

Background: Tau aggregation has been implicated in several neurodegenerative diseases.
Results: Intramolecular disulfide bonds retard tau aggregation by stabilizing conformations that lack β-strand content in subsequences that are aggregation-prone.
Conclusion: Tau self-association may be influenced by the precise conformation of aggregation-prone subsequences.
Significance: This is an important step toward understanding structural features that retard tau aggregation.
Keywords: Aggregation, Alzheimer Disease, Computation, Intrinsically Disordered Proteins, Tau
Abstract
Because tau aggregation likely plays a role in a number of neurodegenerative diseases, understanding the processes that affect tau aggregation is of considerable importance. One factor that has been shown to influence the aggregation propensity is the oxidation state of the protein itself. Tau protein, which contains two naturally occurring cysteine residues, can form both intermolecular disulfide bonds and intramolecular disulfide bonds. Several studies suggest that intermolecular disulfide bonds can promote tau aggregation in vitro. By contrast, although there are data to suggest that intramolecular disulfide bond formation retards tau aggregation in vitro, the precise mechanism underlying this observation remains unclear. While it has been hypothesized that a single intramolecular disulfide bond in tau leads to compact conformations that cannot form extended structure consistent with tau fibrils, there are few data to support this conjecture. In the present study we generate oxidized forms of the truncation mutant, K18, which contains all four microtubule binding repeats, and isolate the monomeric fraction, which corresponds to K18 monomers that have a single intramolecular disulfide bond. We study the aggregation propensity of the oxidized monomeric fraction and relate these data to an atomistic model of the K18 unfolded ensemble. Our results argue that the main effect of intramolecular disulfide bond formation is to preferentially stabilize conformers within the unfolded ensemble that place the aggregation-prone tau subsequences, PHF6* and PHF6, in conformations that are inconsistent with the formation of cross-β-structure. These data further our understanding of the precise structural features that retard tau aggregation.
Introduction
Tau is an intrinsically disordered protein (IDP)2 that is found in neurons of the central nervous system (1). Although tau normally serves as a modulator of neuronal stability (2), it can form insoluble aggregates, called neurofibrillary tangles, that are rich in cross-β-structure (3). Moreover, there are data to suggest that neurofibrillary tangle formation is correlated with the loss of microtubules and the interruption of organelle transport along the neuron leading to neuronal dysfunction and death (4–7). Hence, neurofibrillary tangles may represent toxic species that lead directly to neuronal death and dysfunction. In addition, it has also been argued that soluble oligomeric tau aggregates are neurotoxic. Several indirect lines of evidence are consistent with this hypothesis. For example, this conjecture is consistent with a comparison of cases of frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) with cases of early onset Alzheimer disease. FTDP-17 is thought to be caused by mutations in tau and is characterized by neuronal loss (8). However, the brains of FTDP-17-afflicted patients contain one-tenth the number of neurofibrillary tangles compared with brains of Alzheimer disease patients, as detected using the phospho-tau antibody, AT8 (9). In addition, soluble tau oligomers have been shown to contribute to neuronal dysfunction in animal models (10–12). In this paradigm, the formed aggregates may be an attempt by the cell to sequester the more toxic oligomeric species (11). Nevertheless, regardless of the precise form of toxic species, it seems clear that aggregated forms of tau protein play a role in disease pathogenesis.
Deciphering the process underlying the formation of ordered tau aggregates is problematic because tau is intrinsically disordered in solution (14). Although folded proteins typically sample a relatively small and homogeneous set of thermally accessible states, IDPs sample a relatively heterogeneous set of conformations during their biological lifetime (15). As a result, unlike folded proteins, experimental measurements on IDPs are typically more difficult to interpret. Because IDPs adopt many different conformations in solution, most experimental measurements on IDPs correspond to ensemble averages over a relatively large set of dissimilar conformations (16, 17). Hence, techniques that are typically used to characterize folded proteins are of limited use when applied to disordered proteins. Nonetheless, any comprehensive description of an IDP requires one to enumerate its thermodynamically accessible conformations and their relative stabilities. It is only with such data that one can develop a comprehensive understanding of the aggregation process. Indeed, knowledge of the thermally accessible states of the protein can facilitate the interpretation of experimental observations on IDPs.
