

Abstract

Unsymmetrical dialkyl ketones can be directly prepared by the nickel-catalyzed reductive coupling of carboxylic acid chlorides or (2-pyridyl)thioesters with alkyl iodides or benzylic chlorides. A wide variety of functional groups are tolerated by this process, including common nitrogen protecting groups and C-B bonds. Even hindered ketones flanked by tertiary and secondary centers can be formed. The mechanism is proposed to involve the reaction of a (L)Ni(alkyl)2 intermediate with the carboxylic acid derivative.
Besides being frequently found in natural products, ketones are a nexus for organic synthesis.1 Methods to chemoselectively convert ketones to a variety of functional groups have long been important to organic synthesis, but recently it has also become important for the chemoselective linking of functionalized fragments in vitro and in vivo.2
The most often used method for the synthesis of ketones from carboxylic acid derivatives is acylation of a carbon nucleophile, usually a pre-formed organometallic reagent.1 The synthesis of functionalized dialkyl ketones and hindered dialkyl ketones requires protecting group manipulations and more reactive nucleophiles. The development of transition metal catalysts to mediate these couplings has enabled the use of less reactive carbon nucleophiles,3 such as alkyltin,4 alkylzinc,5,6 dialkylzinc,7 or alkylboron8 derivatives. While these methods have allowed for the synthesis of more functionalized ketones than previously possible, the synthesis of hindered ketones remains challenging9 and acidic protons, such as N-H protons on a biotin precursor, must still be protected.6 A second, less developed route is the coupling of a nucleophilic acyl group with an organic halide (Figure 1).10
Figure 1.

The majority of methods couple a nucleophile and an electrophile, while this work couples two electrophiles.
Both of these approaches rely upon a nucleophilic carbon reagent, which limits commercial availability of these reagents11 and/or limits functional-group compatibility. A method that utilizes more readily available starting materials would be an attractive alternative, especially in discovery efforts.
A potential solution for the synthesis of dialkyl ketones is the direct reductive acylation of alkyl halides, but few reports of such processes exist in the literature.5a,12 The NiCl2/Zn-mediated acylation of alkyl iodides with substituted pyridyl carboxylates was reported by Mukaiyama 30 years ago, but access to the esters required extra steps, an excess of R-I (3 equiv) was needed, and a reaction with a more hindered ester provided a low yield.12c Other studies with either Pd/Zn5a or Ni/e-12e–f found that primary benzylic chlorides could be acylated in good yield, but an excess of benzyl halide was required to overcome competing dimerization and no hindered substrates were reported.
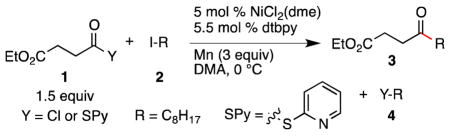
We report here a new catalyst (NiCl2 and 4,4′-di-tert-butyl-2,2′-bipyridine) that is effective for the acylation of alkyl iodides with acid chlorides and (2-pyridyl)thioesters. Even relatively hindered ketones can by synthesized with a nearly equimolar ratio of reactants (1.5 to 1:1 ratio). Functional group tolerance is excellent.
Our previous studies on reductive cross-coupling methods had demonstrated the importance of ligands, especially bipyridine and terpyridine, in preventing the formation of dimeric products, avoiding side reactions of alkyl halides, and improving cross-selectivity.13 4,4′,4″-tri-tert-butyl-2,2′:6′,2″-terpyridine produced large amounts of alkyl dimer product (Entry 3), but the formation of these side products was minimized when 4,4′-di-tert-butyl-2,2′-bipyridine was used as a ligand (Table 1, Entries 1 and 3). As noted by Mukaiyama, reactions conducted without a ligand provided little ketone product (Entry 2).12a
Table 1.
