

Background: PP2Cm phosphatase was recently identified as the branched-chain α-ketoacid dehydrogenase complex (BCKDC) phosphatase (BDP).
Results: The 2.4 Å BDP structure reveals a central β-sandwich with two bound metal ions in the active-site cleft.
Conclusion: Unlike related pyruvate dehydrogenase phosphatases, BDP strictly depends on Mn2+, and not Mg2+, for phosphatase activity.
Significance: The structural/biochemical results validate PP2Cm as the BCKDC phosphatase.
Keywords: Metabolic Diseases, Phosphatase, PP2A, Protein Phosphatase, X-ray Crystallography, Branched-chain α-Ketoacid Dehydrogenase Complex, Central β-Sandwich Fold, Maple Syrup Urine Disease, Mitochondrial Protein Phosphatases, Mn2+-dependent Phosphatases, PP2Cm Mitochondrial Phosphatase
Abstract
The branched-chain α-ketoacid dehydrogenase phosphatase (BDP) component of the human branched-chain α-ketoacid dehydrogenase complex (BCKDC) has been expressed in Escherichia coli and purified in the soluble form. The monomeric BDP shows a strict dependence on Mn2+ ions for phosphatase activity, whereas Mg2+ and Ca2+ ions do not support catalysis. Metal binding constants for BDP, determined by competition isothermal titration calorimetry, are 2.4 nm and 10 μm for Mn2+ and Mg2+ ions, respectively. Using the phosphorylated decarboxylase component (p-E1b) of BCKDC as a substrate, BDP shows a specific activity of 68 nmol/min/mg. The Ca2+-independent binding of BDP to the 24-meric transacylase (dihydrolipoyl transacylase; E2b) core of BCKDC results in a 3-fold increase in the dephosphorylation rate of p-E1b. However, the lipoyl prosthetic group on E2b is not essential for BDP binding or E2b-stimulated phosphatase activity. Acidic residues in the C-terminal linker of the E2b lipoyl domain are essential for the interaction between BDP and E2b. The BDP structure was determined by x-ray crystallography to 2.4 Å resolution. The BDP structure is dominated by a central β-sandwich. There are two protrusions forming a narrow cleft ∼10 Å wide, which constitutes the active site. The carboxylate moieties of acidic residues Asp-109, Asp-207, Asp-298, and Asp-337 in the active-site cleft participate in binding two metal ions. Substitutions of these residues with alanine nullify BDP phosphatase activity. Alteration of the nearby Arg-104 increases the Km for p-E1b peptide by 60-fold, suggesting that this residue is critical for the recognition of the native p-E1b protein.
Introduction
The human branched-chain α-ketoacid dehydrogenase complex (BCKDC)4 is a member of the highly conserved mitochondrial α-ketoacid dehydrogenase complexes comprising the BCKDC, the pyruvate dehydrogenase complex (PDC), and the α-ketoglutarate dehydrogenase complex (1–3). The BCKDC catalyzes the oxidative decarboxylation of branched-chain α-ketoacids derived from the branched-chain amino acids leucine, isoleucine, and valine (2) according to Reaction 1.
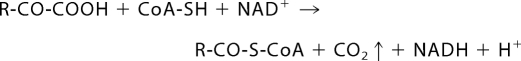 |
The 4 × 106-dalton BCKDC is organized around a cubic core comprising 24 lipoyl-bearing dihydrolipoyl transacylase (E2b) subunits. Multiple copies of branched-chain α-ketoacid decarboxylase/dehydrogenase (E1b), dihydrolipoamide dehydrogenase (E3), the BCKD kinase (BDK), and the BCKD phosphatase (BDP) are associated with this core (1). The E1b and E2b components are specific for the BCKDC, whereas the E3 component is common among the three α-ketoacid dehydrogenase complexes (1). The overall reaction (Reaction 1) catalyzed by BCKDC is the sum of individual reaction steps catalyzed by each of the three catalytic components (E1b, E2b, and E3), which are linked through substrate channeling (1).
The activity of BCKDC is regulated post-translationally by a phosphorylation (inactivation)/dephosphorylation (reactivation) cycle under various dietary conditions and hormonal stimuli (1, 4). Rats fed low-protein diets show increased phosphorylation of the hepatic BCKDC (5). Starvation and diabetes increase BCKDC activity in skeletal muscle by decreasing phosphorylation of the enzyme complex (6). The state of phosphorylation in BCKDC inversely correlates with the level of BDK in tissues regulated by extracellular stimuli (7, 8). The molecular mechanism for the acute inactivation of BCKDC through phosphorylation by the E2b-bound BDK has been elucidated (9). The presence of a bulky phosphoryl group at Ser-292 (phosphorylation site 1) of the E1α subunit renders the phosphorylation loop harboring the phosphoserine residue disordered. The disordered loop conformation interrupts substrate channeling between E1b and E2b, leading to the inactivation of BCKDC (9).
Despite advances in understanding the negative regulation of BCKDC by BDK, little is known regarding the dephosphorylation and reactivation of phosphorylated BCKDC by BDP, the reciprocal step of the reversible phosphorylation. A putative “BDP” protein of 460 kDa in size capable of removing the phosphoryl group from phosphorylated E1b (phospho-E1b) was previously isolated from bovine kidney (10), and the 33-kDa catalytic subunit of this phosphatase was subsequently purified (11). This phosphatase subunit catalyzed the dephosphorylation of phospho-E1b independent of metal ions but was unable to hydrolyze p-nitrophenyl phosphate (pNPP) and has never been cloned. It was not until 2 decades later that a novel PP2C-type mitochondrial phosphatase (PTMP) with a 20% amino acid sequence identity to the PDPs was identified in the NCBI human genome database (12). PTMP was shown to be imported into mitochondria and to specifically dephosphorylate phospho-E1b but not phospho-E1p of the PDC in vitro. A recent landmark study by the Wang group showed that the same PTMP, termed protein phosphatase 2Cm (PP2Cm) in that study, co-immunoprecipitated with BCKDC subunits, including the E2b (13). PP2Cm knock-out mice manifested elevated branched-chain amino acid concentrations and the intermediate maple syrup urine disease (MSUD) phenotype (13), a genetic disorder caused by deficiency of the human BCKDC (2). The predicted variant form of MSUD presented by PP2Cm knock-out mice was recently confirmed by the first case of an MSUD patient carrying a homozygous p.His139fs null mutation in the PP2Cm protein (14). The above results, taken together, provide direct evidence that PP2Cm is the endogenous BDP component of BDKDC and document the occurrence of type 5 MSUD caused by deficiency in BDP (2).
PP2C serine/threonine phosphatases, including mitochondrial pyruvate dehydrogenase phosphatase (PDP) of PDC and BDP (i.e. PP2Cm) of BCKDC, belong to the PPM family of Mn2+/Mg2+-dependent phosphatases (15). PP2C phosphatases not only impart the reversal of protein kinase cascades activated by stress but also play a role in cell differentiation and growth as well as cell survival, apoptosis, and metabolism (16). In addition to causing MSUD (13, 14), there is evidence to suggest that loss of PP2Cm (i.e. BDP) is a significant contributor to the pathogenesis of heart failure, linking branched-chain amino acid catabolism to cardiac pathophysiology (17). As for the PDP of PP2C phosphatases, there are two isoforms, PDP1 and PDP2, which positively regulate PDC activity (18). PDP1 is a heterodimer of one catalytic and one regulatory subunit; the catalytic subunit of PDP1 (PDP1c) is a Mg2+-dependent PP2C phosphatase. PDP1 activity is markedly stimulated by forming a Ca2+-dependent complex between PDP1c and the inner lipoyl domain (L2) of E2p (19). In contrast, monomeric PDP2 is not regulated by Ca2+ ions and is instead stimulated by biological polyamine (20). The rat PDP1c structure showed a structural core consisting of a central β-sandwich flanked on both sides by loops and α-helices. This fold is very similar to that of human PP2Ca phosphatase, despite a low level of sequence identity between these two PP2C phosphatases (21). In the PDP1c structure, there are two Mg2+ ions in the active site and a putative lipoyl moiety-binding pocket for binding to the L2 domain of E2p.
To understand the structure and function of BDP as a regulatory component of BCKDC, we have expressed the BDP of human BCKDC in the soluble form and characterized its interactions with the E2b scaffold. We show that the strictly Mn2+-dependent BDP phosphatase activity is markedly enhanced upon binding to the lipoyl-bearing domain (LBD) and its C-terminal linker region of E2b. Surprisingly, unlike PDP1c, the lipoyl prosthetic group is not required for the BDP-E2b interactions. The crystal structure of human BDP was determined at 2.4 Å resolution. The BDP structure reveals the conserved organization of PP2C phosphatases and the presence of two bound metal ions in the active site. Based on this BDP structure, site-directed mutagenesis of metal-binding and catalytic residues in the active site was performed. The results support the critical role of these amino acid residues in mediating BDP phosphatase activity.
EXPERIMENTAL PROCEDURES
Materials
Except for those indicated at the first mention, all reagents were obtained from Sigma-Aldrich at the highest grade available.
Protein Expression, Purification, and Site-directed Mutagenesis
Human recombinant BDP phosphatase was cloned into a pSUMO vector (where SUMO represents small ubiquitin modifier) (Lifesensors, Malvern, PA) C-terminal to the SUMO sequence, with the linker region harboring the tobacco etch virus (TEV) protease recognition site (LENLYFQ↓G; the arrow shows the cleavage site). This construct is referred to as SUMO-BDP and contains the entire mature BDP sequence (residues Asp-30 to Ala-373) without the putative mitochondrial targeting sequence (residues 1–29). For crystallization purposes, an additional vector was constructed containing both N-terminal (residues 30–83) and C-terminal (residues 362–373) truncations and referred to as ΔN83/F361X SUMO-BDP. The expression plasmids for BDP were transformed into Escherichia coli BL21 competent cells containing an expression plasmid (pGroESL) (a gift from Anthony Gatenby (DuPont)) for bacterial chaperonins GroEL and GroES. The co-transformed overexpressed chaperonins significantly increase the yield of SUMO-BDP by promoting proper folding and assembly of the target protein (22). LB culture media were supplemented with 1 mm MnCl2 to further augment the yield of SUMO-BDP (12). Cells with added kanamycin and chloramphenicol for antibiotic selection were grown at 37 °C for 2–3 h until A600 reached 0.6. The expression of wild-type and mutant BDP proteins at 23 °C was induced by adding IPTG to a final concentration of 0.3 mm, and the culture was continued to incubate for 5 h. The cells were lysed by sonication in 20 mm Tris-HCl, pH 7.9, 100 mm NaCl, 5 mm β-mercaptoethanol, 0.625% Triton X-100, 10% glycerol, 1 mm PMSF, and 1 mm benzamidine; the SUMO-BDP phosphatase proteins were immobilized on a Ni2+-NTA column (Qiagen, Valencia, CA) and eluted with 200 mm imidazole. The concentrated combined fractions were applied to a Superdex S-200 16/60 size exclusion column (GE Healthcare), which had been equilibrated with a buffer containing 20 mm Tris-HCl, pH 7.5, 100 mm KCl, 5% (v/v) glycerol, and 10 mm β-mercaptoethanol. Peak SUMO-BDP phosphatase fractions were loaded on a Resource Q ion exchange column (GE Healthcare) after diluting the SUMO-BDP with 20 mm Tris-HCl, 50 mm NaCl, 5% glycerol, and 10 mm β-mercaptoethanol. SUMO-BDP was eluted using a 0–600 mm NaCl gradient in the same buffer. The C-terminal His6-tagged wild-type and mutant E2b domain constructs were produced as described previously (23). Site-directed mutagenesis was carried out using the QuikChange kit (Stratagene, La Jolla, CA), according to the manufacturer's instructions. All mutations were confirmed by DNA sequencing.