The results of disulfide-trapping experiments combined with recently constructed atomistic models of the unfolded state of tau protein provide a unique opportunity to probe structural features that affect the propensity of tau to aggregate (18–21). As we explain below, oxidation of tau leads to conformers that have aggregation properties that differ from the wild-type protein. Therefore, although the precise role that disulfide bonded forms of tau play in vivo is not clear, we can use these data to deduce general structural features of the unfolded ensemble that affect tau aggregation.
Isoforms of tau that contain all four microtubule-binding repeats (4R) contain two naturally occurring cysteines at positions 291 and 322 (numbering based on the longest isoform) (22). When tau is exposed to oxidizing conditions, a series of inter- and intramolecular disulfide bonds are formed (20, 23, 24). To probe the role that intermolecular disulfide-bonded species play in the aggregation process, tau mutants that only contain one cysteine residue have been studied. Oxidized forms of this protein are necessarily constrained to form intermolecular disulfide bonds and therefore serve as a vehicle to probe the role that intermolecular disulfide bonds play in fibril formation. Aggregation studies with these species suggest that intermolecular disulfide bond formation facilitates entry into the aggregation cascade (18, 19, 25–27). Additional aggregation experiments using wild-type tau protein, which contains two cysteine residues, have been less straightforward to interpret because the wild-type protein can form both intramolecular and intermolecular disulfide bonds. Furukawa and co-workers attempted to separate monomer peaks (corresponding to conformers with an intramolecular disulfide bond) and dimer peaks (corresponding to species with intermolecular disulfide bonds) from the oxidized wild-type protein using gel filtration chromatography; however, these efforts were unsuccessful (20).
Some experiments suggest that the formation of intramolecular disulfide bonds between the two naturally occurring cysteines leads to conformations that are relatively aggregation-resistant (19, 28). One possible explanation for these results is that the formation of intermolecular disulfide bonds is important for tau self-association. Because conformers that have an intramolecular disulfide bond cannot form intermolecular disulfide bonds (i.e. tau only has 2 cysteines), they do not aggregate. In other words, it may be that the only role of intramolecular disulfide bonds is to stabilize (monomeric) forms of the protein that cannot form intermolecular disulfide bonds. However, recent studies demonstrate that tau (4R) mutants, which do not contain any cysteine residues, still aggregate and form some fibrils over the course of a day (24). Consequently, aggregation can occur in the absence of intermolecular disulfide bond formation. In light of this, the role of intramolecular disulfide bonds is more complex than simply stabilizing protein monomers.
Others have hypothesized that intramolecular disulfide bonds lead to the formation of “compact monomers” that cannot form extended structures capable of forming paired helical filaments (19, 25, 26, 28–31). Nevertheless, there are few if any data to support the notion that compaction alone fully explains the effect of intramolecular disulfide bonds have on tau aggregation kinetics. Moreover, in contrast to the previous studies, it has recently been argued that fibrils can form from structures that contain intramolecular disulfide bonds (20). Consequently, the precise effect that intramolecular disulfide bonds have on the conformational preferences of tau has yet to be elucidated.
In the present study we generate oxidized forms of a 4R truncation mutant of tau protein, K18, isolate the monomeric fraction that contains only intramolecular disulfide bonds, and quantitatively assess its aggregation potential. These observations are then interpreted in light of an atomistic ensemble for the unfolded state of K18 (21). The combination of disulfide trapping experiments and the existence of an atomistic conformational ensemble for K18 provide new insights into how intramolecular disulfide bonds influence the conformational distribution of states, leading to a new ensemble with distinct aggregation properties.