Reaction Optimization and Control Reactions.a
 | ||||
|---|---|---|---|---|
| entry | Y | change in conditions | yield 3 (%)b | yield 4 (%) |
| 1 | Cl | None | 89 | 3 |
| 2 | Cl | No dtbpy | 8 | 7 |
| 3 | Cl | terpyridine in place of dtbpy | 27c | <1 |
| 4 | Cl | 1 equiv 1 instead of 1.5 equiv | 69 | 3 |
| 5 | Cl | 1.5 equiv 2, 1 equiv 1 | 56 | 2 |
| 6 | Cl | No nickel | 6d | 5 |
| 7 | Cl | No nickel, no dtbpy | 8d | 4 |
| 8 | Cl | No Mn | 0d | 25 |
| 9 | Cl | Zn in place of Mn | 41 | 1 |
| 10 | Spy | None | 37 | 43 |
| 11 | Spy | Zn in place of Mn | 53 | 25 |
| 12 | Spy | Pre-stir catalyst with Zn for 1 h | 71 | 10 |
| 13 | Spy | As in Entry 12, but 1 equiv 1 | 71 | 6 |
dtbpy = 4,4′-di-tert-butyl-2,2′-bipyridine. Terpyridine = 4,4′,4″-tri-tert-butyl-2,2′:6′,2″-terpyridine. Reactions were run on 0.5 mmol scale in 2 mL of N,N-dimethylacetamide (DMA). See Supporting Information for details.
Yield determined by GC analysis, uncorrected.
Alkyl dimer was the primary side-product (54% with respect to iodooctane). Alkyl dimer was not a significant byproduct in Entry 1.
>50% of R-I starting material remained.
Reactions run with a slight excess of acid chloride at 0 °C provided higher yields of product, presumably because decomposition of the acid chloride is a side reaction (Entries 4 and 5 vs. Entry 1). Reactions conducted without nickel or reductant did not produce significant product, consistent with a nickel-mediated process (Entries 6–8). Finally, reactions in which zinc was the reductant instead of manganese resulted in product, but with a diminished yield (Entry 9).
While acid chlorides are readily available from carboxylic acids, in some cases more stable starting materials are advantageous. While aryl thioesters and carboxylic acid esters produced only small amounts of product, (2-pyridyl)thioesters were promising substrates (Entry 10). Yields of 3 were diminished by the competitive formation thioether 4 (Y = (2-pyridyl)S-) by liberated (2-pyridyl)thiolate. Changing to zinc as the reductant,14 and ensuring complete coordination of the nickel catalyst before adding substrate allowed an improvement in yield (Entries 11–13).
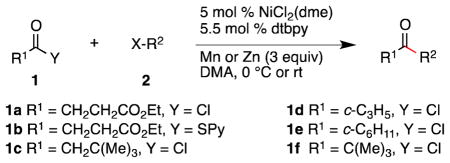
We next applied these conditions to the synthesis of a variety of ketone skeletons (Table 2). Carboxylic acid derivatives which are mono-, di-, and tri-substituted all couple with high efficiency with both primary and secondary organic halides. This is significant because the formation of relatively congested tertiary/secondary ketones has presented a challenge for most of the methods reported previously. Due to competing dimerization, benzylic chlorides provided lower yields than unactivated alkyl iodides (Entries 3 vs. 4 and 7 vs. 8).
Table 2.
Scope of Ketone Synthesisa
 | ||||
|---|---|---|---|---|
| entry | 1 | 2 (R2-X) | product | yield (%)b |
| 1 | 1a | I-C8H17 (2a) |
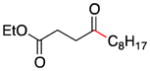 3 |
86 |
| 2c | 1b | 2a | 74 | |
| 3 | 1a |
2c |
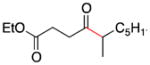 4 |
91 |
| 4d | 1a |
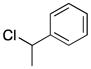 2d |
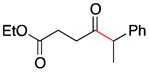 5 |
49e |
| 5 | 1c | 2c |
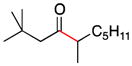 6 |
82 |
| 6 | 1d |
2e |
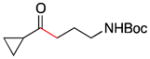 7 |
78 |
| 7 | 1e | 2c |
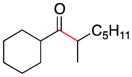 8 |
72 |
| 8f | 1e | 2d |
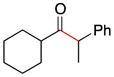 9 |
33 |
| 9 | 1f | 2e |
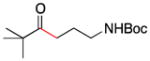 10 |
91 |
| 10 | 1f | 2c |
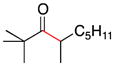 11 |
67 |
General conditions: 1.5 mmol of 1, 1.0 mmol of 2, 0.05 mmol NiCl2(dme), 0.055 mmol 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy), 3 mmol Mn, in 2 mL of DMA at 0 °C for 13–30 h.