Analytical Ultracentrifugation
SUMO-BDP was dialyzed into a buffer comprising 50 mm Tris, pH 7.5, and 100 mm NaCl. Centrifugation cells were prepared by sandwiching an Epon dual sectored centerpiece between two sapphire windows in a cell housing. Sample (∼390 ml) was introduced into the sample sector, and an equal volume of reference buffer was placed in the reference sector. The samples were placed in an An50-Ti rotor, and the rotor was inserted into the Beckman Optima XL-I (Beckman-Coulter) centrifuge, followed by evacuation of the centrifugation chamber. The samples were allowed to equilibrate at the experimental temperature (20 °C) for 2 h and then were accelerated to 50,000 rpm. The interference optical system was used to acquire data on the time-dependent concentration profiles of the sedimenting proteins. The data were analyzed by the freeware program Sedfit, using the c(s) distribution (24, 25) and systematic noise subtraction (26). The weighted average of the sedimentation coefficient of the dominant c(s) peak and the knowledge of the buffer conditions and refined frictional ratio of the protein were used to arrive at a molar mass for this material. Obtaining the molar mass of a majority species in a heterogeneous mixture is known to be reliable using this method (27). The freeware program SEDNTERP (28) was used to estimate the solution density, solution viscosity, and protein partial specific volume.
Phosphatase Activity Assays with pNPP as Substrate
The assay was modified from that described previously (29). The assay mixture in 0.5 ml contained 20 mm freshly prepared artificial substrate pNPP, 20 mm Tris-HCl, pH 7.5, 7 mm MnCl2, and 1 μg of SUMO-BDP. Increased A410 due to the production of p-nitrophenol was monitored at 30 °C in a Cary-100 Bio UV-visible spectrophotometer (Agilent Technologies, Santa Clara, CA). Non-enzymatic rates were subtracted from the absorbance rates obtained with the addition of SUMO-BDP. Changes in A410 were converted to the rates of nmol/min/mg, using a molar extinction coefficient of 18,000 m−1·cm−1. To determine the metal ion requirement, the purified SUMO-BDP was exhaustively dialyzed with three changes against 50 mm HEPES, pH 7.5, 100 mm KCl, 5% glycerol, 5 mm EDTA, and 5 mm EGTA. The residual EDTA or EGTA was removed by a final dialysis step against a buffer without these metal chelators. MgCl2 or CaCl2 instead of MnCl2 was added to the pNPP assay mixture. Lipoylated forms of LBD84, LBD99, LBD-SBD152, K44A E2b, or E2b protein, obtained by in vitro lipoylation (30), at a 3:1 molar excess to SUMO-BDP (monomer/monomer) were added to the assay mixture. Enzyme activity data were fitted with the Prism software program (GraphPad, Inc.) using non-linear analysis for Michaelis-Menten kinetics.
Phosphatase Activity Assays with Native Substrate Phospho-E1b
The E1b (31), E2b (30), and BDK (32) components of mammalian BCKDC were expressed in E. coli and purified as described previously. The E1b-E2b-BDK subcomplex was reconstituted by mixing His6-tagged E1b (18 μm), lipoylated E2b (2.15 μm), and MBP-BDK (101 nm) in 0.9 ml of a buffer containing 30 mm HEPES, pH 7.5, 2 mm MgCl2, 2 mm DTT, 0.1 mm EGTA, 5% glycerol. The phosphorylation reaction was initiated by the addition of 0.1 mm [γ-32P]ATP, followed by incubation at 37 °C for 30 min. The E1b-E2b-BDK subcomplex was dissociated by incubating the subcomplex in 1 m NaCl for 1 h. The 32P-labeled His6-tagged E1b (p-E1b) was extracted with Ni2+-NTA resin in 1 m NaCl, 50 mm HEPES, pH 7.5, 5% glycerol and eluted with the same buffer containing 250 mm imidazole. A PD-10 gel filtration column equilibrated in the same buffer was used to remove imidazole.
The dephosphorylation reaction of p-E1b was carried out in microcentrifuge tubes as described previously (33). To initiate the dephosphorylation reaction, SUMO-BDP (40 nm) was added to 50 mm MOPS, pH 7.5, 7 mm MnCl2, 100 mm NaCl, 5% glycerol containing 32P-labeled p-E1b (1.1 μm) with or without lipoylated E2b (134 nm) in a final volume of 0.05 ml. After incubation at 37 °C for 2 min, the reaction was stopped by the addition of 0.1 ml of 20% (w/v) trichloroacetic acid, followed by incubation at room temperature for 10 min. After centrifugation at 13,000 rpm for 3 min, 50-μl aliquots of the supernatant were added to 3 ml of scintillation mixture (Research Products International, Mount Prospect, IL) and counted to determine the released radioactivity.
Phosphatase Activity Assays Using Phosphoheptapeptide as Substrate
SUMO-BDP (1.5 μm) phosphatase activity was measured using a 10 μm concentration of a Ser(P)-292-containing heptapeptide based on the E1b-α sequence (NH2-Gly-His-His-Ser(P)-Thr-Ser-Asp-COOH) (synthesized at the institutional protein chemistry core) in 96-well microtiter plates in a final volume of 25 μl. Buffer conditions were as follows: 50 mm HEPES, pH 7.5, 7 mm MnCl2, and 100 mm KCl. Care was taken to avoid any phosphate contamination from either the buffer or protein solutions. Solutions were pipetted into the plates and allowed to react for 10 min at 22 °C. Reactions were terminated by the addition of 50 μl of malachite green (Enzo Life Sciences, Plymouth Meeting, PA) solution into the microtiter plates. The incubation was continued for 15 min at room temperature before measuring the absorbance at 620 nm in a Biotech microplate reader. Standard curves were run using a standard phosphate-containing buffer. Phosphate released was calculated using the equation, phosphate released = (A620 − y axis intercept)/slope of the standard curve. In general, 1 nmol of phosphate yields an absorbance of 0.1 unit at 620 nm in the standard assay. However, in the microtiter plate assay reported here, we routinely achieved an absorbance of 0.45–0.5 for the same quantity of phosphate. These absorbance levels are consistent with those previously reported (34).
Pull-down Assays for Wild-type and Mutant E2b
Lipoylated wild-type, K44A, or L420X E2b (315 nm) and SUMO-BDP (2.7 μm) were mixed in 20 mm Tris-HCl, pH 7.5, 50 mm NaCl, 5% glycerol, and the mixtures were shaken in the cold room for 15–20 min. Protein mixtures were applied onto Ni2+-NTA columns, and the bound proteins were eluted with the Tris-HCl buffer containing 250 mm imidazole. Trichloroacetic acid in a final 50% concentration was added to each tube, and the precipitated pellets were washed with acetone. The pellets were dissolved in 150 μl of 8 m urea and subjected to SDS-PAGE analysis.
Pull-down Assays for Wild-type and Mutant LBD99 Constructs
Pull-down assays were performed essentially as described above except for the following changes. Lipoylated wild-type or mutant LBD99 construct (C-terminal His6-tagged) (120 μg) and non-tagged BDP (25 μg) were incubated in 50 mm HEPES, pH 7.5, with 50 mm NaCl and 5% glycerol. Bound proteins were eluted with the same HEPES buffer containing 250 mm imidazole. Approximately 20% of the final eluted fraction was used of the SDS-PAGE analysis.
Crystallization of ΔN83/F361X SUMO-BDP
Crystals were obtained at 20 °C using the hanging drop vapor-diffusion method by mixing 3 μl of SUMO-BDP solution (27–30 mg/ml in 20 mm Tris-HCl, pH 7.5, 100 mm KCl, 5% glycerol, and 0.285 mm MnCl2) or SUMO-BDP solution without MnCl2 with 3 μl of well solution (19% (w/v) PEG 3350, 0.2 m MgCl2, 0.1 m Tris-HCl, pH 8.5, and 20 mm β-mercaptoethanol), followed by suspending the mixture over a reservoir of 0.5 ml of the precipitant solution. Dilution seeding was used to induce nucleation. Crystals were transferred to a fresh soaking solution (20% PEG 3350, 0.1 m Tris-HCl, pH 8.5, 0.2 m MgCl2) with or without 0.3 mm MnCl2 for 18–24 h. The crystals were serially transferred to a cryosolution containing 25% glycerol and flash-cooled in liquid nitrogen.
Structure Determination and Refinement
All x-ray diffraction data for BDP were collected at beamline 19-ID at the Advanced Photon Source, Argonne National Laboratories. For determination of the SUMO-BDP structure, diffraction data were collected from a single crystal that was grown in 0.2 m MgCl2 with no MnCl2 present. The crystal had the symmetry of space group P3121, and the dmin of the diffraction was 2.4 Å. The phase problem was solved using the molecular replacement protocols available in Phaser (35); the crystal structure of a similar construct of human PP2Cκ (Protein Data Bank accession code 2iq1), stripped of its heterogens, was used as the search model. The structure was refined using PHENIX (36), and Coot (37) was used between refinement cycles for the manual adjustment of the model. The final model had excellent geometry and residual statistics (Table 1).
TABLE 1.
Data collection and refinement statistics
| SUMO-BDP, native | SUMO-BDP, Mn2+ only | |
|---|---|---|
| Data collection | ||
| Space group | P3121 | P3121 |
| Wavelength (Å) | 0.97934 | 1.89029 |
| Resolution range (Å) | 50–2.38 | 37.8–2.55 |
| (2.42–2.38) | (2.59–2.55) | |
| Unique reflections | 22,429 (1,096) | 35,548 (1,572)a |
| Multiplicity | 6.1 (5.3) | 3.5 (2.4) |
| Data completeness (%) | 99.9 (99.7) | 98.3 (86.4) |
| Rmerge (%)b,c | 8.4 (83.6) | 8.7 (57.0) |
| I/σ(I) | 21.5 (2.0) | 20.3 (2.0) |
| Wilson B value (Å2) | 42.0 | 42.0 |
| Refinement | ||
| Resolution range (Å) | 43.9–2.38 | |
| (2.49–2.38) | ||
| No. of reflections Rwork/Rfree | 21,286/1,146 | |
| (2,610/133) | ||
| Data completeness (%) | 99.6 (99.0) | |
| Atoms (non-hydrogen protein/solvent/ions) | 2,040/65/3 | |
| Rwork (%) | 17.7 (26.8) | |
| Rfree (%) | 20.5 (30.6) | |
| r.m.s. deviation bond length (Å) | 0.008 | |
| r.m.s. deviation bond angle (degrees) | 1.1 | |
| Mean B value (Å2) (protein/solvent/ions) | 51.5/47.2/44.8 | |
| Ramachandran plot (%) (favored/additional/disallowed)d | 98.4/1.6/0.0 | |
| Maximum likelihood coordinate error (Å) | 0.61 | |
| Missing residues | 84–89, 248–250, 350–360 | |
a Bijvoet pairs were kept separate for data processing.
b Data for the outermost shell are given in parentheses.
c Rmerge = 100 ΣhΣi|Ih,i − 〈Ih〉|/ΣhΣiIh,i, where the outer sum (h) is over the unique reflections and the inner sum (i) is over the set of independent observations of each unique reflection.
d As defined by the validation suite MolProbity (36).