EXPERIMENTAL PROCEDURES
Reagents
Low molecular weight heparin was purchased from Santa Cruz Biotechnology. Thioflavin T (ThT) was purchased from Acros Organics. All other chemicals and reagents were purchased from Invitrogen, BD Biosciences, or Sigma. K18 was expressed as described below using a technique modified from that of Barghorn et al. (32). The purity of the proteins was analyzed by SDS-PAGE, and the protein concentrations were determined by absorbance at 214 nm.
Expression and Purification of K18
DNA coding K18 was cloned in a pRK172 plasmid and transformed into Escherichia coli BL21-Gold (DE3) strain (Agilent Technologies) for expression. Transformed cells were grown at 37 °C in 500 ml of ZYM-5052 autoinduction medium (33) with 100 μg/ml ampicillin at 225 rpm. After a 12-h incubation the cells were collected by centrifugation at 3900 × g for 25 min at 4 °C. The cell pellet was resuspended in 50 ml of lysis buffer (20 mm MES, pH 6.8, 1 mm EDTA, 0.2 mm MgCl2, 5 mm DTT, 1 mm PMSF, 10 μg/ml leupeptin, 2 mm benzamidine), and the cells were subsequently reharvested by centrifugation. The washed cell pellet was then resuspended in 15 ml of lysis buffer and sonicated using an S-4000 Ultrasonic Processor (Misonix). The cells were sonicated in 3-s bursts followed by 10-s pauses under ice-cold conditions. This was carried out until the cells had received ∼10,000 J. The cell homogenate was centrifuged at 3900 × g for 25 min at 4 °C. The resulting supernatant was made up to 500 mm NaCl, boiled for 20 min, and then centrifuged at 3900 × g for 25 min at 4 °C. The cell lysate was dialyzed into 20 mm MES, pH 6.8, 50 mm NaCl, 1 mm EDTA, 1 mm MgCl2, 2 mm DTT, and 0.1 mm PMSF.
A large scale purification of recombinant K18 was performed at 25 °C on a Bio-Rad DuoFlow work station. The cell lysate was loaded onto a 5-ml HiTrap SP FF cation exchange column (Amersham Biosciences) at a flow rate of 5 ml/min. The column was then washed with 15 ml of 20 mm MES, pH 6.8, 50 mm NaCl, 1 mm EDTA, 1 mm MgCl2, 2 mm DTT, and 0.1 mm PMSF until the A280 base line became stable. The K18 protein was eluted with a salt gradient (50-1000 mm NaCl over 25 ml) in the same buffer. One-milliliter fractions were collected and analyzed on SDS-PAGE. The fractions containing K18 were pooled and dialyzed into storage buffer (20 mm MES, pH 6.8, 50 mm NaCl, 1 mm EDTA, 1 mm MgCl2, 2 mm DTT, and 0.1 mm PMSF) and stored at −20 °C. Further purification was carried out by size exclusion chromatography. This involved 0.5 ml of sample being injected onto a Superdex 75 30/300 GL column (Amersham Biosciences) at a flow rate of 1 ml/min. The protein sample was eluted with PBS, pH 7.4, over 2 column volumes. One-milliliter fractions were collected, and those containing K18 species were stored at −20 °C. The purity of the resulting protein was established by SDS-PAGE, and subsequent densitometry was carried out using Molecular Imaging software version 4.0 (Kodak).
Generating Oxidized Forms of K18
Purified K18 was dialyzed against 100 mm Tris, pH 8.4, in the presence of 2 μm CuSO4, which acted as an oxidizing agent. This was then incubated at 37 °C for 8 h with shaking at 150 rpm (following the protocols described in Ref. 34). The formation of disulfide bonds was confirmed by SDS-PAGE, size exclusion chromatography, and mass spectroscopy. Mass spectroscopy was carried out on samples in 4-hydroxy-3,5-dimethoxycinnamic (in 70% ACN and 30% 0.1% TFA) on a AB Sciex 480 Plus MALDI TOF/TOF in linear mode with 6500 laser intensity. The spectra were then calibrated using an external calibration file and confirmed the presence of the disulfide bond in the oxidized K18 monomer.
