

Background: Cyclodextrin (CD) mobilizes stored cholesterol and delays neurodegeneration in Niemann-Pick C (NPC) mice.
Results: 0.1 mm CD increased, whereas 1 mm decreased, ER cholesterol; 10 mm CD was neurotoxic.
Conclusion: CD liberated cholesterol from lysosomes of NPC1-deficient neurons and glia and modulated cholesterol homeostasis.
Significance: A therapeutic dose of CD for NPC patients is indicated.
Keywords: Astrocytes, Cholesterol Regulation, Lysosomes, Neurodegenerative Diseases, Neurons
Abstract
Niemann-Pick C (NPC) disease is an inherited, progressive neurodegenerative disorder caused by mutations in the NPC1 or NPC2 gene that result in an accumulation of unesterified cholesterol in late endosomes/lysosomes (LE/L) and impaired export of cholesterol from LE/L to the endoplasmic reticulum (ER). Recent studies demonstrate that administration of cyclodextrin (CD) to Npc1−/− mice eliminates cholesterol sequestration in LE/L of many tissues, including the brain, delays neurodegeneration, and increases lifespan of the mice. We have now investigated cholesterol homeostasis in NPC1-deficient cells of the brain in response to CD. Primary cultures of neurons and glial cells from Npc1−/− mice were incubated for 24 h with 0.1 to 10 mm CD after which survival and cholesterol homeostasis were monitored. Although 10 mm CD was profoundly neurotoxic, and altered astrocyte morphology, 0.1 and 1 mm CD were not toxic but effectively mobilized stored cholesterol from the LE/L as indicated by filipin staining. However, 0.1 and 1 mm CD altered cholesterol homeostasis in opposite directions. The data suggest that 0.1 mm CD releases cholesterol trapped in LE/L of neurons and astrocytes and increases cholesterol availability at the ER, whereas 1 mm CD primarily extracts cholesterol from the plasma membrane and reduces ER cholesterol. These studies in Npc1−/− neurons and astrocytes establish a dose of CD (0.1 mm) that would likely be beneficial in NPC disease. The findings are timely because treatment of NPC disease patients with CD is currently being initiated.
Introduction
Dysregulation of cholesterol metabolism in the brain has been implicated in neurodegenerative disorders such as Alzheimer disease, Huntington disease, Smith-Lemli-Opitz syndrome, and Niemann-Pick type C (NPC)2 disease. NPC disease is a fatal, progressive, autosomal recessive disorder caused by mutations in the NPC1 or NPC2 gene (1, 2). The symptoms of NPC disease, including progressive neurodegeneration as well as liver and lung disease, typically begin in childhood and cause premature death (reviewed in Ref. 3). The NPC1 and NPC2 proteins act sequentially in releasing cholesterol derived from endocytosed LDL from late endosomes/lysosomes (LE/L) (4). Thus, in NPC1- or NPC2-deficient cells unesterified cholesterol and other lipids are sequestered in LE/L so that cholesterol transport from LE/L to the plasma membrane (5) and endoplasmic reticulum (ER) (6) is attenuated. The ER cholesterol concentration dictates the processing of sterol response element-binding protein-2 (SREBP2), which transcriptionally regulates the expression of many genes involved in cellular cholesterol homeostasis (7). Thus, when the ER cholesterol content exceeds ∼5% of total lipids (6), the expression of genes involved in cholesterol accretion is reduced whereas cholesterol storage as cholesteryl esters (CE), and expression of genes involved in cholesterol efflux, are increased. Consequently, cholesterol homoestasis is impaired in NPC1-deficient cells.
A treatment for NPC disease has been elusive. However, Liu et al. (8) reported recently that a single injection of 2-hydroxypropyl-β-cyclodextrin (CD) into 7-day-old Npc1−/− mice delayed neurodegeneration and significantly prolonged survival. Moreover, serial injections of CD further increased lifespan of Npc1−/− as well as Npc2−/− mice (9, 10). The CD treatments reduced cholesterol accumulation in LE/L, increased the amount of CE, and decreased cholesterol synthesis in vivo in livers and brains of Npc1−/− mice (8). CD also decreased the amounts of mRNAs encoding SREBP2 and its target genes and increased mRNAs involved in cholesterol efflux in several tissues (8, 9). CD binds cholesterol with high affinity (11), and a high dose of CD (5–10 mm for 10 min) is often used experimentally to extract cholesterol from the plasma membrane of cells but can cause cell death (12). Lower doses of CD, however, appear to enter cells by bulk-phase endocytosis and bypass functions of NPC1/NPC2 so that sequestered cholesterol is mobilized from the LE/L (13). Although CD crosses the blood-brain barrier of mice inefficiently, some (∼0.2%) plasma CD does enter the brain (14).
These findings indicate that CD might be useful for treatment of NPC patients. CD is relatively non-toxic and is used in humans for drug delivery (12). In 2010, the FDA approved peripheral injection of CD for “compassionate use” in NPC patients and direct delivery of CD to the brain is being considered currently. Nevertheless, many questions remain concerning the mechanism by which CD acts in the brain, the organ most profoundly affected in NPC disease. Because previous studies with CD were performed in intact Npc1−/− mice (8, 9, 14), the types of brain cells affected by CD were unknown. Thus, we have established the dose of CD that is tolerated by neurons and glial cells from Npc1−/− mice and have investigated mechanisms by which CD produces beneficial effects on cholesterol homeostasis in these cells.