For calculation of the anomalous difference Fourier maps, the diffraction data were collected from a crystal that had been exposed to 0.3 mm MnCl2 (in the presence of 0.2 m MgCl2; see above); soaking at higher than 0.3 mm MnCl2 resulted in Mn2+ ion precipitation. Long-wavelength (1.89 Å) incident X-rays were employed for data collection. This crystal had the same symmetry as the one described above, but it was not sufficiently isomorphous to the native crystal for facile phase calculation. Therefore, the refined SUMO-BDP model was used as a molecular replacement model to determine the phases for the long-wavelength data set. Once molecular replacement was accomplished, the phases could be calculated from the correctly placed model, facilitating the calculation of the anomalous difference Fourier maps.
Electrostatic calculations were carried out using the program APBS (38), with a protein dielectric of 1 and positive and negative ion concentrations held at 0.15 m. All structure figures were made using PyMOL (Schrödinger, LLC).
Isothermal Titration Calorimetry (ITC) (24–28)
The wild-type and mutant SUMO-BDP proteins were exhaustively dialyzed (three changes) against 2 liters of dialysis buffer containing 50 mm HEPES, pH 7.5, and 100 mm KCl, 5% glycerol, 5 mm EDTA, and 5 mm EGTA. The residual EDTA or EGTA was removed by a final dialysis step in a buffer without these metal chelators. The MnCl2 or MgCl2 solution (400–600 μm) was placed in the syringe and injected in 8-μl increments into the reaction cell containing 1.4 ml of 35–40 μm SUMO-BDP at 20 °C in a VP-ITC microcalorimeter (MicroCal, Northampton, MA). The binding of Mn2+ to SUMO-BDP was very strong, exhibiting a thermogram (not shown) and isotherm (see below) in which the transition from high to low heats of injection was very abrupt. Determining the dissociation (binding) constant (Kd) from such an isotherm is problematic (39). To ameliorate this difficulty, we performed competition titrations (40, 41) in which SUMO-BDP was first saturated with 2–10-fold molar ratios of the competitor, Mg2+. All of the ITC data (six experiments) were globally analyzed using SEDPHAT, utilizing a model in which it is assumed that Mg2+ and Mn2+ compete for the same binding site on SUMO-BDP (42). Error intervals were calculated in this program using the “error projection” method (43, 44). The concentrations of wild-type and ΔN83/F361X SUMO-BDP proteins were determined by absorbance at 280 nm using calculated molar extinction coefficients (m−1·cm−1) of 37,930 and 19,940, respectively.
RESULTS
Monomeric State and Mn2+-dependent Phosphatase Activity of BDP
The mature human mitochondrial BDP (residues 30–372) was expressed as an N-terminal SUMO-tagged fusion and purified on Ni2+-NTA, S-200, and Resource Q columns. To ascertain the oligomerization state of the protein, sedimentation velocity analytical ultracentrifugation was performed (Fig. 1). Although the presence of multiple aggregated forms of the protein is evident in this analysis, one species is responsible for the majority of the signal, which facilitated the estimation of its molar mass: 53,500 g/mol. This result is consistent with the monomeric form of SUMO-BDP (calculated molar mass = 50,922 g/mol). The SUMO moiety was retained for its ability to render BDP more soluble, but this small domain is without effect on the phosphatase activity of BDP (data not shown). For crystallization purposes, the full-length mature BDP was truncated in the N-terminal region (resides 30–83) and at the C-terminal end (residues 361–372); the variant construct is termed ΔN83/F361X SUMO-BDP. The truncated regions were predicted to occur in a random coil structure based on BLAST sequence alignments with the catalytic subunit of human pyruvate dehydrogenase phosphatase 1 (PDP1c) and the rat PDP1c sequence (18). This truncated SUMO-BDP protein was also analyzed using sedimentation velocity (Fig. 1). The predominant species had an estimated molar mass of 44,100 g/mol, which also corresponded well to a monomer (calculated monomer molar mass = 43,493 g/mol). The dialyzed metal ion-free SUMO-BDP assayed using pNPP as a substrate showed an absolute dependence on Mn2+ ions for phosphatase activity, with an optimal concentration of 7 mm and an apparent Km for the cation of 5.1 ± 0.18 mm (Table 2). The high Km for Mn2+ ion explains the relatively high concentration of 10 mm MnCl2 used to assay for BDP activity in the earlier study (12). The related PDP1c phosphatase also shows a dependence on high metal ion concentrations, with a Km of 3.4 mm for MgCl2 (18). Significantly, when Mn2+ ions were replaced by either Mg2+ or Ca2+ ions, BDP completely lacked pNPP phosphatase activity (Table 2). When BDP phosphatase activity was assayed at 7 mm MnCl2, the addition of 20 mm MgCl2 or CaCl2 resulted in 67 and 78% inhibition, respectively (data not shown). Also with pNPP as substrate, the present SUMO-BDP preparation exhibited the highest phosphatase activity at pH 7.5 in the range of pH 5.5–8.0 studied (data not shown). At 20 mm pNPP concentration, SUMO-BDP exhibits a specific activity of 14.8 μmol/min/mg, which is 4-fold higher than previously reported (3.6 μmol/min/mg) for the same phosphatase, termed PTMP (12). The truncated SUMO-BDP variant ΔN83/F361X is catalytically active, showing a Mn2+-dependent specific activity of 8.9 μmol/min/mg, or 60% of that for wild-type SUMO-BDP (data not shown). The Km values for pNPP and Mn2+ are 7.7 ± 0.6 and 8.9 ± 0.2 mm, respectively, for the truncated construct, and Mg2+ and Ca2+ ions are catalytically inactive, similar to those of the wild-type SUMO-BDP. The results suggest that the ΔN83/F361X used for crystallographic studies is similar to the wild-type BDP enzyme.
FIGURE 1.

Size distributions of wild-type and ΔN83/F361X SUMO-BDP. Size c(s) distributions are shown for three different concentrations: low (dotted lines), medium (dashed lines), and high (solid lines); see below for concentration values. The distributions were normalized by the total signal present between 2 and 20 S. A, wild-type SUMO-BDP (low = 4.0 μm, medium = 11.5 μm, high = 35 μm); B, ΔN83/F361X SUMO-BDP (low = 5.7 μm, medium = 16 μm, high = 49 μm).
TABLE 2.
Kinetic parameters of wild-type SUMO-BDP and mutant ΔΝ83/F361X SUMO-BDP proteins with pNPP as substrate
SUMO-BDP and ΔΝ83/F361X SUMO-BDP proteins were expressed in growth medium supplemented with 1 mm MnCl2. The purified enzymes were exhaustively dialyzed against 50 mm HEPES, pH 7.5, 100 mm KCl, 5% glycerol, 5 mm EDTA, and 5 mm EGTA. The residual EDTA and EGTA were removed by a second dialysis step in the absence of the metal chelators. The phosphatase activity of SUMO-BDP (33 nm) and ΔΝ83/F361X SUMO-BDP (35 nm) were assayed spectrophotometrically at 30 °C in a Tris-HCl buffer using varying concentrations of pNPP (0.95 mm to 20 mm) as a substrate; increase in A410 nm was monitored. The kcat is equivalent to a specific activity of 14.8 μmol/min/mg for SUMO-BDP and 8.7 μmol/min/mg for ΔΝ83/F361X SUMO-BDP.
| SUMO-BDP | Metals | Substrate | kcat | Km (pNPP) | Km (metal) | kcat/ Km (pNPP) | kcat/ Km (metal) |
|---|---|---|---|---|---|---|---|
| s−1 | mm | mm | m−1s−1 | m−1s−1 | |||
| Wild type | MnCl2 | pNPP | 43.2 ± 2 | 4.4 ± 0.1 | 5.1 ± 0.2 | 9,818 ± 410 | 8,470 ± 430 |
| Wild type | MgCl2 | pNPP | 0 | ||||
| Wild type | CaCl2 | pNPP | 0 | ||||
| ΔN83/F361X | MnCl2 | pNPP | 25.5 ± 1 | 7.7 ± 0.6 | 8.9 ± 0.2 | 3,311 ± 120 | 2,865 ± 130 |
| ΔN83/F361X | MgCl2 | pNPP | 0 | ||||
| ΔN83/F361X | CaCl2 | pNPP | 0 |
E2b Binds to BDP and Enhances BDP-mediated Dephosphorylation of Phospho-E1b Substrate
To confirm that BDP is the endogenous phosphatase component of BCKDC, we measured phosphatase activity of SUMO-BDP with p-E1b as a substrate. The phosphorylation of E1b was catalyzed by a reconstituted E1b-E2b-BDK subcomplex using [γ-32P]ATP as substrate. His-tagged 32P-labeled p-E1b was dissociated from the 24-meric E2b scaffold in 1 m NaCl and purified on Ni2+-NTA resin. SUMO-BDP catalyzes the release of the 32P-phosphoryl group from p-E1b with a kcat of 17.2 min−1 (equivalent to a specific activity of 68 nmol/min/mg) and a Km of 57.8 μm for p-E1b (Table 3). Pull-down assays were employed to dissect the binding of SUMO-BDP to lipoylated wild-type, non-lipoylated K44A mutant E2b 24-mers, and lipoylated L420X mutant E2b trimers, taking advantage of the N-terminal His6 tag present in SUMO-BDP. The relatively weak binding did not allow for the detection of BDP-E2b interactions by ITC. At high monomer ratios of E2b/SUMO-BDP, significant amounts of His6-tagged SUMO-BDP and wild-type E2b, K44A mutant E2b, or L420X mutant E2b were retained by Ni2+-NTA resin. A close to 1:1 subunit stoichiometry was obtained with the L420X E2b trimer (Fig. 2A). The cubic E2b core of BCKDC is made up of eight trimeric basal units through homologous trimer-trimer interactions, which are abrogated in the L420X E2b variant forming trimers (45). The pull-down data suggest that a saturating concentration of SUMO-BDP can bind to the 24-meric E2b core at the molar ratio of BDP/E2b = 24:1. Because the K44A mutant E2b cannot be lipoylated, the results indicate that the lipoyl prosthetic group of E2b is not needed for the SUMO-BDP-E2b interaction. On the other hand, SUMO-BDP phosphatase activity based on the rate of release of 32P from p-E1b is enhanced 3-fold in the presence of lipoylated wild-type E2b (Fig. 2B). The specific activity with p-E1b as substrate is 2 orders of magnitude lower than that obtained with the artificial substrate of pNPP. With the non-lipoylated K44A E2b variant, in which the lipoylation site is abolished, SUMO-BDP phosphatase activity is still increased 2.5-fold over the basal activity without wild-type or mutant E2b. This result shows that the lipoyl moiety is not essential for the E2b-mediated stimulation of BDP phosphatase activity.