Thioflavin T Assay
Aggregation was induced by incubating K18 at a concentration of 50 μm in PBS, pH 7.4, in the presence of 12 μm heparin and 1% protease inhibitor mixture (1 mm PMSF, 1 mm EDTA, 1 mm EGTA, 1 μg/ml leupeptin, and 1 μg/ml pepstatin). 200-μl aliquots were incubated in a 96-well plate and incubated at 37 °C with shaking at 120 rpm. The aggregation time course was monitored using the conditions described above with the addition of 25 μm ThT. Fluorescence was monitored every 20 min over the course of 8 days using a Fluoroskan Ascent (Thermo Scientific) with an excitation wavelength of 450 nm and an emission wavelength of 485 nm.
Dynamic Light Scattering
Dynamic light scattering experiments were carried out using a DynaPro Nanostar (Wyatt Technology). Base-line measurements with the buffer alone (without protein) did not show any scatter. Samples of K18 (concentration 50 mm in PBS, pH 7.4, 12 μm heparin, and 1% protease inhibitor mixture), containing intermolecular disulfide bond and K18 solution containing a mixture of inter- and intramolecular disulfide bonds were incubated at 37 °C with shaking at 225 rpm for 5 h. Every hour a sample was taken and assessed by dynamic light scattering. The samples (50 μl) were placed in a disposable UV cuvette and measured using a function that accumulated 10 measurements/sample. The size distribution plots, the x axis showing a distribution of estimated particle radius (nanometers), and the y axis showing the relative intensity of the scattered light (percentage of mass) were analyzed and prepared with the software Dynamics V7.0.0.94 (Wyatt Technology).
Generating Ensemble of Structures for K18
Because we have previously described our method for the construction of an ensemble for K18 in detail (21), we only repeat the salient features of the Bayesian weighting (BW) method here. The BW method can be divided into two steps: (i) generation of a structural library that represents energetically favorable conformations of the protein; (ii) estimating the weights of these conformations and calculating their uncertainties in a computationally efficient manner.
We generated a set of energetically favorable structures for K18 by first dividing the protein into overlapping segments eight residues long. A local sequence size of eight residues was chosen for the size of the peptides used in the segment simulations, which is approximately the size of the average persistence length of a polypeptide (35). The sequence of K18 was divided into 26 peptides of eight residues each, with an overlap of three residues between adjacent segments.
A structural library for K18 was obtained by independently sampling and joining peptide conformations of local segments of the K18 sequence. This scheme is comparable with the structure generation methods in statistical coil algorithms. However, instead of building sequence structures one residue at a time, the sequence is extended by independently sampling and adding one peptide segment at a time. Starting with the N-terminal segment, each subsequent segment structure is sampled independently from the replica exchange molecular dynamics trajectory and aligned by the backbone atoms of the three overlapping residues. An individual K18 conformation is constructed as a Protein Data Bank file, created with duplicate atoms erased and residues renumbered. Structures were minimized to remove bad contacts using 1000 steps of steepest descent minimization followed by 1000 steps of adopted basis Newton-Raphson minimization. In the end a conformational library of 30,000 structures was constructed. To reduce the size of the structural library to a number that could be easily run with the BW algorithm, we used a simple pruning algorithm that reduced the number of structures to 300, which largely captured the diversity seen in the larger initial set of structures.
Once the structural library is constructed, the BW method for calculating a posterior distribution assigns a probability to each possible choice of weights as a way of quantifying uncertainty in the ensemble. The BW probability density function, ƒW⃗|M⃗(w⃗|m⃗), is calculated using Bayes' theorem,
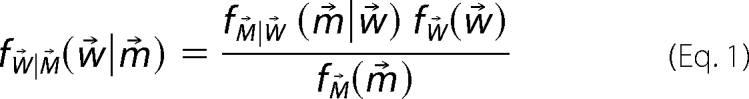 |
where w⃗ = {mi}i=1n denotes the set of population weights for n structures in the structure library and m⃗ = {mi}i=1k denotes the set of k experimental measurements. To calculate Equation 1, we must specify a likelihood function, ƒM⃗|W⃗(m⃗|w⃗), and a prior distribution, ƒW⃗(w⃗). This prior distribution, ƒW⃗(w⃗), is chosen to represent a priori knowledge about the weights, w⃗, and can be estimated from the potential energies of the structures in the structural library.