EXPERIMENTAL PROCEDURES
Materials
2-Hydroxypropyl-β-cyclodextrin (H107), poly-d-lysine hydrobromide, trypsin (type XII-S, bovine pancreas), and filipin were from Sigma. DMEM, Ham's F12 medium, NeurobasalTM medium, FBS, B-27 supplement, 0.25% trypsin-EDTA, Hoechst 33258 pentahydrate, DNase I (amplification grade), oligo(dT)12–18 primer, SuperScript® II, Platinum® qPCR SuperMix-UDG, and SYBR Green I were from Invitrogen. Culture flasks (75-cm2), 96-well plates, and 100 × 20 mm culture dishes were from BD Falcon (BD Biosciences), and 60 × 15 mm culture dishes were from Corning, Inc. (Corning, NY). [1-14C]Oleic acid was from PerkinElmer Life Sciences, and sodium [3H]acetate and CytoScintTM were from MP Biomedicals (Solon, OH). PCR-grade dNTPs (deoxynucleoside triphosphates) were from Roche Applied Sciences.
Npc1+/+ and Npc1−/− Mice
A breeding colony of Balb/cNctr-NpcN/+ mice from Jackson Laboratories (Bar Harbor, ME) was maintained at the University of Alberta under temperature-controlled conditions with a 12-h light:12-h dark cycle. Npc1+/− mice were used for breeding. Mice were supplied with 9% fat breeders' diet (Purina LabDiet, Richmond, IN) and water ad libitum. The Npc1 genotype was determined by PCR analysis of genomic DNA from tail clippings using REDExtract-N-AmpTM tissue PCR kit (Sigma) (23). Npc1+/+ littermates were used as controls. All experiments were approved by the University of Alberta Animal Welfare Committee.
Culture of Cortical Astrocytes and Microglia
Glial cells were isolated from cerebral cortices of 1- to 3-day-old Npc1+/+ and Npc1−/− mice (23, 24) and plated at a density of 2 cortices/75-cm2 flask or one cortex/100 × 20 mm dish. Cells were maintained at 37 °C and 5% CO2 in DMEM containing 10% FBS. The medium was replaced every 3–5 days. After 3–4 weeks, confluent glial cells were washed in phosphate-buffered saline, harvested with 0.125% trypsin and replated at a density of 1:3 in DMEM containing 10% FBS. The cultures were highly enriched (>90%) in astrocytes (24) and were used within 7–14 days. Microglia were isolated from confluent glial cultures by mild trypsinization (25).
Culture of Cerebellar Granule Neurons
Dissociated cerebellar granule neurons from 7- to 8-day-old mice (26) were resuspended in NeurobasalTM medium containing 2% B27, 0.5 mm glutamine and 20 mm KCl and then passed through a 40-μm cell strainer to remove clumped cells. Cells were plated in 96-well plates (31,250 cells/well), 100 × 20 mm dishes (2.1 × 106 cells/dish), 75-cm2 flasks (3.1 × 106 cells/flask) or 60 × 15 mm dishes (1.0 × 106 cells/dish) coated with 10 μg/ml poly-d-lysine, and maintained at 37 °C in 5% CO2.
Cyclodextrin Treatment
A sterilized stock solution of CD (H107, Sigma) was prepared in water. Astrocytes were given fresh DMEM containing 10% FBS 24 h prior to experiments. Medium was replaced with DMEM containing indicated amounts of CD without serum. Vehicle-treated cells were given an equivalent volume of water.
Staining with Filipin and Hoechst Dye
Neurons and astrocytes were cultured for 7 days in 96-well plates and then washed and fixed for 15 min in 4% (w/v) paraformaldehyde. For assessment of cell death by apoptosis, cells were stained at room temperature for 12 min with 0.5 μg/ml Hoechst 33258. Stained nuclei were counted, and cells containing shrunken or fragmented nuclei were scored as apoptotic (24). The number of apoptotic cells was calculated as % of total number of neurons. For qualitative assessment of cholesterol distribution cells were stained for 1.5 h with 0.15 mg/ml filipin (23). Cells were examined with a Leica DM IRE2 fluorescence microscope with an excitation wavelength of 351 nm for Hoechst and filipin.
Radiolabeling of Cholesterol and CE
Neurons and astrocytes were cultured for 7 days in 60 × 15 mm dishes prior to CD treatment. For cholesterol labeling, cells were incubated with 5 μCi/ml [3H]acetate and 100 μm sodium acetate for 4 h. For cholesteryl esterification assays, astrocytes or neurons were incubated for 4 or 24 h, respectively, with 0.2 μCi/ml [14C]oleate, 100 μm oleic acid, 0.5% bovine serum albumin ± 2 μg/ml Sandoz 58-035 (Sigma). Lipids were extracted with chloroform/methanol (2:1), washed with methanol/water (1:1), and separated by thin-layer chromatography in the solvent system heptane/isopropyl ether/acetic acid/isopropanol (65:35:4:2). Bands corresponding to cholesterol, CE, and phospholipids were scraped, and radioactivity was quantified. Protein concentrations were determined using the BCATM protein assay (Thermo Scientific, Rockford, IL).
RNA Isolation and Real-time qPCR
Neurons were cultured for 7 days in 75-cm2 flasks, whereas astrocytes were cultured for 14 days in 100 × 20 mm dishes. Total RNA was isolated using the RNeasy mini kit (Invitrogen) and stored at −80 °C. RNA was treated with DNase I, then cDNA was synthesized from 1.25 μg total RNA using oligo(dT)12–18 random primers and Superscript II reverse transcriptase according to the manufacturer's instructions. qPCR reactions were performed with Platinum® Quantitation PCR supermix, SYBR Green I, and 250 nmol of gene-specific primers. qPCR analysis was performed on a Rotor-Gene 3000 instrument (Montreal Biotech, Montreal, QC). Transcripts were quantified relative to GAPDH mRNA with Rotor-Gene 6.0.19 software and a standard curve. Primers (supplemental Table S1) were synthesized by IDT Technologies (San Diego, CA).