TABLE 3.
Kinetic parameters of SUMO-BDP with p-E1b as substrate
SUMO-BDP phosphatase activity was measured at 37 °C in 50 mm MOPS, pH 7.5, 7 mm MnCl2, and 100 mm NaCl with increasing concentrations of 32P-labeled p-E1b and E2b prepared as described under “Experimental Procedures.” The kcat is equivalent to a specific activity of 68 nmol/min/mg.
| Metal | Substrate | kcat | Km (E1b) | kcat/Km (E1b) |
|---|---|---|---|---|
| min−1 | μm | μm−1min−1 | ||
| MnCl2 | p-E1b | 17.2 ± 0.88 | 57.8 ± 4.8 | 0.29 ± 0.035 |
FIGURE 2.

SUMO-BDP phosphatase activity with 32P-labeled phospho-E1b as substrate and interactions with E2b. A, pull-down assays of SUMO-BDP with lipoylated wild type, non-lipoylated K44A and lipoylated trimers. His6-tagged SUMO-BDP was incubated with a monomer excess of wild-type or mutant E2b. E2b bound to His6-tagged SUMO-BDP was extracted with Ni2+-NTA resin, eluted with imidazole, and analyzed by SDS-PAGE. B, SUMO-BDP phosphatase activity in the absence and presence of non-lipoylated K44A E2b or lipoylated wild type. Error bars, S.E.
To locate the E2b region(s) required for interactions with SUMO-BDP, different E2b domain constructs were engineered and expressed (Fig. 3A). SUMO-BDP activity was assayed with pNPP as substrate in the absence and presence of a different lipoylated E2b domain construct (Fig. 3B). The LBD84 construct (residues 1–84), containing the entire LBD without the C-terminal linker region (Fig. 3A), has no effect on BDP activity. BDP activity is stimulated about 2-fold in the presence of the LBD99 (residues 1–99) construct containing the LBD domain and most of the linker region connecting LBD and the subunit-binding domain (SBD); the latter domain binds E1b or E3. A similar degree of increased BDP activity was obtained with the LBD-SBD152 construct harboring both LBD and SBD domains and the complete linker region. These results indicate that the linker region (residues 85–99) C-terminal to LBD in E2b is essential for the stimulation of SUMO-BDP activity. Additional stimulation of SUMO-BDP activity to about 3-fold was observed with the full-length E2b 24-mer compared with the LBD-SBD152 construct. The additional stimulated BDP activity probably results from the multivalence effect (i.e. avidity) (46) associated with the 24-meric E2b core.
FIGURE 3.

Interactions of SUMO-BDP with E2b and E2b domains. A, lipoylated full-length E2b and E2b domain constructs. The full-length E2b contains an LBD, an SBD, and a core domain connected by linker regions. The His6 tag is present at the C termini of LBD84, LBD99, and LBD152 constructs. The bracket in LBD99 depicts amino acid residues in the C-terminal linker region of LBD. The TEV cleavage site immediately downstream of the SBD was used to generate the LBD-SBD152 protein by protease digestion. B, SUMO-BDP phosphatase activity with pNPP as substrate in the absence and presence of various E2b domain constructs. C, pull-down assays of incubation mixtures containing untagged BDP and lipoylated C-terminal His6-tagged wild-type or mutant LBD99 constructs. The bound (B) and the flow-through (F) fractions from Ni2+-NTA resin extractions were analyzed by SDS-PAGE. Error bars, S.E.
The above results suggest that the C-terminal linker region of the LBD99 construct is necessary for stimulation of BDP activity. Pull-down assays with Ni2+-NTA resin were also employed to locate residues in the C-terminal His6-tagged LBD99 construct that are critical for interactions with SUMO-free untagged BDP. Lipoylated wild-type and mutant LBD99 constructs and BDP at a 1:1 molar ratio (based on the respective monomers) were mixed and applied to a column containing Ni2+-NTA resin. Fractions were collected from the column's flow-through (“flow-through fractions”) and also following application of 200 mm imidazole (“bound fractions”). Both sets of fractions were subjected to SDS-PAGE (Fig. 3C). Untagged BDP and wild-type His6-tagged LBD99 constructs appear exclusively in the bound fractions, supporting the interaction between the two proteins. Substitutions of Glu-87 (E87A) and Asp-88 (D88A) with alanine completely abolish the binding, with untagged BDP present entirely in the flow-through fractions. A similar result was obtained with the LBD domain (residues 1–84) without the linker, indicating the absence of binding to BDP. Alanine substitutions in Glu-86 (E86A), Glu-91 (E91A), and Asp-98 (D98A) do not impede BDP binding to the LBD99 construct, with the former constructs present only in the bound fractions. These results, taken together, establish that residues Glu-87 and Asp-88 in the linker region between LBD and SBD domains of E2b are essential for BDP binding to the E2b core of BCKDC.
Crystal Structure of Human BDP
Full-length human SUMO-BDP was resistant to crystallization despite repeated attempts. Therefore, putatively flexible segments of amino acids (residues 30–83 and 361–373) based on sequence alignment with human PDP1c (18) were deleted from the protein construct. The resulting protein, ΔN83/F361X SUMO-BDP, crystallized readily; the SUMO tag was retained for the solubility it confers on the protein. The crystals diffracted x-rays to a dmin spacing of 2.4 Å, and these data were suitable for structure determination and refinement (see Table 1). Thus, the structural information throughout this paper refers to this truncated version of the protein. Although the SUMO tag was present on the protein used for crystallization, no electron density could be located for it, so it was presumed to be disordered. We therefore refer to the structure below simply as “BDP.” Visible in electron density maps were residues 90–247 and 251–349.
The structure of BDP (Fig. 4) is dominated by two central β-sheets that pack on each other, forming a β-sandwich. Flanking each β-sheet is a pair of antiparallel α-helices. A marked protrusion is proximal to one of the sheets; it consists mainly of α-helices (Fig. 4, A and B). The poorly ordered loop at residues 248–250 occurs at the periphery of this protrusion. A less marked but evident protrusion occurs on the same side of the protein but from the opposite β-sheet; it is made up of mostly loop regions. Between these two protrusions is a narrow cleft ∼10 Å wide. Reports on previous homologous structures of metal-dependent phosphatases identified the cleft as the enzyme's active site (47).
FIGURE 4.

Overall structural features of SUMO-BDP. A, ribbon representation of the structure. The α-helices are shown in blue, whereas the β-strands are drawn in purple. Regions without regular secondary structure are shown in green. A poorly ordered loop between residues 247 and 251 is shown as green dots. The N and C termini of the protein are labeled. The two bound metal ions are shown as gray spheres. B and C, surface and electrostatic surface features of the protein. The electrostatic potential of the protein (see “Experimental Procedures”) is drawn on a van der Waals surface representation. A legend to the color scheme is shown at the bottom of B. The view in B is the same as that in A. The arrows indicate that the view in C is 180° rotated from that in B.
Electrostatic calculations (Fig. 4, B and C) demonstrate that this cleft is dominated by negative electrostatic potential. Other notable features from these calculations are the negative potential found on a flat surface near the cleft (Fig. 4B) and the positive potential in a pair of depressions on the face opposite the flat face (Fig. 4C).
Crystallographic Evaluation of Metal Ion Binding
The dramatically negative electrostatic potential in the active-site cleft is caused by the presence of a cluster of acidic amino acids there. The carboxylate moieties of some of these residues participate in binding two divalent metal ions (Fig. 5, A and B; these cations were not included in the electrostatic calculations). In accordance with precedent in the field (48), we term these cations “M1” and “M2” (Fig. 5A). Given the fact that this crystal was grown in the presence of 0.2 m MgCl2, it is very likely that both metal ions are Mg2+. The carboxylate moieties of three acidic residues (Asp-127, Asp-298, and Asp-337) form inner-sphere contacts with M2; Asp-127 and the main-chain oxygen atom of Gly-128 directly contact M1. Thus, the carboxylate moiety of Asp-127 is a bridging ligand in that it has inner-sphere contacts to both metal ions. None of the carboxylate-cation interactions are bidentate (i.e. a given carboxylate donates only one oxygen atom to ligate either of the metal ions). A third metal ion is present in the structure, about 20 Å distant from the active site. It is bound to two aspartic acid residues (Asp-112 and Glu-143) on the flat, negatively charged surface noted above (Fig. 4B). Its octahedral coordination environment and the presence of 0.2 m MgCl2 in the crystallization and cryoprotection solutions justify its assignment as Mg2+.
FIGURE 5.

Metal interaction residues in the SUMO-BDP active site. Crystals of SUMO-BDP without added MnCl2 were grown by the hanging drop method in 19% (w/v) PEG 3350, 0.2 m MgCl2, 0.1 m Tris-HCl, pH 8.5, and 20 mm β-mercaptoethanol. A, metal ions. Metal ion 1 (M1) and metal ion 2 (M2), both likely Mg2+ ions, are shown as gray spheres. Side chains from the protein are shown in ball-and-stick format, with oxygen atoms in red, nitrogens in blue, and carbons in light blue. Purple dashed lines show inner-sphere contacts from the protein or water molecules (red spheres) to the metal ions, and black dashed lines show apparent hydrogen bonds. Side chains are labeled with their respective residues, and secondary structure is shown faded for clarity. These coloring and labeling conventions hold for the remainder of the structural figures. B, expanded view of the active site.
The coordination of the metal ions would be incomplete if only the carboxylates participated. The coordination spheres of the two metal ions also include water molecules (Fig. 5A). Thus, the coordination geometry of both metal ions is octahedral, with some distortions from ideal angles. One water molecule, which we refer to below as Wat1, serves as an inner-sphere ligand to both metal ions (Fig. 5A).
Some of the acidic residues in the cleft do not make direct contacts to the metal ions; instead, they form apparent hydrogen bonds with water molecules that are inner-sphere contacts. These acidic residues are thus outer-sphere ligands to the metal ions. They are Glu-108 and Asp-109. Of these two, Asp-109 appears to be key because it apparently hydrogen-bonds to inner-sphere water molecules for both metal ions. One of these bonds appears to be bifurcated (Fig. 5B).
An extensive network of hydrogen bonds among water molecule and side chains exists in this active site. Of particular note is the side chain of His-129. Nϵ of its imidazole group is apparently engaged in a bifurcated hydrogen bond with the side-chain carboxylate moiety of Asp-266. On the other hand, Nδ of His-129 is within hydrogen-bonding distance of a water molecule residing near the active-site metals. Hence, this water and/or His-129 appear to be activated to participate in catalysis (see below).