The likelihood function, ƒM⃗|W⃗(m⃗|w⃗), describes the probability of observing the experimental data, m⃗, for a given weight vector, w⃗. In practice a likelihood function is defined for each type of experimental measurement, e.g. residual dipolar coupling (RDC), chemical shift, radius of gyration estimate, etc., yielding separate probability distributions for each type of experiment; e.g. ƒM⃗|W⃗RDC(m⃗|w⃗), ƒM⃗|W⃗R(G)(m⃗|w⃗). The precise form of each likelihood function was described in our prior work (21). The overall likelihood function is a product of the various likelihood functions.
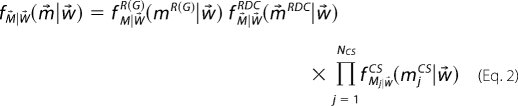 |
A Markov chain Monte Carlo algorithm was used to calculate the needed integrals (21). The posterior density given by Equation 1 can be simulated using Gibbs sampling. A Metropolis-Hastings step was implemented for sampling the weights using a simplicial normal distribution centered at the current weight vector as the proposal distribution. The proposal distribution had an isotropic variance that was tuned during an equilibration period so that about 25% of the steps were accepted.
The tau Markov chain Monte Carlo simulations consisted of a 100-million step equilibration period followed by a 1-billion step sampling period to yield a sample size of 50,000 weight vectors. The running averages for the BW estimates and the posterior expected divergence were monitored to ensure that convergence was achieved. Experimental measurements consisted of NMR chemical shifts, residual dipolar couplings, and small angle x-ray scattering data (36–38). Experimental errors were taken to be 0.1 ppm (39), 1 Hz, (37, 40), and 3 Å (38) for the chemical shifts, RDCs, and radius of gyration, respectively. Errors in the SHIFTX predicted chemical shifts were taken from Neal et al. (41).
Once the ensemble was constructed we wished to determine what structures in the K18 ensemble were capable of forming a disulfide bond between the two naturally occurring cysteine residues (residues 291 and 322 using the numbering from the longest tau isoform). We looked at the Cα–Cα distance between residues 49 and 80 in all structures within the ensemble; i.e. a Cα–Cα distance of ∼6.5 Å is a necessary condition for disulfide cross-link formation (42). Only two structures in the ensemble had a Cα–Cα distance that fulfilled these criteria.
RESULTS
Generating Oxidized Forms of K18
Human neurons contain six isoforms of tau protein that arise from the alternate splicing of exons 2, 3, and 10 of the MAPT (microtubule-associated protein tau) gene located at the locus 17q21 on chromosome 17 (43–47). These isoforms range in length between 352 and 441 amino acids and contain a number of microtubule binding repeats (MTBRs), which are located near the carboxyl terminus of tau and contain either three or four MTBRs. The 130-residue protein, K18, which was used in this study, is a 4R truncation mutant that contains all four MTBRs. Of note, the second (R2) and third (R3) repeats contain hexapeptide sequences (PHF6* and PHF6, respectively) that have been shown to initiate tau aggregation in vitro (48–50).
K18 was purified using was a technique similar to that reported by Barghorn et al. (32) (Fig. 1, lane 1). Purification was carried out in the presence of DTT to prevent disulfide bond formation. SDS-PAGE of the purified protein was carried out after DTT was dialyzed off, and the result is shown in Fig. 1, lane 1. Even though a reducing agent was used during purification, a faint high molecular mass band is present when wild-type (WT) K18 is run under nonreducing conditions (Fig. 1, lane 1) that disappears when the protein is reduced (Fig. 1, lane 4). This suggests that some small amount of intermolecular disulfide bonds form after DTT is removed from the buffer.