Statistical Analysis
Statistical significance of difference was determined by the Student's t test with differences considered significant for p < 0.05.
RESULTS
Cholesterol is sequestered in LE/L of NPC1-deficient cells because the export of LDL-derived cholesterol from LE/L is impaired (15). Qualitative comparison of filipin staining of unesterified cholesterol in primary cultures of cerebellar granule neurons (Fig. 1, A and B), cortical astrocytes (Fig. 1, C and D) and cortical microglia (Fig. 1, E and F) from Npc1+/+ and Npc1−/− mice shows that NPC1 deficiency in all these cells induces an intracellular accumulation of unesterified cholesterol in a punctate distribution typical of that in other Npc1−/− cells. In Npc1+/+ neurons, astrocytes, and microglia, filipin staining was much less pronounced than in Npc1−/− cells (Fig. 1, A–F).
FIGURE 1.

Intracellular cholesterol sequestration in Npc1−/− neurons and glia. Cerebellar granule neurons (A and B), cortical astrocytes (C and D), and cortical microglia (E and F) from Npc1−/− and Npc1+/+ mice were cultured for 7 days. The distribution of unesterified cholesterol was qualitatively assessed by filipin staining. Similar results were obtained in at least three additional experiments.
Low Concentrations of CD Are Not Toxic and Reduce Cholesterol Sequestration in Npc1−/− Neurons and Astrocytes
High concentrations of CD (5–10 mm) typically deplete cells of cholesterol and can cause cell death (11, 12, 16). Nevertheless, CD is more toxic to some cell types than to others (reviewed in Ref. 16). To determine which CD concentrations are tolerated by neurons and astrocytes from Npc1−/− and Npc1+/+ mice, we incubated the cells for 24 h with 0.1–10 mm CD. Phase-contrast microscopy revealed that morphology of Npc1+/+ (Fig. 2, A–C) and Npc1−/− (Fig. 3, A–C) neurons was unaltered by 0.1 or 1 mm CD. However, no Npc1+/+ (Fig. 2D) or Npc1−/− (Fig. 3D) neurons survived after a 24-h incubation with 10 mm CD. Treatment of Npc1+/+ (Fig. 2, E–G and J) or Npc1−/− (Fig. 3, E–G and J) astrocytes with 0.1 or 1 mm CD did not change cell morphology or induce cell death, whereas 10 mm CD markedly altered morphology of Npc1+/+ (Fig. 2H) and Npc1−/− (Fig. 3H) astrocytes, although the detrimental effect in astrocytes was not as profound as in neurons. The number of apoptotic nuclei in the cultured Npc1+/+ and Npc1−/− neurons and astrocytes was quantified by Hoechst staining. Neither 0.1 nor 1 mm CD decreased survival of Npc1+/+ (Fig. 2, I and J) or Npc1−/− (Fig. 3, I and J) neurons. Thus, exposure of neurons and astrocytes to 0.1 and 1 mm CD for 24 h does not alter cell morphology or compromise survival. We performed no further experiments with 10 mm CD because this concentration was neurotoxic.
FIGURE 2.

Low doses (0.1 and 1 mm) of CD are not toxic to Npc1+/+ mouse neurons or astrocytes. Representative phase contrast images of 7-day-old Npc1+/+ cerebellar granule neurons (A–D) and cortical astrocytes (E–H) incubated for 24 h with vehicle or the indicated CD concentration. Cells were fixed with paraformaldehyde and stained with Hoechst dye for assessment of cell death by apoptosis. The number of apoptotic nuclei was quantified as % of total number of neurons (I) and astrocytes (J). Data are means ± S.E. for >750 cells from three independent cell preparations. For CD versus vehicle (Veh), p > 0.05.
FIGURE 3.

Low doses of CD (0.1 and 1 mm) are not toxic to Npc1−/− neurons or astrocytes. Representative phase contrast images of 7-day-old Npc1−/− cerebellar granule neurons (A–D) and cortical astrocytes (E–H) incubated for 24 h with vehicle or the indicated CD concentration. Cells were fixed with paraformaldehyde and stained with Hoechst dye for assessment of cell death by apoptosis. The number of apoptotic nuclei was quantified as % of total number of neurons (I) and astrocytes (J). Data are means ± S.E. for >750 cells from three independent cell preparations. For CD versus vehicle (Veh), p > 0.05.
To determine whether 0.1 and 1 mm CD released sequestered cholesterol from LE/L of Npc1−/− neurons, astrocytes, and microglia, the cells were incubated with CD for 24 h and then examined by filipin staining. The intracellular punctate filipin staining essentially was eliminated in all three types of Npc1−/− cells (Fig. 4). Weak filipin staining, primarily on the cell surface, was evident in Npc1+/+ neurons (Fig. 5, A–C) and astrocytes (Fig. 5, D–F) and was not significantly altered by CD. These data indicate that low doses (0.1 and 1 mm) of CD mobilized LE/L cholesterol in Npc1−/− neurons and glia.
FIGURE 4.

CD reduces cholesterol sequestration in Npc1−/− neurons and glia. Filipin staining of unesterified cholesterol in 7-day-old Npc1−/− cerebellar granule neurons (A–C), astrocytes (D–F), and microglia (G–I) incubated for 24 h with vehicle (Veh) or indicated CD concentration. Shown are representative images from three independent cell preparations.