Ascertaining the identities of the metal ions that can bind in the active site of BDP is important for two reasons. First, we have shown that efficient enzyme activity is dependent on the presence of Mn2+ in solution (Table 2). It thus becomes important to establish whether Mn2+ can bind at the putative active site. Second, at least one related phosphatase, MspP from Mycobacterium smegmatis, binds three metal ions in the active site (48). Therefore, it is necessary to rule out that some of the electron density that we modeled as water molecules is instead a metal ion. To address these questions experimentally, we expressed SUMO-BDP with added 1 mm MnCl2 in the culture medium. We grew BDP crystals in the presence of 0.2 m MgCl2 and 0.3 mm MnCl2; the crystals were then soaked with 0.3 mm MnCl2 in the same crystallization buffer. A higher concentration of MnCl2 in the soaking solution was not used because it resulted in the precipitation of Mn2+. Diffraction data were collected from the Mn2+ ion-soaked BDP crystals using long-wavelength x-rays. Any Mn2+ ions bound to BDP should diffract such x-rays anomalously, allowing us to locate the cations in an anomalous difference Fourier map. The resulting map is shown in Fig. 6. Clearly, only two Mn2+ ions are bound in the active site, not three. The strong peak in the anomalous difference Fourier map at M2 indicates that this site has a pronounced preference for Mn2+ under the crystallographic conditions; that the cation binds at M2 despite the 67-fold molar excess of Mg2+ competitor present in crystallization buffer. Site M1 also has a significant peak associated with it in this map. However, it is less prominent than that at site M2. This fact suggests either that M1 prefers Mg2+ or that there is no strong metal ion preference at this site. Nevertheless, the fact that any Mn2+ binding can occur at both metal positions despite the presence of a large molar excess of a competitor ion is strong evidence that Mn2+ would bind at both sites in the absence of competing divalent metal ions.
FIGURE 6.

Evidence for Mn2+ bound in the active site of SUMO-BDP. A, anomalous signal due to Mn2+ ions. SUMO-BDP expressed in culture media supplemented with 1 mm MnCl2 was crystallized in a Tris buffer containing 0.2 m MgCl2 and 0.3 MnCl2. The crystal was soaked with 0.3 mm MnCl2 in the crystallization buffer for 18–24 h, followed by the collection of long-wavelength x-ray diffraction data. Superimposed on the final refined model, which has been transformed to the coordinate system of a Mn2+-only-containing crystal, is an anomalous difference Fourier map (black mesh) contoured at the 4-σ level. The representation and coloration are as in Fig. 5.
Comparisons with Other Phosphatase Structures
Given that a conserved domain search (49) identified BDP as a member of the PP2C family, it is unsurprising that searching the Protein Data Bank with DALI (50) yields a number of structures for PP2C-like proteins. Among them are PP2Cs from Arabidopsis thaliana (r.m.s. deviation = 1.2 Å for 241 comparable Cα atoms) and Taxoplasma gondii (r.m.s. deviation = 1.2 Å for 236 comparable Cα atoms), but the most prominent of these structures is that of a similar construct of truncated human PP2Cκ (residues 89–351) determined by the New York SGX Research Center for Structural Genomics (51) (Protein Data Bank accession code 2iq1). BDP and PP2Cκ are products of the same gene; we retain different names herein to distinguish between the two crystal structures. The r.m.s. deviation of the PP2Cκ structure compared with the present BDP structure (residues 84–361) is about 0.9 Å. There are several differences between the BDP and PP2Cκ structures. For example, the course of the C terminus of the two proteins is different. In PP2Cκ, the C terminus forms a turn, whereas this turn is not present in BDP (Fig. 7A). The lack of this feature in BDP causes local rearrangements in the structures of a nearby β-strand and α-helix. Further, a loop at the apex of the “marked protrusion” described above is disordered in the current BDP structure but not in PP2Cκ. Conversely, a loop that is present in BDP (residues 178–180) near the flat face of the molecule is not modeled in PP2Cκ.
FIGURE 7.

Comparisons of the BDP structure with PP2Cκ (Protein Data Bank code 2iq1). A, large scale comparison of the two structures. SUMO-BDP is shown in blue, and PP2Cκ is shown in red. The three areas of interest noted under “Results” are circled. At the top left is a loop that is ordered in PP2Cκ but has ambiguous electron density in BDP and was thus left unmodeled. At the bottom left is a loop that was ordered in BDP but not in PP2Cκ. At the right, the deviant courses of the respective C termini can be seen. The two metal ions in BDP are shown as gray spheres for a spatial reference. B, differences in the active site. The BDP residues and metal ions are colored as in Fig. 5. The corresponding residues and the metal ion in PP2Cκ are depicted in pink.
We note that the active sites of the previously reported PP2Cκ and BDP differ significantly. In PP2Cκ, only M1 (modeled as a Mg2+) is present in the active site, and two of the aspartate side chains (Asp-298 and Asp-337) that ligate to M2 are oriented away from the metal-binding site (Fig. 7B). The precipitation buffer for PP2Cκ did not include metal ions. Unfortunately, details on the purification protocol of the protein used for PP2Cκ are not available; therefore, it is not known whether that protein was exposed to metal ions in its purification or storage conditions. Because PP2C-type phosphatases are known to require both metal ions (47), it is likely that the active-site structure in the current report (Fig. 5B) is more relevant than that in PP2Cκ for understanding the catalytic details of the enzyme.
Finally, we compared BDP to another related mammalian phosphatase, PDP1c, which is responsible for the dephosphorylation of the E1p subunit of the PDC. Compared with PDP1c, BDP is a minimalistic phosphatase; the former is decorated with additional structural features not found in BDP. A common phosphatase “core” can be defined (Fig. 8), comprising most of BDP. A comparison of the two structures shows that 223 Cα atoms in this core are comparable and have a 1.3 Å r.m.s. deviation The additional features of PDP1c occur at the N and C termini and at positions (in the BDP numbering) 151, 216, 269, 318, and 332. The most prominent of these extra features of PDP1c is a 60-residue span that includes loops and α-helices. This feature is posited to bind to the lipoic acid bound to the inner lipoyl-bearing domain L2 of the E2p subunit of the PDC (21). Both PDP1c and BDP tether to the respective E2p and E2b cores of PDC and BCKDC. The lipoic acid prosthetic group of the lipoyl-bearing domain is required neither for the E2b stimulation of BDP phosphatase activity nor for the binding of BDP to E2b (Fig. 2, A and B). These results may be explained by the absence of the extra PDP1c structural element (Fig. 8, circled) in BDP.
FIGURE 8.

A structural comparison of BDP with PDP1c. A superposition is shown. SUMO-BDP is shown in blue, and the catalytic subunit of PDP1 (Protein Data Bank accession code 2PNQ, chain A) is shown in magenta. The putative lipoic acid-binding pocket of PDP1c is circled; BDP has no analogous structure.
Metal Ion Binding Measurements with Wild-type SUMO-BDP
ITC was employed to decipher differential binding affinities of SUMO-BDP for Mn2+ and Mg2+ ions. Titrating Mn2+ into exhaustively dialyzed, chelator-treated SUMO-BDP results in an exothermic binding isotherm (Fig. 9). In marked contrast, titrating Mg2+ into SUMO-BDP shows that the binding of this cation is endothermic (Fig. 9). These ITC measurements indicate a molar ratio of ∼1:1 between Mn2+ or Mg2+ ions and SUMO-BDP monomers. Given these results and the anomalous difference Fourier map (Fig. 6), it is probable that the metal ion binding detected in the titrations is occurring at site M2 and that binding of metal at site M1 is below the detection threshold of these experiments.
FIGURE 9.

Metal ion binding to SUMO-BDP determined by isothermal titration calorimetry. SUMO-BDP was expressed in a growth medium supplemented with 1 mm MnCl2. The purified enzyme was exhaustively dialyzed against 50 mm HEPES, pH 7.5, 100 mm KCl, 5% glycerol, 5 mm EDTA, and 5 mm EGTA. The residual EDTA and EGTA were removed by a second dialysis step in the absence of the metal chelators. Individual or competitive titrations of SUMO-BDP with metal ions were performed as described under “Experimental Procedures.” The isotherms for three of the six globally analyzed ITC experiments are shown. They are Mg2+-only (black circles), Mn2+-only (gray squares), and the competition experiment (Mn2+ titrated into a 10:1 mixture of Mg2+ and SUMO-BDP, respectively; light gray diamonds). The results of the global fitting session are shown as the respectively colored lines.
To extend this calorimetric analysis, we used global fitting in the program SEDPHAT (42). Six titrations were simultaneously analyzed: 1) two titrations of Mn2+ into SUMO-BDP, 2) two titrations of Mg2+ into SUMO-BDP, and 3) two competitive titrations of Mn2+ into a mixture containing Mg2+ and SUMO-BDP, with Mg2+ present in molar excesses of either 2- or 10-fold. The competitive titrations were performed because of the difficulty in accurately fitting high-affinity calorimetric titrations (39) caused by the very abrupt transition between the early, high heats of injection and the later, lower heats of injection. The competitive titration allows for the more accurate determination of the very low dissociation constant (see “Experimental Procedures”). As shown in Fig. 9, the competitive binding model fits the data well. In this analysis, the Kd for Mn2+ was 2.4 (0.73 to 5.84) nm (the values in parentheses indicate the 68.3% confidence interval for this quantity). The ΔH (Mn2+) was −4.0 (−4.2 to −3.8) kcal/mol. Thus, ΔH (Mn2+) is well determined by this experiment, but Kd (Mn2+) is small and has a large error interval, even with the competition experiment included. Kd (Mg2+) was 10 (6 to 17) μm, which is more than 4,000-fold higher than Kd (Mn2+), and ΔH (Mg2+) was fitted as 4.6 (4.0 to 5.2) kcal/mol. Implicit in this model is a 1:1 stoichiometry of metal ion and SUMO-BDP; deviations from this assumption were modeled as “incompetent fractions” of SUMO-BDP, with these values ranging from 0 to 0.281. The competitive titration in which Mg2+ was titrated into a mixture of Mn2+ and SUMO-BDP (with Mn2+ present at a molar excess over SUMO-BDP) resulted in an undetectable ΔH (Mg2+) (data not shown), consistent with the inability of Mg2+ to compete off the tight-binding Mn2+ at the M2 site. Interestingly, the ΔH (Mn2+) values were −8.0 kcal/mol in competitive binding experiments (with Mg2+ already bound), which is almost doubled when compared with enthalpy measurements from the Mn2+ titrations alone (−4.0 kcal/mol) (Fig. 9). The combined enthalpy changes in the competition titrations probably reflect both the release of Mg2+ ions and the subsequent binding of Mn2+ ions to the same M2 site.
Structure-guided Site-directed Mutagenesis of Active Site
To confirm that the structural observations had bearing on the catalytic activity of BDP, several residues that ligated metal ions or appeared to be in key positions for catalysis were mutated to alanine. First, the acidic residues (Asp-127, Asp-298, and Asp-337) that serve as inner-sphere ligands to the two metal ions were mutated. None of these mutated proteins exhibited any measurable phosphatase activity using a native phosphorylated peptide as the substrate (Table 4). Asp-109, an outer-sphere ligand to both metal ions, was also mutated to alanine, with the same deleterious result. These observations confirm that the metal ligands are critical for catalysis by BDP.
TABLE 4.