FIGURE 1.

Purified K18. Lanes 1–3 were run under nonreducing conditions, whereas lanes 4–6 were run under reducing conditions. The compact monomer formed upon intramolecular disulfide bonding was purified by size exclusion chromatography. Samples were run under nonreducing (lanes 1–3) and reducing conditions (lanes 4–6). The molecular masses according to the densitometry data are 16.6 kDa for K18 and 15.7 kDa for the more compact monomer formed under oxidizing conditions. The molecular masses of the higher order species in lane 2 range from 24.0 to 47.7 kDa.
Incubation of WT K18 in the presence of 2 μm CuSO4, which acts as an oxidizing agent (34), yields multiple oligomeric species (Fig. 1, lane 2) corresponding to dimers, trimers, and other higher order forms. Because the protein is run under nonreducing conditions, the bands run at different molecular masses depending on their disulfide bonding pattern; i.e. a dimer that contains two intermolecular disulfide bonds will run at a different molecular mass than a dimer that contains only one intermolecular disulfide bond. The monomer fraction was isolated from the mixture of oligomers using size exclusion chromatography and purified (under nonreducing conditions) to at least 95% purity (Fig. 1, lane 3). The presence of a single intramolecular disulfide bond in the monomeric fraction was confirmed by MALDI mass spectroscopy (51). Furthermore, we note that the band corresponding to the monomer fraction is slightly shifted to lower molecular masses, a finding that likely reflects that the disulfide bonded monomer is more compact that the reduced monomeric protein (Fig. 1, lane 3 and lowest band in lane 2). This is supported by the observation that the shift in molecular mass is reversed when the protein is reduced (Fig. 1, lane 6).
Characterizing Aggregation Propensity of WT and Oxidized K18
A ThT assay was used to assess the propensity of both WT and oxidized K18 to form fibrils. Because ThT binds to aggregates containing β-strands, an increase in fluorescence suggests the formation of amyloid fibrils (52, 53). Although the experiment was allowed to run for 8 days, the subsequent plots only contain data for the first 90 h (∼4 days) because there was no change in the fluorescent data from day 4 to day 8. Over the 8-day time course an increase in fluorescence was observed for both WT K18 (see Fig. 2, black) and oxidized K18 (Fig. 2, cyan), with the greatest increase in fluorescence being associated with the oxidized sample, which contains compact monomers and higher order disulfide bonded oligomers (Fig. 1, lane 2). By contrast, the fluorescent reading from the oxidized monomer is nearly flat, suggesting that fibrillar structures are not formed by this species (Fig. 2, magenta). The failure of the monomeric oxidized species to form aggregates, in contrast to the oxidized sample (that contains higher order disulfide bonded forms) argues that the formation of intermolecular disulfide bonds promote tau aggregation. This finding is in agreement with prior studies (18, 19, 25, 27). The fact that aggregation of the fully oxidized solution has a longer lag time than the aggregation of the WT protein is explained by the fact that the fully oxidized protein contains a significant fraction of monomeric species that retard aggregation.
FIGURE 2.

β-Sheet-specific aggregation of K18, oxidized K18, and oxidized K18 monomer monitored by ThT fluorescence. Results represent the average (and S.D., error bars) of three independent samples. The experiment was run for 8 days; however, only data from the first 90 h are shown (∼4 days). There was no change in the ThT fluorescence between days 4 and 8.