FIGURE 5.
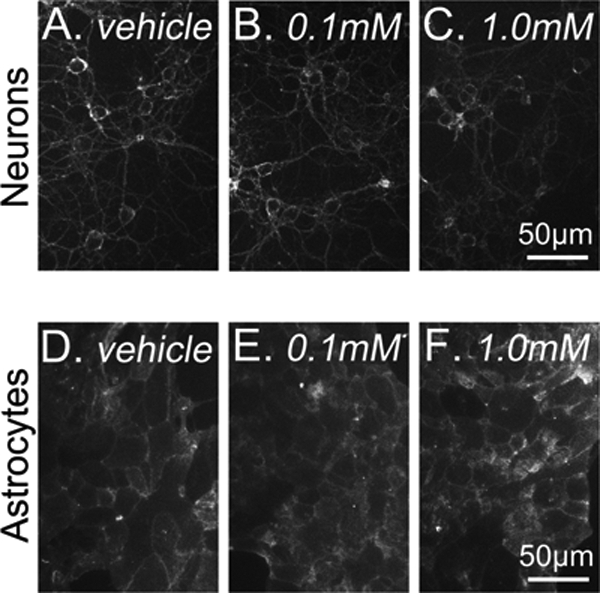
CD does not alter cholesterol distribution in Npc1+/+ neurons or astrocytes. Filipin staining of Npc1+/+ neurons (A–C) and astrocytes (D–F) incubated for 24 h with vehicle (Veh) or indicated CD concentration. Shown are representative images from three independent cell preparations.
Modulation of Cholesterol Metabolism in Npc1−/− Neurons and Astrocytes by CD
Previous studies in Npc1−/− fibroblasts showed that because cholesterol is sequestered in LE/L, the amount of cholesterol transported to the plasma membrane (5), and ER (6) is reduced. Consequently, NPC1 deficiency increases cholesterol synthesis and uptake in several types of cells (15). We, therefore, assessed cholesterol synthesis in Npc1+/+ and Npc1−/− neurons and astrocytes by incubating the cells for 4 h with [3H]acetate. [3H]acetate incorporation into cholesterol was not increased by NPC1 deficiency in neurons (Fig. 6A) or astrocytes (Fig. 6B). Similarly, we previously observed that NPC1 deficiency did not increase [3H]acetate incorporation into cholesterol in mouse sympathetic neurons.3 As an indication that [3H]acetate uptake and the pool size of the acetate precursor of cholesterol were unaffected by the Npc1 genotype, Fig. 6 demonstrates that radiolabeling of phospholipids in neurons and astrocytes was independent of the Npc1 genotype Thus, unlike other types of Npc1−/− cells (15), NPC1 deficiency in cerebellar neurons and cortical astrocytes does not appear to enhance cholesterol synthesis. These observations are consistent with the report of Liu et al. (8) who observed no difference between cholesterol synthesis in brains of Npc1+/+ and Npc1−/− mice.
FIGURE 6.
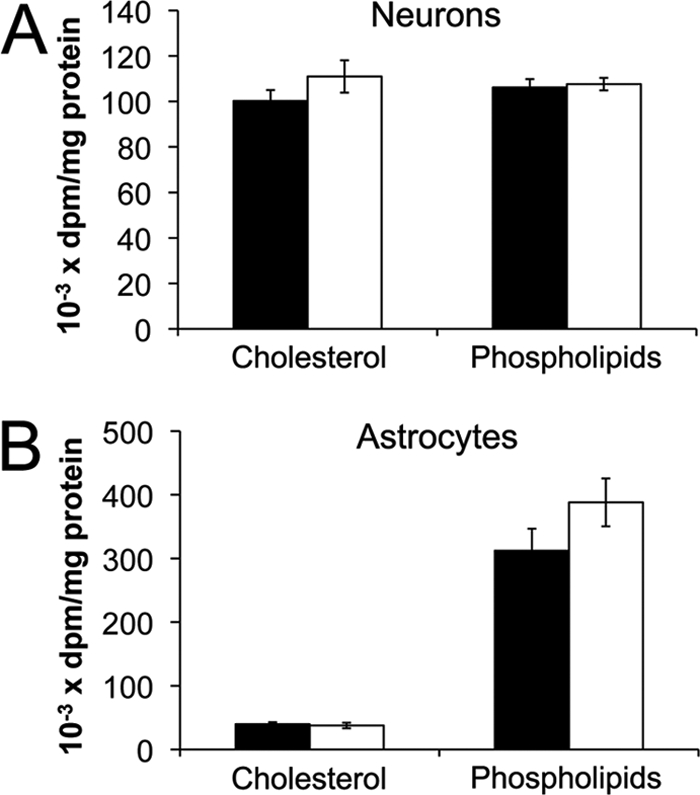
Incorporation of [3H]acetate into cholesterol of Npc1+/+ and Npc1−/− neurons and astrocytes. Neurons (A) and astrocytes (B) from Npc1+/+ (black) and Npc1−/− (white) mice were cultured for 7 days then incubated with [3H]acetate for 4 h. Lipids were extracted, separated by thin-layer chromatography, and radioactivity was quantified in cholesterol and phospholipids. Data for neurons are means ± S.E. of duplicate measurements from three independent experiments, and for astrocytes, data are means ± S.E. from four independent experiments.