Enzyme kinetic parameters assayed with p-S292 heptapeptide for wild-type and mutant SUMO-BDP carrying alanine substitutions in residues coordinated to the Mn2+ ions
The dephosphorylation reaction was carried out at 22 °C in 50 mm HEPES, pH 7.5, 7 mm MnCl2, and 100 mm KCl with increasing concentrations of Ser(P)-292 peptide. The kcat for wild-type SUMO-BDP is equivalent to a specific activity of 85 nmol/min/mg.
| SUMO-BDP | kcat | Km (Ser(P)-292) | kcat/Km (Ser(P)-292) |
|---|---|---|---|
| min−1 | μm | μm−1min−1 | |
| WT | 21.5 ± 1.2 | 3.1 ± 0.16 | 6.9 ± 0.64 |
| R104A | 21.9 ± 0.8 | 189 ± 15.6 | 0.116 ± 0.012 |
| H129A | 23.2 ± 1.4 | 5.2 ± 0.08 | 4.4 ± 0.48 |
| D109A | 0 | ||
| D127A | 0 | ||
| D298A | 0 | ||
| D337A | 0 |
The configuration of the active site of BDP suggests that other residues besides those that ligate metal ions can be important for catalysis. Among these are Arg-104 and His-129. As mentioned above, the guanidinium moiety of Arg-104 appears to be positioned to interact with the phosphate group of the substrate. His-129 interacts with a water molecule near the metal ions and with Asp-266 (Fig. 5B). Mutation of Arg-104 to Ala reduces the catalytic efficiency (kcat/Km) of this mutant BDP toward the peptide substrate to <2% of the wild type (Table 4). This reduction is entirely due to a 61-fold increase in Km (with kcat remaining unchanged) relative to the wild-type enzyme, suggesting that this residue's side chain is important for substrate binding. However, the mutation of His-129 to Ala causes only a small change in the catalytic efficiency. The imidazolium group of His-129 is therefore only marginally involved in catalysis, or another group assumes its catalytic role when it is absent.
The substrate mimic pNPP is commonly used to monitor the catalytic activity of phosphatases (29, 47). Hence, we also used this assay to measure the activities of wild-type and variant BDP proteins carrying substitutions in residues coordinated to the Mn2+ ion (Table 5). As before, acidic residues (Asp-109, Asp-127, Asp-298, and Asp-337) that were tested and that are inner- or outer-sphere ligands to the metal ions were indispensable for BDP activity using the pNPP assay.
TABLE 5.
Enzyme kinetic parameters (kcat and Km) assayed with pNPP for wild-type and mutant SUMO-BDP carrying alanine substitutions in residues coordinated to the Mn2+ ions
SUMO-BDP phosphatase activity was assayed at 30 °C in 20 mm Tris-HCl, pH 7.5, 7 mm MnCl2 with 20 mm pNPP as a substrate.
| SUMO-BDP | kcat | Km (pNPP) | kcat/Km (pNPP) |
|---|---|---|---|
| s−1 | mm | m−1s−1 | |
| WT | 43.1 ± 1.6 | 4.4 ± 0.11 | 9,818 ± 414 |
| D109A | 0 | ||
| D127A | 0 | ||
| G128A | 44.3 ± 1.1 | 4.0 ± 0.09 | 11,075 ± 465 |
| D298A | 0 | ||
| D337A | 0 |
Other key residues were also mutated, and the activities of the resulting proteins with respect to pNPP were monitored. The roles of Arg-104 and His-129 seem to be different in the context of the pNPP assay. Table 6 shows that the R104A mutation had only a relatively small (29%) reduction in kcat, with no change in Km toward pNPP when compared with the wild-type enzyme. In contrast, the H129A mutation had drastic effects in the pNPP assay, with the kcat and catalytic efficiency (kcat/Km) of this mutant reduced to only 7 and 6%, respectively, of the wild-type BDP, without an effect on the Km of this variant (Table 6).
TABLE 6.
Enzyme kinetic parameters assayed with pNPP for wild-type and mutant SUMO-BDP carrying alanine substitutions in putative catalytic residues
SUMO-BDP phosphatase activity was assayed at 30 °C in 20 mm Tris-HCl, pH 7.5, 7 mm MnCl2 with 20 mm pNPP as a substrate.
| SUMO-BDP | kcat | Km (pNPP) | kcat/Km (pNPP) |
|---|---|---|---|
| s−1 | mm | m−1s−1 | |
| WT | 43.1 ± 1.6 | 4.4 ± 0.11 | 9,818 ± 414 |
| R104A | 30.7 ± 0.9 | 4.2 ± 0.14 | 7,309 ± 389 |
| H129A | 3.07 ± 0.04 | 5.6 ± 0.08 | 548 ± 55 |
| D266A | 45.2 ± 1.3 | 3.5 ± 0.09 | 12,914 ± 524 |
DISCUSSION
Until recently, investigation into regulation of BCKDC by reversible phosphorylation has largely focused on the kinase component BDK because the phosphatase remained the elusive component of this α-ketoacid dehydrogenase complex. The direct evidence that the PP2Cm phosphatase is the long-sought-after BDP component of mammalian BCKDC was obtained using the PP2Cm−/− mouse model. This model provided, for the first time, the opportunity to study BDP at the molecular level (13). In the present work, the availability of soluble BDP allowed for the characterization of this phosphatase using both biochemical and structural approaches. The binding of this mitochondrial phosphatase to E2b and the resulting activation of phosphatase activity verifies that PP2Cm is the BDP of the mammalian BCKDC. The research in this work regarding the structure and function of this enzyme may have implications for developing novel therapeutic approaches to cancer, diabetes, neural disorders, and cardiovascular diseases (16).
In this study, three different substrates were used to assay for BDP phosphatase activity: pNPP, phosphopeptide, and the native phospho-E1b protein. The specific activity of BDP obtained with pNPP at 14.8 μmol/min/mg (kcat = 43.2 s−1) (Table 5) is the highest among the three substrates used. The high specific activities against pNPP have been reported for other PP2C phosphatases (35, 52, 53). The specific activity assayed with the phosphopeptide substrate of 85 nmol/min/mg (kcat = 21.5 min−1) (Table 4) is 2 orders of magnitude lower than that obtained with pNPP. Similarly, the present BDP preparation showed a specific activity of 68 nmol/min/mg (kcat = 17.2 min−1) when assayed using the native phospho-E1b as the substrate in the presence of E2b. This value probably represents the closest estimate of the reaction rate in the native BCKDC and is 17-fold higher than that of 4 nmol/min/mg protein reported previously (12). The latter specific activity was 1 order of magnitude lower than that of 35 nmol/min/mg for recombinant PDP1c (18). This prompted these authors to suggest that the previous BDP (i.e. PTMP) preparation might not have been optimally folded or that post-translational modification or other protein factors may be required for robust phosphatase activity. In this work, BDP was expressed as an N-terminal SUMO fusion in the presence of co-transformed bacterial chaperonins GroEL and GroES. The higher specific activity of our preparation might be due to the increased solubility imparted by the SUMO tag. More importantly, the finding that the SUMO-BDP specific activity is comparable with PDPs with the current BDP preparation supports the notion that the PP2Cm phosphatase is the bona fide phosphatase component of BCKDC.
ITC has been a sensitive, reliable method to measure the binding of Mn2+ and other divalent metal-ions to proteins, including phosphatases (54–57). In the present study, the significant enthalpy changes resulting from metal binding allow measurements of differential binding affinities between Mn2+ and Mg2+ ions. Presumably, these metals compete for binding at the M2 site. The inability to detect heat changes associated with the M1 site binding may be associated with the low-affinity of this site for both Mn2+ and Mg2+ ions. It is noteworthy that the Kd and Km of BDP for Mn2+ ions differ by 6 orders of magnitude at 2.4 nm and 4.4 mm, respectively, the latter being determined with pNPP as a substrate for phosphatase activity in either the Tris or HEPES buffer. A similar disparity between low Kd and high Km values was observed in a PPM phosphatase, tPphA from Thermosynechococcus elongates (54). Both BDP in this study and the Thermosynechococcus tPphA phosphatase bind Mn2+ ions at more than one site. It can therefore be speculated that the high Km values compared with Kd reflect the concentrations required to saturate the low affinity binding sites for the optimal phosphatase activity of these enzymes. It cannot be ruled out, however, that the difference between Km and Kd for Mn2+ ions may be exaggerated due to the faster kcat using the pNPP substrate; i.e. with pNPP, the classic Michaelis-Menten kinetics is much further away from rapid equilibrium, resulting in inflation in the complex kinetic constant Km when compared with the thermodynamic parameter Kd.
BCKDC and PDC of the mammalian mitochondrial α-ketoacid dehydrogenase complexes are both regulated by reversible phosphorylation in response to hormonal and dietary stimuli (1, 58, 59), and the regulation by the post-translational modification is perturbed in disease states, such as cancer and type 2 diabetes (60–63). BCKDC and PDC differ in the complements of their respective phosphatases; BCKDC contains only one known phosphatase, BDP (13), whereas PDC harbors two phosphatase isoforms (i.e. PDP1 and PDP2) (18). Despite the fact that BDP and the PDPs belong to the PP2C family of phosphatases, there are distinct biochemical properties differentiating these mitochondrial phosphatases of α-ketoacid dehydrogenase complexes. 1) BDP is strictly dependent on Mn2+ for phosphatase activity; the enzyme exhibits no phosphatase activity in the presence of increasing MgCl2 or CaCl2 (Table 2). High concentrations of Mg2+ and Ca2+ ions actually inhibit Mn2+-dependent BDP phosphatase activity, presumably by competing with Mn2+ ions for binding to the same metal-binding sites. These results confirm the metal ion requirement for this phosphatase reported previously (12). The same study showed that the human PTMP (i.e. BDP) when expressed with Mn2+ ion in the culture medium was 50-fold higher than the activity of the protein expressed without Mn2+ ions. It was suggested that the Mn2+ ions may be required for proper folding of this phosphatase, similar to other PP2C phosphatases expressed in E. coli (52). Therefore, in the present study, 1 mm MnCl2 was routinely added to culture media. In contrast, PDP1 and PDP2 rely on Mg2+ ions for phosphatase activity, and PDP1 has additional requirements for Ca2+ ions that promote its binding to the L2 domain of E2p for optimal phosphatase activity (64). 2) BDP functions as a monomer (Fig. 1), whereas PDP1 is a heterodimer with a catalytic subunit (PDP1c) and a regulatory subunit (PDP1r). Upon Ca2+-mediated binding of PDP1 to E2p, the L2 domain forms a functional PDP1c-Ca2+-L2 complex, with a concomitant release of the inhibitory PDP1r subunit (64). 3) BDP phosphatase activity is enhanced by 3-fold upon binding to E2b at a 1:1 subunit stoichiometry. Surprisingly, the lipoyl prosthetic group in the LBD domain is dispensable for BDP-LBD interactions. In PDP1, non-lipoylated L2 domain is incapable of binding to the PDP1c catalytic subunit to enhance PDP1 phosphatase activity (64). The difference in the lipoyl prosthetic group requirement between the two phosphatases may be explained by the lack of a putative lipoic acid-binding pocket on BDP (Fig. 8).