Although the monomeric species does not form fibrillar structures, it is possible that the protein does form soluble oligomers. To explore how the formation of intramolecular disulfide bonds affects the formation of soluble oligomeric species, as opposed to fibrils, we measured the dynamic light scattering for each of the different K18 species during for the first 5 h after incubating protein samples with heparin, which initiates aggregation. This period corresponds to the early portion of the aggregation pathway, before any significant increase in ThT fluorescence is observed (Fig. 2). At time 0, solutions with WT K18, oxidized K18, and the oxidized K18 monomer contain oligomers that have hydrodynamic radii below 3 nm (Fig. 3). After 5 h, however, WT K18 forms oligomeric species that have hydrodynamic radii that range from 10 nm to ∼2000 nm (Fig. 3, top panel). By contrast, solutions containing oxidized K18 and the oxidized monomer are relatively devoid of species with high hydrodynamic radii (Fig. 3, middle and bottom panels). The similarity between the data from the oxidized K18 species and its monomeric fraction is not surprising in light of the fact that the solution containing oxidized K18 is composed largely of oxidized monomers (Fig. 1, lanes 2 and 3). Moreover, as is shown in Fig. 2, the ThT fluorescence for oxidized K18 does not plateau until approximately 50 h. These data argue that both the formation of soluble oligomers and fibrillar species is delayed in conformers that contain an intramolecular disulfide bond.
FIGURE 3.

Soluble oligomer formation as assessed by dynamic light scattering for WT K18 (black, top panel), oxidized K18 (cyan, middle panel), and the oxidized K18 monomer fraction (magenta, bottom panel). Solid lines represent data at time 0 h, dashed lines correspond to measurements at 5 h.
Correlating Structure with Aggregation Data
Previous observations have suggested that intramolecular disulfide bonds between the two naturally occurring cysteines in tau protein lead to a compact structure that is unable to form extended β-structure, a hallmark of tau fibrils (19, 28). Nevertheless, this qualitative hypothesis does not provide insight into the precise structural features that explain why intramolecular disulfide bonds prevent aggregation. To gain insight into the particular conformational constraints that affect tau self-association, we interpret our aggregation data in light of an atomistic model of the K18 unfolded ensemble, which models the thermally accessible states of the protein.
In general, we model an IDP as a set of interconverting, structurally diverse conformers. We say that an ensemble is fully specified when one is given a predefined set of conformers that represent the dominant thermally accessible states, and their associated weights, or relative stabilities. In the case of K18, exposing the protein to oxidizing conditions yields a series of disulfide bonded species, as schematically shown in Fig. 4. The monomeric fraction is of particular interest because conformers that place the two naturally occurring cysteine residues in positions that are consistent with the formation of a disulfide bond will be stabilized when the protein is oxidized (Fig. 4). Hence, the oxidized monomeric fraction will be enriched with these structures, even if the initial ensemble (prior to oxidation) contains a small number of conformers that are consistent with intramolecular disulfide bond formation.
FIGURE 4.

Schematic representing how exposure to oxidizing conditions traps conformers with intramolecular and intermolecular disulfide bonds. The thiol groups of the two naturally occurring cysteine residues are explicitly shown. Most notably, the monomeric fraction from the oxidized solution contains conformers that have appropriately positioned cysteine residues.
In a previous work we constructed an ensemble for reduced K18 using a BW formalism (21). The method combines data from experiment (typically NMR chemical shifts, residual dipolar couplings, and small angle x-ray scattering data) (36–38) with an efficient conformational search algorithm to construct and evaluate different ensembles that represent the unfolded state of a disordered protein. To be precise, the BW algorithm yields a posterior probability distribution over all possible ways of assigning weights to structures within a predefined structural library, yielding a wealth of information about the different “ensembles” that model the protein of interest. The Bayes' estimate, w⃗B, corresponds to the mean (or expected value) over the entire posterior distribution and is intended to represent the relative weight, or stability, of each structure in the structural library. Together the Bayes' estimate, w⃗B, and the structural library correspond to a reasonable choice for the protein ensemble.
Although it is important to demonstrate that any potential ensemble yields calculated observables that agree with experiment, it is important to note that agreement with experiment alone is not sufficient to ensure that the constructed ensemble is correct (21). This is because the number of experimental constraints used to construct the ensemble is typically much smaller than the protein degrees of freedom. Hence, there are typically many different ensembles that one can construct that agree with a given set of experimental data (21). In light of this, it is important to develop quantitative measures of one's uncertainty in the underlying ensemble. One advantage of the BW method is that it provides a built in parameter called the posterior divergence, also called the uncertainty parameter, 0 ≤ σw⃗(B) ≤ 1, which provides a quantitative measure of how correct the resulting ensemble is. This metric is akin to the S.D. of Gaussian distribution and therefore measures the “spread” of the posterior density function. When σw⃗(B) = 0, we can be reasonably certain that the ensemble is correct; however, when σw⃗(B) ≠ 1 we cannot say with certainty that the ensemble is accurate. Nevertheless, in this latter case we can provide precise confidence intervals, using the posterior distribution, for calculated values of interest.