Studies in intact Npc1−/− mice suggest that CD can release cholesterol from LE/L and increase the amount of ER cholesterol available for regulation of cholesterol homeostasis (8, 17). To determine whether CD mobilized cholesterol from LE/L to the ER and decreased cholesterol synthesis, in primary cultures of Npc1−/− neurons and astrocytes, the cells were incubated for 28 h with vehicle alone, or with 0.1 or 1 mm CD; [3H]acetate was included for the final 4 h. In Npc1−/− neurons (Fig. 7A) and astrocytes (Fig. 7C) 0.1 mm CD significantly decreased [3H]cholesterol by 28 and 37%, respectively. In contrast, [3H]cholesterol was not decreased by 0.1 mm CD in Npc1+/+ astrocytes (Fig. 7D) and was even slightly increased in Npc1+/+ neurons (Fig. 7B), supporting the hypothesis that 0.1 mm CD increases ER cholesterol in Npc1−/− cells. If, on the other hand, CD had extracted cholesterol from the plasma membrane and ER, one would have expected cholesterol synthesis to be increased. Accordingly, the incorporation of [3H]acetate into cholesterol was increased by 1 mm CD in Npc1−/− and Npc1+/+ neurons (Fig. 7, A and B) and astrocytes (Fig. 7, C and D). Importantly, CD did not significantly alter [3H]acetate incorporation into phospholipids (Fig. 7), implying that [3H]acetate uptake and the intracellular pool size of acetate were not affected by CD. Thus, 0.1 mm and 1 mm CD induce opposite effects on cholesterol synthesis in both neurons and astrocytes.
FIGURE 7.

CD modulates [3H]acetate incorporation into cholesterol in neurons and astrocytes. Neurons and astrocytes were incubated for 28 h with vehicle (dark gray), 0.1 mm CD (light gray), or 1 mm CD (white); [3H]acetate was included for the final 4 h. Lipids were separated by thin-layer chromatography, and radioactivity was quantified in cholesterol and phospholipids. Data for neurons are means ± S.E. from duplicate determinations of three independent cell preparations. The experiments with Npc1+/+ neurons were performed independently of those with Npc1−/− neurons and therefore are not comparable. For astrocytes, the data are means ± S.E. from four independent cell preparations. The experiments with Npc1+/+ and Npc1−/− astrocytes were performed concurrently. *, p < 0.05; ***, p < 0.001.
Cholesterol Esterification in Astrocytes and Neurons
Excess ER cholesterol can be esterified to CE by the ER enzyme acyl-CoA:cholesterol acyltransferase (ACAT) (18). NPC1 deficiency in fibroblasts markedly reduced CE formation via ACAT because the substrate, cholesterol, is trapped in LE/L (6, 15). We, therefore, tested our hypothesis that 0.1 mm CD mobilizes cholesterol from LE/L to the ER in Npc1−/− cells by examining cholesterol esterification in an assay that measures ER cholesterol availability. Npc1−/− and Npc1+/+ astrocytes were incubated with 0.1 or 1 mm CD for 28 h; [14C]oleate ± the ACAT inhibitor Sandoz 58-035 were added for the last 4 h. In Npc1−/− astrocytes, 0.1 mm CD increased [14C]oleate incorporation into CE by 2.6-fold (Fig. 8A); the increase in esterification was abolished by Sandoz 58-035, implying that ACAT was responsible for the increased esterification. These data suggest that 0.1 mm CD increased ER cholesterol. In contrast, 1 mm CD dramatically reduced [14C]CE in Npc1−/− astrocytes (Fig. 8A) consistent with a reduction in ER cholesterol. Moreover, [14C]oleate incorporation into phospholipids of Npc1−/− (Fig. 8B) and Npc1+/+ (Fig. 8D) astrocytes was not significantly different and was unaltered by Sandoz 58-035, indicating that CD did not affect [14C]oleate uptake or the intracellular pool size of oleate. In contrast, in Npc1+/+ astrocytes, neither 1 mm nor 0.1 mm CD significantly altered the amount of [14C]CE (Fig. 8C), consistent with the idea that CD mobilized only small amounts of cholesterol from LE/L to the ER because cholesterol was not sequestered in LE/L of Npc1+/+ cells. These studies provide further evidence that 0.1 mm CD releases cholesterol trapped in LE/L of Npc1−/− astrocytes and increases cholesterol availability at the ER. In addition, the observation that 1 mm CD reduced cholesterol esterification in Npc1−/− astrocytes suggests that 1 mm CD extracted cholesterol from the cells and reduced ER cholesterol.
FIGURE 8.

CE formation in astrocytes. [14C]oleate incorporation into CE and phospholipids was quantified in Npc1−/− and Npc1+/+ astrocytes. Cells were incubated for 28 h with vehicle (dark gray), 0.1 mm CD (light gray), or 1 mm CD (white); [14C]oleate ± Sandoz 58-035 were included for the final 4 h. Lipids were separated, and radioactivity was measured in CE and phospholipids. Data are means ± S.E. from three independent cell preparations. *, p < 0.05; **, p < 0.01.
Cholesterol esterification was also examined in Npc1+/+ and Npc1−/− neurons. In comparison with astrocytes, very little cholesterol was esterified in cerebellar granule neurons of either Npc1 genotype (Fig. 8 versus Fig. 9). Moreover, 0.1 mm CD did not increase 14C incorporation into CE or phospholipids in Npc1−/− neurons and Sandoz 58-035 did not reduce the basal radiolabeling of CE (Fig. 9A). In addition, the small amount of CE formed in neurons was independent of NPC1 because [14C]CE formation in Npc1+/+ and Npc1−/− neurons was similar, albeit barely detectable (Fig. 9, A and B). Consistent with these findings, the level of ACAT1 mRNA in the neurons was only ∼10% of that in astrocytes and did not depend on the Npc1 genotype (Fig. 9C). We conclude that (i) cholesterol esterification is far less active in cerebellar granule neurons than in astrocytes, (ii) 0.1 mm CD mobilizes cholesterol sequestered in LE/L of Npc1−/− astrocytes but not neurons and increases cholesterol availability at the ER for esterification by ACAT in Npc1−/− astrocytes but not neurons, and (iii) 1 mm CD reduces CE formation in Npc1−/− astrocytes presumably by reducing ER cholesterol availability.