Yield of isolated, purified product.
1 Equiv of thioester 1b was used in place of acid chloride 1a and Zn was used in place of Mn.
2 Equiv of 1a was used.
Product contained a small amount of diethyl succinate, yield of 5 determined by NMR.
Run at 30 °C instead of 0 °C.
Although reductive methods are anticipated to have improved functional-group compatibility when compared to the use of organometallic reagents, no previous reports have investigated this concept in depth. Our results appear below in Table 3.
Table 3.
Functional-Group Compatibilitya
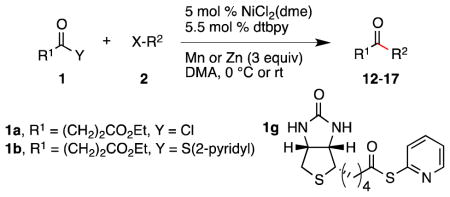
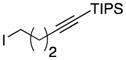
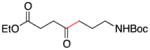
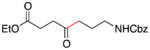
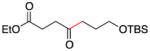
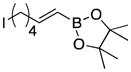
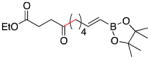
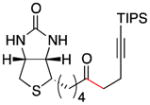
Conditions as in Table 1.
Yield of isolated, purified product.
1 Equiv of thioester 1b was used in place of acid chloride 1a and Zn was used in place of Mn.
The (2-pyridyl)thioester of biotin (1g) and Zn were used in this reaction.
Average of two runs.
Alkyne, protected nitrogen, and silyl ethers were all well tolerated (Entries 1–4). Vinyl boron reagents are useful for subsequent Suzuki coupling reactions and product 16 is obtained without loss of boron or loss of the stereochemical integrity of the olefin.
Functionalized (2-pyridyl)thioesters can be synthesized directly from free carboxylic acids by di-(2-pyridyl)disulfide and triphenylphosphine,15 and the products are stable to aqueous workup and chromatography on silica gel. As an example of the utility of this approach, the thioester of biotin (1g) was synthesized in one step (82% yield). This thioester was then coupled with alkynyl iodide 2f to form ketone 17 (Scheme 1). Although the yield of product is modest, ketone 17 contains three orthogonal reactivities for ligation (1) the ketone can be coupled with O-alkyl amines (oxime formation2), (2) the alkyne can be coupled with organic azides (Huisgen [3+2] cycloaddition2), and (3) the bicyclic urea portion could be used for binding to streptavidin.16 The reaction of organozinc reagents with a biotin-precursor required N-benzyl protection.6b Functionalized biotin constructs have many applications in bioorganic chemistry and biochemistry. However, the typical amide or ester bond linkages can be susceptible to peptidases, which limits utility in vivo.2,17 No biotin conjugates which replace the amide or ester with a ketone have been reported previously.
Scheme 1.