We show that the segment of the LBD C-terminal linker region containing acidic residues Glu-87 and Asp-88 is necessary and essential for BDP binding to the LBD domain of the E2b core (Fig. 3). We also reported previously that the same C-terminal linker of LBD containing the Asp-Ser-Glu-Glu-Asp-Val-Val-Glu (residues 84–91) sequence is critical for the BDK interaction with the LBD domain (23). In parallel, an acidic cluster in the C-terminal linker region of the L2 domain participates in the tethering of PDP1 to the E2p core of PDC (64). Therefore, despite the different requirements for divalent metal ions, the clusters of acidic residues in the linker regions of lipoyl domains, which are necessary for BDP and PDP binding to their respective BCKDC and PDC cores, are conserved. It is conceivable that BDP and BDK interact with different sets of determinants on the LBD domain. However, the recognition of the phosphatase and kinase by the same acidic residues in the C-terminal linker region of LBD may serve as a “gating switch” that prevents simultaneous binding of these two proteins to the same E2b chain of the BCKDC core. The existence of BDP in the monomeric form is incompatible with the “hand-over-hand” movement proposed for the BDK-related pyruvate dehydrogenase kinase on the PDC core for efficient phosphorylation of the bound E1p components (64). Therefore, the BDP of BCKDC may carry out the dephosphorylation of p-E1b immobilized on the SBD domain of individual E2b subunits on the E2b core through dynamic on-off binding to different LBD domains, as a mechanism to increase the efficiency of the dephosphorylation reaction.
The structure of BDP reveals a fold similar to other PP2C-type phosphatases (Figs. 4 and 8). As in these other enzymes, BDP binds two metal ions, and we demonstrated that two Mn2+ ions can bind in the active site despite an overwhelming presence of the inhibitor cation Mg2+, which is necessary for crystallization (Figs. 6 and 9). Mutations to any inner-sphere metal ligand destroy the activity of the enzyme (Table 5), confirming the catalytic essentiality of these residues and presumably of the metal ions. In the native context, an arginine residue (Arg-104) appears to be very important for substrate binding (Table 6). Our kinetic results, coupled with the structural characterization of the BDP, illustrate that the choice of substrate is key for probing the catalytic mechanism of this enzyme (and enzymes in general). The structure pointed out two residues whose side chains appear to be poised to assist catalysis in BDP: Arg-104 and His-129. However, the effects of mutations of these residues on the activity of the enzyme are substrate-dependent. Mutation of Arg-104 to Ala has only a small effect on the activity on pNPP but a more profound effect on BDP activity toward the native-like phosphopeptide (Table 4). Conversely, the side chain of His-129 appears to be very important for the catalytic activity of BDP on pNPP but not on the phosphopeptide. Thus, the enzyme may be using different mechanisms to catalyze the dephosphorylation of these two chemically variant substrates. Given the non-native structure of pNPP, we feel that the results using the phosphopeptide are more physiologically relevant. We thus conclude that Arg-104 is important for substrate binding and that the side chain of His-129 is not performing an essential role in catalysis. Previously, the structurally homologous enzyme PP2Ca was proposed to use the analogous histidine side chain as a proton donor for the leaving, negatively charged oxyanion (48); we observed no evidence of this role in the context of E1b-derived phosphopeptide substrate. Interestingly, just N-terminal to the phosphorylated serine on the substrate peptide are two histidine residues, raising the possibility that one of them could substitute as a proton donor in the absence of the side chain of His-129.
What then is the catalytic mechanism of BDP? For guidance, we may use the close correspondence between the active sites of BDP and PP2Ca. In the latter enzyme, two metal ions are bound, activating the equivalent of Wat1 for attack on the phosphorus atom of the scissile phosphate, with the expulsion of the oxyanion of the phosphoserine (48). It seems likely that the same attack takes place in the active site of BDP. In PP2Ca, the resulting oxyanion is apparently protonated by His-62 in PP2Ca (i.e. His-62 is a general acid). However, as noted above, we posit that BDP must accomplish protonation via an alternative route; perhaps imidizolium moieties in the substrate participate. In a structural study of MspP, a mycobacterial phosphatase with homology to PP2C phosphatases (29), the phosphoryl group of the substrate may directly contact the metal ions as inner-sphere ligands. Interestingly, kinetic studies on MspP also do not support a proton donor role for the equivalent histidine in that active site; it was also proposed in that enzyme that the substrate itself could act as a general acid (48). Notably, MspP apparently binds three metal ions, raising the possibility that its mechanism differs substantially from the eukaryotic enzymes.
An important question regarding BDP is why the enzyme prefers Mn2+ over any other divalent cations. Our structural and kinetic studies do not provide a clear answer to this conundrum. Structural experiments using bound substrate mimetics or products may be able to begin to address this problem. However, structural differences alone may not provide the question's ultimate resolution, which likely includes the physicochemical properties of the metal ions, the active site, and the substrate.
Acknowledgments
The x-ray data are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source, which is operated by the University of Chicago at Argonne, LLC, for the United States Department of Energy, Office of Biological and Environmental Research under Contract DE-AC02–06CH11357. We are indebted to Brittany Young for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants DK26758, DK62306, and DK92921. This work was also supported by Welch Foundation Grant I-1286.
The atomic coordinates and structure factors (code 4DA1) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- BCKDC
- branched-chain α-ketoacid dehydrogenase complex
- BDK
- branched-chain α-ketoacid dehydrogenase kinase
- BDP
- branched-chain α-ketoacid dehydrogenase phosphatase
- E1b
- branched-chain α-ketoacid decarboxylase/dehydrogenase
- p-E1b
- phospho-E1b
- E2b
- dihydrolipoyl transacylase
- E3
- dihydrolipoamide dehydrogenase
- MSUD
- maple syrup urine disease
- PDC
- pyruvate dehydrogenase complex
- E2p
- dihydrolipoyl transacetylase
- PDP
- pyruvate dehydrogenase phosphatase
- PDP1c
- catalytic subunit of PDP
- PP2C
- protein phosphatase 2C
- PP2Cm
- PP2C phosphatase of mitochondrial matrix residence
- PTMP
- PP2C-type mitochondrial phosphatase
- pNPP
- p-nitrophenyl phosphate
- LBD
- lipoyl-bearing domain; subunit-binding domain
- SUMO
- small ubiquitin-like modifier
- TEV
- tobacco etch virus
- ITC
- isothermal titration calorimetry
- r.m.s
- root mean square.
REFERENCES
- 1. Reed L. J., Damuni Z., Merryfield M. L. (1985) Regulation of mammalian pyruvate and branched-chain α-keto acid dehydrogenase complexes by phosphorylation-dephosphorylation. Curr. Top. Cell Regul. 27, 41–49 [DOI] [PubMed] [Google Scholar]
- 2. Chuang D. T., Shih V. E. (2001) Maple syrup urine disease (branched-chain ketoaciduria). in The Metabolic and Molecular Basis of Inherited Disease (Scriver C. R., Beaudet A. L., Sly W. S., Valle D., Childs B., Kinzler K. W., Vogelstein B., eds) pp. 1971–2006, McGraw-Hill, New York [Google Scholar]
- 3. Yeaman S. J. (1989) The 2-oxo acid dehydrogenase complexes. Recent advances. Biochem. J. 257, 625–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harris R. A., Bowker-Kinley M. M., Wu P., Jeng J., Popov K. M. (1997) Dihydrolipoamide dehydrogenase-binding protein of the human pyruvate dehydrogenase complex. DNA-derived amino acid sequence, expression, and reconstitution of the pyruvate dehydrogenase complex. J. Biol. Chem. 272, 19746–19751 [DOI] [PubMed] [Google Scholar]
- 5. Harris R. A., Paxton R., Goodwin G. W., Powell S. M. (1986) Regulation of the branched-chain 2-oxo acid dehydrogenase complex in hepatocytes isolated from rats fed on a low-protein diet. Biochem. J. 234, 285–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Paul H. S., Adibi S. A. (1982) Role of ATP in the regulation of branched-chain α-keto acid dehydrogenase activity in liver and muscle mitochondria of fed, fasted, and diabetic rats. J. Biol. Chem. 257, 4875–4881 [PubMed] [Google Scholar]
- 7. Zhao Y., Denne S. C., Harris R. A. (1993) Developmental pattern of branched-chain 2-oxo acid dehydrogenase complex in rat liver and heart. Biochem. J. 290, 395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang Y. S., Chuang D. T. (1999) Down-regulation of rat mitochondrial branched-chain 2-oxoacid dehydrogenase kinase gene expression by glucocorticoids. Biochem. J. 339, 503–510 [PMC free article] [PubMed] [Google Scholar]
- 9. Wynn R. M., Kato M., Machius M., Chuang J. L., Li J., Tomchick D. R., Chuang D. T. (2004) Molecular mechanism for regulation of the human mitochondrial branched-chain α-ketoacid dehydrogenase complex by phosphorylation. Structure 12, 2185–2196 [DOI] [PubMed] [Google Scholar]
- 10. Damuni Z., Humphreys J. S., Reed L. J. (1986) A potent, heat-stable protein inhibitor of branched-chain α-keto acid dehydrogenase-phosphatase from bovine kidney mitochondria. Proc. Natl. Acad. Sci. U.S.A. 83, 285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Damuni Z., Reed L. J. (1987) Purification and properties of the catalytic subunit of the branched-chain α-keto acid dehydrogenase phosphatase from bovine kidney mitochondria. J. Biol. Chem. 262, 5129–5132 [PubMed] [Google Scholar]
- 12. Joshi M., Jeoung N. H., Popov K. M., Harris R. A. (2007) Identification of a novel PP2C-type mitochondrial phosphatase. Biochem. Biophys. Res. Commun. 356, 38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu G., Sun H., She P., Youn J. Y., Warburton S., Ping P., Vondriska T. M., Cai H., Lynch C. J., Wang Y. (2009) Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J. Clin. Invest. 119, 1678–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oyarzabal A., Martinez-Pardo M., Merinero B., Navarrette R., Desviat L. R., Ugarte M., Rodriquez-Pombo P. (2011) PPM1K defective gene in a mild variant maple syrup urine disease (MSUD) patient with a paternal uniparental isodisomy of chromosome 4. J. Inherit. Metab. Dis. 34 (Suppl 3), S49–S286 [Google Scholar]
- 15. Shi Y. (2009) Serine/threonine phosphatases. Mechanism through structure. Cell 139, 468–484 [DOI] [PubMed] [Google Scholar]
- 16. Lu G., Wang Y. (2008) Functional diversity of mammalian type 2C protein phosphatase isoforms. New tales from an old family. Clin. Exp. Pharmacol. Physiol. 35, 107–112 [DOI] [PubMed] [Google Scholar]