FIGURE 9.

CE formation in neurons. [14C]oleate incorporation into CE and phospholipids was quantified in the following: Npc1−/− neurons incubated with vehicle (dark gray) or 0.1 mm CD (light gray) ± Sandoz 58-035 (A) and Npc1+/+ neurons incubated with vehicle (B). Data are means ± S.E. from three independent experiments. C, qPCR analysis of ACAT1 mRNA relative to GAPDH mRNA in Npc1+/+ (black) and Npc1−/− (white) neurons, and astrocytes were incubated for 24 h with vehicle. Data are means ± S.E. from duplicate analyses of three independent cell preparations. p > 0.05.
Expression of Genes of Cholesterol Homeostasis
When the ER cholesterol content of fibroblasts exceeds ∼5% of total lipids (6), the expression of genes of cholesterol synthesis and uptake, including SREBP2 and its target genes encoding 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) and LDLR, decreases. In addition, expression of genes that encode two ATP cassette-binding (ABC) proteins involved in cholesterol efflux from cells (ABCA1 and ABCG1) increases. Conversely, when ER cholesterol drops below the 5% threshold, the expression of genes of cholesterol synthesis and uptake increases, whereas mRNAs involved in cholesterol efflux decrease. We, therefore, compared the expression of genes of cholesterol homeostasis in neurons and astrocytes from Npc1+/+ and Npc1−/− mice. In neurons, as in whole brain (14), levels of mRNAs encoding SREBP2, HMGCR and LDLR were not affected significantly by the Npc1 genotype (Fig 10A). However, in astrocytes, SREBP2 and HMGCR mRNAs were reduced by NPC1 deficiency (Fig 10B). Although ABCA1 mRNA was not changed by NPC1 deficiency in either neurons or astrocytes, ABCG1 mRNA was significantly decreased in both neurons and astrocytes (Fig 10, A and B).
FIGURE 10.

qPCR analysis of mRNAs involved in cholesterol metabolism in Npc1+/+ and Npc1−/− neurons and astrocytes. mRNA levels were quantified relative to GAPDH mRNA in vehicle-treated (24 h) neurons (A) and astrocytes (B) from Npc1+/+ (black) and Npc1−/− (white) mice. LDLR, low density lipoprotein receptor; SREBP, sterol response element-binding protein; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; ABC, ATP-binding cassette. Data are means ± S.E. of triplicate analyses from three independent experiments. *, p < 0.05; **, p < 0.001.
We also quantified mRNAs involved in cholesterol metabolism in neurons and astrocytes in response to CD. In Npc1−/− neurons, SREBP2 mRNA was significantly decreased by 0.1 mm CD but increased by 1 mm CD (Fig 11A). Correspondingly, HMGCR mRNA was decreased by 0.1 mm CD and increased by 1 mm CD. In Npc1−/− neurons, mRNAs encoding LDLR, ABCA1 and ABCG1 were not significantly changed by 0.1 mm CD (Fig 11A), whereas 1 mm CD markedly increased LDLR and HMGCR mRNAs and decreased ABCA1 and ABCG1 mRNAs. These observations provide additional support for a model in which 0.1 mm CD mobilizes cholesterol from LE/L to the ER in Npc1−/− neurons. In contrast, in addition to releasing cholesterol from LE/L, the predominant action of 1 mm CD appears to be cholesterol removal from the plasma membrane/ER; thus, the small amount of LE/L cholesterol that reaches the ER appears to be insufficient to compensate for the larger amount of cholesterol removed from the Npc1−/− neurons. In Npc1+/+ neurons, 0.1 mm CD did not alter SREBP2, HMGCR, or LDLR mRNAs, whereas 1 mm CD robustly increased levels of these mRNAs (Fig 11B); furthermore, ABCA1 and ABCG1 mRNAs were markedly attenuated (Fig 11B). Thus, because cholesterol is not sequestered in LE/L of Npc1+/+ cells, it appears that the small amount of cholesterol released from LE/L by 0.1 mm CD was insufficient to raise ER cholesterol content because more cholesterol had been removed from the cell surface.
FIGURE 11.

qPCR analysis of mRNAs involved in cholesterol synthesis and efflux in neurons (A and B) and astrocytes (C and D) from Npc1−/− (A and C) and Npc1+/+ (B and D) mice. Cells were incubated for 24 h with vehicle (dark gray), 0.1 mm CD (light gray), or 1 mm CD (white), and mRNA levels were quantified relative to GAPDH mRNA. Gene abbreviations are defined in the legend to Fig 10. Data are means ± S.E. from triplicate analyses of three independent experiments. *, p < 0.05; ** p < 0.01; ***, p < 0.001.