Proposed Catalytic Cycle for Ketone Synthesis
Based upon our observations and those of Mukaiyama,12a Amatore, Jutand, and Périchon,12f a hypothesized mechanism is presented in Scheme 1. We propose that (dtbpy)NiIICl2 (21) is reduced to (dtbpy)Ni0 (18), which then reacts preferentially with the alkyl iodide (R′-I) to form (dtbpy)NiII(R′)(I) (19).12f Rapid disproportionation then forms (dtbpy)NiI2 (21) and (dtbpy)Ni(R′)2 (20).18 Dialkyl complex 20 can react with carboxylic acid derivative 1 to form ketone product 3 and regenerate 19. Yamamoto reported that (bpy)Ni(Et)2 reacted with EtC(O)Cl to form (bpy)Ni(Et)(Cl) and EtC(O)Et in 68% yield in only 10 min.18a While thioesters were not examined in that study, an ester and an anhydride were reported to react slowly with the related complex (Et3P)2Ni(Me)2 to form ketone products in good yield. The mechanism of the formation of 3 and complex 19 from complex 20 and an acid chloride 1 is unclear and is under investigation in our laboratories.
In conclusion, the direct reductive coupling of both carboxylic acid chlorides and (2-pyridyl)thioesters with alkyl iodides and benzylic chlorides produces unsymmetrical dialkylketones in good to excellent yield. The simple protocol and high functional-group tolerance will enable the synthesis of multifunctional products without recourse to extensive protecting group manipulations.
Supplementary Material
Acknowledgments
Financial support from the NIH is gratefully acknowledged (GM097243-01). Ruja Shrestha (University of Rochester) is thanked for running several reactions and for assistance in characterization.
Footnotes
Supporting Information Available. Detailed experimental procedures and full characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Dieter RK. Tetrahedron. 1999;55:4177. [Google Scholar]; (b) Lawrence JN. Perkin Trans. 1998;1:1739. [Google Scholar]; (c) Singh J, Satyamurthi N, Aidhen I. J Prakt Chem. 2000;342:340. [Google Scholar]
- 2.Ketones form stable linkages with hydrazide, hydroxylamino, and thiosemicarbazide groups under physiological conditions: Stephanopoulos N, Francis MB. Nat Chem Biol. 2011;7:876. doi: 10.1038/nchembio.720.Kalia J, Raines RT. Curr Org Chem. 2010;14:138. doi: 10.2174/138527210790069839.Sletten EM, Bertozzi CR. Angew Chem, Int Ed. 2009;48:6974. doi: 10.1002/anie.200900942.
- 3.An alternative strategy is the use of functionalized RMgX: Scheiper B, Bonnekessel M, Krause H, Fürstner A. J Org Chem. 2004;69:3943. doi: 10.1021/jo0498866.
- 4.(a) Milstein D, Stille JK. J Am Chem Soc. 1978;100:3636. [Google Scholar]; (b) Li H, Yang H, Liebeskind LS. Org Lett. 2008;10:4375. doi: 10.1021/ol8018456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Sato T, Naruse K, Enokiya M, Fujisawa T. Chem Lett. 1981;10:1135. [Google Scholar]; (b) Negishi E-i, Bagheri V, Chatterjee S, Luo F-T, Miller JA, Stoll AT. Tetrahedron Lett. 1983;24:5181. [Google Scholar]; (c) Tamaru Y, Ochiai H, Sanda F, Yoshida Z-i. Tetrahedron Lett. 1985;26:5529. [Google Scholar]; (d) Harada T, Kotani Y, Katsuhira T, Oku A. Tetrahedron Lett. 1991;32:1573. [Google Scholar]; (e) Asaoka M, Kosaka A, Tanaka M, Ueda T, Houkawa T, Takei H. Perkin Trans. 1997;1:2949. [Google Scholar]; (f) Wang D, Zhang Z. Org Lett. 2003;5:4645. doi: 10.1021/ol035801w. [DOI] [PubMed] [Google Scholar]; (g) Yus M, Ortiz R. Eur J Org Chem. 2004;2004:3833. [Google Scholar]
- 6.Fukuyama T, Tokuyama H. Aldrichimica Acta. 2005;37:87.for references on industrial uses, see: Mori Y, Seki M. Adv Synth Catal. 2007;349:2027.