- 17. Lu G., Ren S., Korge P., Choi J., Dong Y., Weiss J., Koehler C., Chen J. N., Wang Y. (2007) A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev. 21, 784–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang B., Gudi R., Wu P., Harris R. A., Hamilton J., Popov K. M. (1998) Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J. Biol. Chem. 273, 17680–17688 [DOI] [PubMed] [Google Scholar]
- 19. Turkan A., Gong X., Peng T., Roche T. E. (2002) Structural requirements within the lipoyl domain for the Ca2+-dependent binding and activation of pyruvate dehydrogenase phosphatase isoform 1 or its catalytic subunit. J. Biol. Chem. 277, 14976–14985 [DOI] [PubMed] [Google Scholar]
- 20. Karpova T., Danchuk S., Huang B., Popov K. M. (2004) Probing a putative active site of the catalytic subunit of pyruvate dehydrogenase phosphatase 1 (PDP1c) by site-directed mutagenesis. Biochim. Biophys. Acta 1700, 43–51 [DOI] [PubMed] [Google Scholar]
- 21. Vassylyev D. G., Symersky J. (2007) Crystal structure of pyruvate dehydrogenase phosphatase 1 and its functional implications. J. Mol. Biol. 370, 417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wynn R. M., Davie J. R., Cox R. P., Chuang D. T. (1992) Chaperonins groEL and groES promote assembly of heterotetramers (α2β2) of mammalian mitochondrial branched-chain α-keto acid decarboxylase in Escherichia coli. J. Biol. Chem. 267, 12400–12403 [PubMed] [Google Scholar]
- 23. Chuang J. L., Wynn R. M., Chuang D. T. (2002) The C-terminal hinge region of lipoic acid-bearing domain of E2b is essential for domain interaction with branched-chain α-keto acid dehydrogenase kinase. J. Biol. Chem. 277, 36905–36908 [DOI] [PubMed] [Google Scholar]
- 24. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schuck P., Perugini M. A., Gonzales N. R., Howlett G. J., Schubert D. (2002) Size-distribution analysis of proteins by analytical ultracentrifugation. Strategies and application to model systems. Biophys. J. 82, 1096–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schuck P., Demeler B. (1999) Direct sedimentation analysis of interference optical data in analytical ultracentrifugation. Biophys. J. 76, 2288–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hedl M., Rodwell V. W. (2004) Enterococcus faecalis mevalonate kinase. Protein Sci. 13, 687–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laue T. M., Shah B. D., Ridgeway R. M., Pelletier S. L. (1992) Computer-aided interpretation of analytical sedimentation data for proteins. in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S. E., Rowe A. J., Horton J. C. eds.) pp. 90–125, The Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 29. Bellinzoni M., Wehenkel A., Shepard W., Alzari P. M. (2007) Insights into the catalytic mechanism of PPM Ser/Thr phosphatases from the atomic resolution structures of a mycobacterial enzyme. Structure 15, 863–872 [DOI] [PubMed] [Google Scholar]
- 30. Chuang J. L., Davie J. R., Wynn R. M., Chuang D. T. (2000) Production of recombinant mammalian holo-E2 and E3 and reconstitution of functional branched-chain α-keto acid dehydrogenase complex with recombinant E1. Methods Enzymol. 324, 192–200 [DOI] [PubMed] [Google Scholar]
- 31. Wynn R. M., Davie J. R., Song J. L., Chuang J. L., Chuang D. T. (2000) Expression of E1 component of human branched-chain α-keto acid dehydrogenase complex in Escherichia coli by cotransformation with chaperonins GroEL and GroES. Methods Enzymol. 324, 179–191 [DOI] [PubMed] [Google Scholar]
- 32. Davie J. R., Wynn R. M., Meng M., Huang Y. S., Aalund G., Chuang D. T., Lau K. S. (1995) Expression and characterization of branched-chain α-ketoacid dehydrogenase kinase from the rat. Is it a histidine-protein kinase? J. Biol. Chem. 270, 19861–19867 [DOI] [PubMed] [Google Scholar]
- 33. Wynn R. M., Kato M., Chuang J. L., Tso S. C., Li J., Chuang D. T. (2008) Pyruvate dehydrogenase kinase-4 structures reveal a metastable open conformation fostering robust core-free basal activity. J. Biol. Chem. 283, 25305–25315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harder K. W., Owen P., Wong L. K., Aebersold R., Clark-Lewis I., Jirik F. R. (1994) Characterization and kinetic analysis of the intracellular domain of human protein tyrosine phosphatase β (HPTP β) using synthetic phosphopeptides. Biochem. J. 298, 395–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Irmler A., Forchhammer K. (2001) A PP2C-type phosphatase dephosphorylates the PII signaling protein in the cyanobacterium Synechocystis PCC 6803. Proc. Natl. Acad. Sci. U.S.A. 98, 12978–12983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems. Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Leavitt S., Freire E. (2001) Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr. Opin. Struct. Biol. 5, 560–566 [DOI] [PubMed] [Google Scholar]
- 40. Sigurskjold B. W. (2000) Exact analysis of competition ligand binding by displacement isothermal titration calorimetry. Anal. Biochem. 277, 260–266 [DOI] [PubMed] [Google Scholar]
- 41. Velazquez-Campoy A., Kiso Y., Freire E. (2001) The binding energetics of first- and second-generation HIV-1 protease inhibitors. Implications for drug design. Arch. Biochem. Biophys. 390, 169–175 [DOI] [PubMed] [Google Scholar]
- 42. Houtman J. C., Brown P. H., Bowden B., Yamaguchi H., Appella E., Samelson L. E., Schuck P. (2007) Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT. Application to adaptor protein complexes in cell signaling. Protein Sci. 16, 30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bevington P. R., Robinson D. K. (1992) Data Reduction and Error Analysis for the Physical Sciences, 2nd Ed., WCB/McGraw Hill, Boston [Google Scholar]
- 44. Padrick S. B., Deka R. K., Chuang J. L., Wynn R. M., Chuang D. T., Norgard M. V., Rosen M. K., Brautigam C. A. (2010) Determination of protein complex stoichiometry through multisignal sedimentation velocity experiments. Anal. Biochem. 407, 89–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kato M., Wynn R. M., Chuang J. L., Brautigam C. A., Custorio M., Chuang D. T. (2006) A synchronized substrate-gating mechanism revealed by cubic-core structure of the bovine branched-chain α-ketoacid dehydrogenase complex. EMBO J. 25, 5983–5994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Levin M. D., Shimizu T. S., Bray D. (2002) Binding and diffusion of CheR molecules within a cluster of membrane receptors. Biophys. J. 82, 1809–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Das A. K., Helps N. R., Cohen P. T., Barford D. (1996) Crystal structure of the protein serine/threonine phosphatase 2C at 2.0 Å resolution. EMBO J. 15, 6798–6809 [PMC free article] [PubMed] [Google Scholar]
- 48. Jackson M. D., Fjeld C. C., Denu J. M. (2003) Probing the function of conserved residues in the serine/threonine phosphatase PP2Cα. Biochemistry 42, 8513–8521 [DOI] [PubMed] [Google Scholar]
- 49. Marchler-Bauer A., Anderson J. B., Chitsaz F., Derbyshire M. K., DeWeese-Scott C., Fong J. H., Geer L. Y., Geer R. C., Gonzales N. R., Gwadz M., He S., Hurwitz D. I., Jackson J. D., Ke Z., Lanczycki C. J., Liebert C. A., Liu C., Lu F., Lu S., Marchler G. H., Mullokandov M., Song J. S., Tasneem A., Thanki N., Yamashita R. A., Zhang D., Zhang N., Bryant S. H. (2009) CDD. Specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 37, D205–D210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Almo S. C., Bonanno J. B., Sauder J. M., Emtage S., Dilorenzo T. P., Malashkevich V., Wasserman S. R., Swaminathan S., Eswaramoorthy S., Agarwal R., Kumaran D., Madegowda M., Ragumani S., Patskovsky Y., Alvarado J., Ramagopal U. A., Faber-Barata J., Chance M. R., Sali A., Fiser A., Zhang Z. Y., Lawrence D. S., Burley S. K. (2007) Structural genomics of protein phosphatases. J. Struct. Funct. Genomics 8, 121–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Komaki K., Katsura K., Ohnishi M., Guang Li M., Sasaki M., Watanabe M., Kobayashi T., Tamura S. (2003) Molecular cloning of PP2Cη, a novel member of the protein phosphatase 2C family. Biochim. Biophys. Acta 1630, 130–137 [DOI] [PubMed] [Google Scholar]
- 53. Marley A. E., Sullivan J. E., Carling D., Abbott W. M., Smith G. J., Taylor I. W., Carey F., Beri R. K. (1996) Biochemical characterization and deletion analysis of recombinant human protein phosphatase 2C α. Biochem. J. 320, 801–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Su J., Schlicker C., Forchhammer K. (2011) A third metal is required for catalytic activity of the signal-transducing protein phosphatase M tPphA. J. Biol. Chem. 286, 13481–13488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Clugston S. L., Yajima R., Honek J. F. (2004) Investigation of metal binding and activation of Escherichia coli glyoxalase I. Kinetic, thermodynamic, and mutagenesis studies. Biochem. J. 377, 309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Baldwin E. T., Sarver R. W., Bryant G. L., Jr., Curry K. A., Fairbanks M. B., Finzel B. C., Garlick R. L., Heinrikson R. L., Horton N. C., Kelley L. L., Mildner A. M., Moon J. B., Mott J. E., Mutchler V. T., Tomich C. S., Watenpaugh K. D., Wiley V. H. (1998) Cation binding to the integrin CD11b I domain and activation model assessment. Structure 6, 923–935 [DOI] [PubMed] [Google Scholar]
- 57. Desrosiers D. C., Sun Y. C., Zaidi A. A., Eggers C. H., Cox D. L., Radolf J. D. (2007) The general transition metal (Tro) and Zn2+ (Znu) transporters in Treponema pallidum. Analysis of metal specificities and expression profiles. Mol. Microbiol. 65, 137–152 [DOI] [PubMed] [Google Scholar]
- 58. Harris R. A., Huang B., Wu P. (2001) Control of pyruvate dehydrogenase kinase gene expression. Adv. Enzyme Regul. 41, 269–288 [DOI] [PubMed] [Google Scholar]
- 59. Harris R. A., Joshi M., Jeoung N. H., Obayashi M. (2005) Overview of the molecular and biochemical basis of branched-chain amino acid catabolism. J. Nutr. 135, 1527S-1530S [DOI] [PubMed] [Google Scholar]
- 60. Michelakis E. D., Webster L., Mackey J. R. (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 99, 989–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Newgard C. B., An J., Bain J. R., Muehlbauer M. J., Stevens R. D., Lien L. F., Haqq A. M., Shah S. H., Arlotto M., Slentz C. A., Rochon J., Gallup D., Ilkayeva O., Wenner B. R., Yancy W. S., Jr., Eisenson H., Musante G., Surwit R. S., Millington D. S., Butler M. D., Svetkey L. P. (2009) A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 9, 311–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hwang B., Jeoung N. H., Harris R. A. (2009) Pyruvate dehydrogenase kinase isoenzyme 4 (PDHK4) deficiency attenuates the long-term negative effects of a high-saturated fat diet. Biochem. J. 423, 243–252 [DOI] [PubMed] [Google Scholar]
- 63. Wang T. J., Larson M. G., Vasan R. S., Cheng S., Rhee E. P., McCabe E., Lewis G. D., Fox C. S., Jacques P. F., Fernandez C., O'Donnell C. J., Carr S. A., Mootha V. K., Florez J. C., Souza A., Melander O., Clish C. B., Gerszten R. E. (2011) Metabolite profiles and the risk of developing diabetes. Nat. Med. 17, 448–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Turkan A., Hiromasa Y., Roche T. E. (2004) Formation of a complex of the catalytic subunit of pyruvate dehydrogenase phosphatase isoform 1 (PDP1c) and the L2 domain forms a Ca2+ binding site and captures PDP1c as a monomer. Biochemistry 43, 15073–15085 [DOI] [PubMed] [Google Scholar]