In Npc1−/− and Npc1+/+ astrocytes, CD altered the levels of mRNAs of cholesterol-responsive genes less profoundly than in neurons (Fig 11, C and D). In Npc1−/− astrocytes, the mRNAs involved in cholesterol accretion (SREBP2, HMGCR, and LDLR) were not changed by 0.1 mm CD, whereas 1 mm CD modestly increased HMGCR and SREBP2 mRNAs (Fig. 11C). In addition, ABCA1 and ABCG1 mRNAs were increased by 0.1 mm but not 1 mm CD (Fig. 11C), suggesting that astrocytes preferentially increase cholesterol efflux in response to 0.1 mm CD. In Npc1+/+ astrocytes, 1 mm CD modestly reduced ABCA1 and ABCG1 mRNAs (Fig. 11D). These observations indicate that 0.1 mm CD mobilizes cholesterol from LE/L to the ER of Npc1−/− neurons thereby decreasing mRNAs involved in cholesterol synthesis rather than increasing mRNAs involved in cholesterol efflux. On the other hand, Npc1−/− astrocytes appear to respond to 0.1 mm CD primarily by increasing mRNAs involved in cholesterol efflux rather than by decreasing mRNAs of cholesterol accretion.
DISCUSSION
The remarkable recent findings of Dietschy and co-workers (8, 17, 19) on the benefit of CD in Npc1−/− mice indicate the potential for using CD to treat NPC patients. Because the previous studies were performed in intact mice, we investigated the CD dose response of primary neurons and glial cells from brains of Npc1−/− mice. We show that cholesterol homeostasis in Npc1−/− neurons and astrocytes depends critically on the CD concentration. Although both 1 mm and 0.1 mm CD reduced cholesterol storage in LE/L of Npc1−/− neurons and astrocytes, these CD concentrations exerted opposite effects on cholesterol homeostasis. Thus, 0.1 mm CD released cholesterol trapped in LE/L and increased cholesterol availability at the ER for CE formation as well as for reducing cholesterol synthesis and modulating expression of genes of cholesterol metabolism. On the other hand, 1 mm CD decreased CE formation, reduced mRNAs required for cholesterol efflux and increased cholesterol synthesis and mRNAs involved in cholesterol synthesis and uptake. Therefore, although both 0.1 and 1 mm CD mobilized LE/L cholesterol in Npc1−/− cells, the overriding effect of 1 mm CD on cholesterol homeostasis appears to be cholesterol extraction from the plasma membrane and depletion of ER cholesterol.
The response of Npc1−/− astrocytes and neurons to CD was distinct in several ways. First, 10 mm CD was profoundly toxic to neurons but less deleterious to astrocytes. Second, ACAT-mediated cholesterol esterification was far more active in astrocytes than in neurons, and CD markedly altered CE formation in astrocytes but not in neurons. Third, low concentrations of CD altered levels of mRNAs involved in cholesterol accretion (SREBP2, HMGCR, and LDL) to a greater extent in neurons than in astrocytes. Fourth, 0.1 mm CD increased mRNAs of genes involved in cholesterol efflux in astrocytes but not in neurons. Because glial cells are the major cell type in the brain and play key roles in brain cholesterol metabolism (reviewed in Ref. 20), it was possible that the CD-mediated increase in survival of Npc1−/− mice/neurons was due to correction of the cholesterol trafficking defect in astrocytes. However, recent studies demonstrate that NPC1 deficiency in neurons rather than astrocytes is the primary factor underlying the neurodegeneration (21, 22).
CD is relatively non-toxic and is currently being administered to some NPC patients. Thus, our studies are timely and imply that the amount of CD to which neurons and astrocytes are exposed is crucial. Continuous exposure of these cells to CD concentrations of 1 mm or higher is likely to be detrimental. Interestingly, despite the half-life of CD in the brain being only ∼6.5 h, a single injection of 40 mg CD/Kg body weight into Npc1−/− mouse brains resulted in a brain CD concentration of ∼0.1–0.4 mm, which suppressed cholesterol synthesis for >6 days (14). Moreover, the ED50 of CD required for a positive therapeutic effect in the brain is ∼0.5 mg/kg. These CD concentrations are similar to the 0.1 mm concentration that was beneficial in the current studies. One limitation to the use of CD for treatment of NPC patients is that transport of CD from the plasma into the brain is inefficient: only ∼0.2% of plasma CD enters the brain (14). Thus, because liver disease often occurs in NPC patients, a possible treatment regimen might be simultaneous administration of CD both peripherally and directly into the brain so that CD concentrations in liver and brain would be similar and in the range of 0.1–0.2 mm.
Supplementary Material
Acknowledgments
We thank Randy Nelson and Russ Watts for excellent technical assistance and Robert B. Campenot and Dennis E. Vance for helpful discussions.
This work was supported by the Ara Parseghian Medical Research Foundation and Canadian Institutes for Health Research (to J. E. V.) as well as graduate studentships from the Natural Sciences and Engineering Research Council of Canada and the Alberta Heritage Foundation for Medical Research (to K. B. P.).

This article contains supplemental Table S1.
B. Karten and J. E. Vance, unpublished data.
- NPC
- Niemann-Pick type C
- ABC
- ATP-binding cassette protein
- ACAT
- acyl-CoA:cholesterol acyltransferase
- CD
- 2-hydroxypropyl-β-cyclodextrin
- CE
- cholesteryl esters
- HMGCR
- 3-hydroxy-3-methylglutaryl-CoA reductase
- LE/L
- late endosomes/lysosomes
- LDLR
- LDL receptor
- SREBP
- sterol-response element-binding protein
- qPCR
- quantitative PCR.
