 10.1016/s0960-9822(02)01019-9. [DOI] [PubMed] [Google Scholar]
- Samoff E, Koumans EH, Markowitz LE, Sternberg M, Sawyer MK, Swan D, et al. Association of Chlamydia trachomatis with persistence of high-risk types of human papillomavirus in a cohort of female adolescents. Am J Epidemiol. 2005;162:668–675. doi: 10.1093/aje/kwi262. [DOI] [PubMed] [Google Scholar]
- Silins I, Ryd W, Strand A, Wadell G, Tornberg S, Hansson BG, et al. Chlamydia trachomatis infection and persistence of human papillomavirus. Int J Cancer. 2005;116:110–115. doi: 10.1002/ijc.20970. [DOI] [PubMed] [Google Scholar]
- Strnad P, Gönczy P. Mechanisms of procentriole formation. Trends Cell Biol. 2008;18:389–396. doi: 10.1016/j.tcb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Strnad P, Leidel S, Vinogradova T, Euteneuer U, Khodjakov A, Gönczy P. Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev Cell. 2007;13:203–213. doi: 10.1016/j.devcel.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenilman JM. Chlamydia and cervical cancer: a real association? JAMA. 2001;285:81–83. doi: 10.1001/jama.285.1.81. [DOI] [PubMed] [Google Scholar]