- 7.Zhang Y, Rovis T. J Am Chem Soc. 2004;126:15964. doi: 10.1021/ja044113k. [DOI] [PubMed] [Google Scholar]
- 8.(a) Tatamidani H, Kakiuchi F, Chatani N. Org Lett. 2004;6:3597. doi: 10.1021/ol048513o. [DOI] [PubMed] [Google Scholar]; (b) Yu Y, Liebeskind LS. J Org Chem. 2004;69:3554. doi: 10.1021/jo049964p. [DOI] [PubMed] [Google Scholar]; (c) Zhang Z, Lindale MG, Liebeskind LS. J Am Chem Soc. 2011;133:6403. doi: 10.1021/ja200792m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metal-catalyzed coupling of tertiary carboxylic acid derivatives with organometallic reagents to form 3°/1°-dialkyl ketones: 4a, 8b, 5g, 12d; 3°/2°-dialkyl ketones - 5d, and 5e.
- 10.Schmink JR, Krska SW. J Am Chem Soc. 2011;133:19574. doi: 10.1021/ja2064318. [DOI] [PubMed] [Google Scholar]
- 11.Ten times more alkyl iodides are commercially available than alkyl organometallic reagents – 70 AlkylZnBr vs. 958 alkyl iodides.
- 12.(a) Habeeb JJ, Tuck DG. Chem Commun. 1976:696. [Google Scholar]; (b) Shono T, Nishiguchi I, Ohmizu H. Chem Lett. 1977;6:1021. [Google Scholar]; (c) Onaka M, Matsuoka Y, Mukaiyama T. Chem Lett. 1981;10:531. [Google Scholar]; (d) d’Incan E, Sibille S, Périchon J, Moingeon M-O, Chaussard J. Tetrahedron Lett. 1986;27:4175. [Google Scholar]; (e) Marzouk H, Rollin Y, Folest JC, Nédélec JY, Périchon J. J Organomet Chem. 1989;369:C47. [Google Scholar]; (f) Amatore C, Jutand A, Périchon J, Rollin Y. Monatsh Chem. 2000;131:1293. [Google Scholar]
- 13.(a) Everson DA, Shrestha R, Weix DJ. J Am Chem Soc. 2010;132:920. doi: 10.1021/ja9093956. [DOI] [PubMed] [Google Scholar]; (b) Prinsell MR, Everson DA, Weix DJ. Chem Commun. 2010;46:5743. doi: 10.1039/c0cc01716g. [DOI] [PubMed] [Google Scholar]; (c) Shrestha R, Weix DJ. Org Lett. 2011;13:2766. doi: 10.1021/ol200881v. [DOI] [PubMed] [Google Scholar]
- 14.Eichman CC, Stambuli JP. J Org Chem. 2009;74:4005. doi: 10.1021/jo900385d. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi S, Iimori T, Izawa T, Ohno M. J Am Chem Soc. 1981;103:2406. [Google Scholar]
- 16.Trifunctional substrates containing biotin have been used to find drug targets, for radiothereapy approaches, for the study of cell function. See: Tsai CS, Liu PY, Yen HY, Hsu TL, Wong CH. Chem Commun. 2010;46:5575. doi: 10.1039/c0cc00345j.Adam GC, Sorensen EJ, Cravatt BF. Mol Cell Proteomics. 2002;1:828. doi: 10.1074/mcp.t200007-mcp200.
- 17.For a summary of the utility of biotin conjugates, the challenges associated with hydrolysis of amide linkers, and the synthesis of a biotin-derived alkyne without an amide or ester likange, see: Corona C, Bryant BK, Arterburn JB. Org Lett. 2006;8:1883. doi: 10.1021/ol060458r.
- 18.(a) Yamamoto T, Kohara T, Yamamoto A. Bull Chem Soc Jpn. 1981;54:2010. [Google Scholar]; (b) Yamamoto T, Kohara T, Osakada K, Yamamoto A. Bull Chem Soc Jpn. 1983;56:2147. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.