REFERENCES
- 1. Carstea E. D., Morris J. A., Coleman K. G., Loftus S. K., Zhang D., Cummings C., Gu J., Rosenfeld M. A., Pavan W. J., Krizman D. B., Nagle J., Polymeropoulos M. H., Sturley S. L., Ioannou Y. A., Higgins M. E., Comly M., Cooney A., Brown A., Kaneski C. R., Blanchette-Mackie E. J., Dwyer N. K., Neufeld E. B., Chang T. Y., Liscum L., Strauss J. F., 3rd, Ohno K., Zeigler M., Carmi R., Sokol J., Markie D., O'Neill R. R., van Diggelen O. P., Elleder M., Patterson M. C., Brady R. O., Vanier M. T., Pentchev P. G., Tagle D. A. (1997) Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science 277, 228–231 [DOI] [PubMed] [Google Scholar]
- 2. Naureckiene S., Sleat D. E., Lackland H., Fensom A., Vanier M. T., Wattiaux R., Jadot M., Lobel P. (2000) Identification of HE1 as the second gene of Niemann-Pick C disease. Science 290, 2298–2301 [DOI] [PubMed] [Google Scholar]
- 3. Vanier M. T., Millat G. (2003) Niemann-Pick disease type C. Clin. Genet. 64, 269–281 [DOI] [PubMed] [Google Scholar]
- 4. Wang M. L., Motamed M., Infante R. E., Abi-Mosleh L., Kwon H. J., Brown M. S., Goldstein J. L. (2010) Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 12, 166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wojtanik K. M., Liscum L. (2003) The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem. 278, 14850–14856 [DOI] [PubMed] [Google Scholar]
- 6. Radhakrishnan A., Goldstein J. L., McDonald J. G., Brown M. S. (2008) Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. 8, 512–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brown M. S., Goldstein J. L. (1997) The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340 [DOI] [PubMed] [Google Scholar]
- 8. Liu B., Turley S. D., Burns D. K., Miller A. M., Repa J. J., Dietschy J. M. (2009) Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse. Proc. Natl. Acad. Sci. U.S.A. 106, 2377–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ramirez C. M., Liu B., Taylor A. M., Repa J. J., Burns D. K., Weinberg A. G., Turley S. D., Dietschy J. M. (2010) Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the niemann-pick type C1 mouse and markedly prolongs life. Pediatr. Res. 68, 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davidson C. D., Ali N. F., Micsenyi M. C., Stephney G., Renault S., Dobrenis K., Ory D. S., Vanier M. T., Walkley S. U. (2009) Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One 4, e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ohvo H., Slotte J. P. (1996) Cyclodextrin-mediated removal of sterols from monolayers: Effects of sterol structure and phospholipids on desorption rate. Biochemistry 35, 8018–8024 [DOI] [PubMed] [Google Scholar]
- 12. Kiss T., Fenyvesi F., Bácskay I., Váradi J., Fenyvesi E., Iványi R., Szente L., Tósaki A., Vecsernyés M. (2010) Evaluation of the cytotoxicity of β-cyclodextrin derivatives: Evidence for the role of cholesterol extraction. Eur. J. Pharm. Sci. 40, 376–380 [DOI] [PubMed] [Google Scholar]
- 13. Rosenbaum A. I., Zhang G., Warren J. D., Maxfield F. R. (2010) Endocytosis of β-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc. Natl. Acad. Sci. U.S.A. 107, 5477–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aqul A., Liu B., Ramirez C. M., Pieper A. A., Estill S. J., Burns D. K., Liu B., Repa J. J., Turley S. D., Dietschy J. M. (2011) Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J. Neurosci. 31, 9404–9413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liscum L., Faust J. R. (1987) Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J. Biol. Chem. 262, 17002–17008 [PubMed] [Google Scholar]
- 16. Zidovetzki R., Levitan I. (2007) Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions, and control strategies. Biochim. Biophys. Acta 1768, 1311–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu B., Ramirez C. M., Miller A. M., Repa J. J., Turley S. D., Dietschy J. M. (2010) Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J. Lipid Res. 51, 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chang C. C., Huh H. Y., Cadigan K. M., Chang T. Y. (1993) Molecular cloning and functional expression of human acyl-coenzyme A:cholesterol acyltransferase cDNA in mutant Chinese hamster ovary cells. J. Biol. Chem. 268, 20747–20755 [PubMed] [Google Scholar]
- 19. Liu B., Li H., Repa J. J., Turley S. D., Dietschy J. M. (2008) Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid Res. 49, 663–669 [DOI] [PubMed] [Google Scholar]
- 20. Vance J. E., Hayashi H. (2010) Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim. Biophys. Acta 1801, 806–818 [DOI] [PubMed] [Google Scholar]
- 21. Yu T., Shakkottai V. G., Chung C., Lieberman A. P. (2011) Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum. Mol. Genet. 20, 4440–4451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lopez M. E., Klein A. D., Dimbil U. J., Scott M. P. (2011) Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J. Neurosci. 31, 4367–4378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karten B., Hayashi H., Francis G. A., Campenot R. B., Vance D. E., Vance J. E. (2005) Generation and function of astroglial lipoproteins from Niemann-Pick type C1-deficient mice. Biochem. J. 387, 779–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hayashi H., Campenot R. B., Vance D. E., Vance J. E. (2004) Glial cell lipoproteins stimulate axon growth of central nervous system neurons in compartmented cultures. J. Biol. Chem. 279, 14009–14015 [DOI] [PubMed] [Google Scholar]
- 25. Peake K. B., Campenot R. B., Vance D. E., Vance J. E. (2011) Activation and function of primary microglia from Niemann-Pick type C-deficient mice. Biochim. Biophys. Acta. 1812, 1121–1129 [DOI] [PubMed] [Google Scholar]
- 26. Michikawa M., Yanagisawa K. (1998) Apolipoprotein E4 induces neuronal cell death under conditions of suppressed de novo cholesterol synthesis. J. Neurosci. Res. 54, 58–67 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.