Application of the BW method to a set of 300 diverse conformers (Fig. 5A) yielded an ensemble that has calculated ensemble averages that agree with the NMR data (21) (Fig. 5, B and C). In addition, the average radius of gyration of the ensemble is 36 ± 0.6 Å, compared with the experimental value of 38 ± 3 Å. For this ensemble the uncertainty parameter is nonzero (σw⃗(B) = 0.33); however, we can provide confidence intervals for calculated observables that quantify our uncertainty.
FIGURE 5.

A, alignment of the 300 structures representing the K18 ensemble under conditions that do not favor the formation of disulfide bonds. B and C, calculated Cα chemical shifts (B) and RDCs (C) show good agreement with experiment.
The experiments discussed in the preceding sections suggest that disulfide bonded K18 monomers are aggregation-resistant. To understand how the formation of intramolecular disulfide bonds leads to aggregation-resistant conformations, we determined what structures within our K18 ensemble are capable of forming disulfide bonds. To this end, we computed the Cα–Cα distance between residues 291 and 322 (numbering based on the longest isoform) in all structures within the ensemble; i.e. Cα atoms of the naturally occurring cysteine residues. Because a Cα–Cα distance of ∼6.5 Å is a necessary condition for disulfide cross-link formation (42), this allows us to identify structures that can potentially form intramolecular disulfide bonds.
An analysis of the Bayes' ensemble suggests that only 2.1% of structures in the unfolded ensemble (90% confidence interval, 0.2–2.9%) can form disulfide bonds, and all of this probability is concentrated in two conformers (Fig. 6, A and B). To assess the aggregation potential of these structures we focus on the aggregation-prone hexapeptide sequences, PHF6* and PHF6, that are known to be minimal interaction motifs that can initiate the formation of β-rich tau aggregates in vitro (49, 50). We hypothesize that structures that place these hexapeptides in β-strand (or extended) conformations are more likely to form cross-β-structure with other tau monomers (49, 50, 54). Secondary structure analysis with STRIDE (13) of these structures suggests that both of these conformers have <5% extended structure. More importantly, no residue in either PHF6* or PHF6 adopts extended structure in these structures, which, again, constitute conformers in the ensemble that could form disulfide bonds.
FIGURE 6.

Backbone traces from representative structures from the Bayes' ensemble. A and B, Cα atoms of residues 291 and 322 are denoted as green spheres. Two structures have Cα–Cα interatomic distances that are consistent with disulfide bond formation between residues 291 and 322. The structures shown in A and B have Cα–Cα distances of 5.4 and 6.6 Å, respectively. C and D, two structures from the Bayes' ensemble place the PHF6* (red) and PHF6 (yellow) in extended conformations.
It may be that the potential disulfide bonded structures shown in Fig. 6, A and B, are lacking extended structure because the conformational sampling algorithm that generated the 300 structures in the ensemble only generates structures that are devoid of secondary structure. In other words, it is possible that the absence of extended structure in the conformations shown in Fig. 6, A and B, is a consequence of how we chose structures to be in the ensemble. Therefore, to determine whether the Bayes' ensemble contains any structures that contain extended structure in the PHF6* and PHF6 hexapeptides, we searched for structures in the ensemble that placed either the PHF6* or PHF6 hexapeptides in solvent-exposed and extended conformations. Two such structures are shown in Fig. 6, C and D. In the structure shown in Fig. 6C the PHF6* region adopts a fully extended strand that is part of a β-hairpin. Similarly, in the structure in Fig. 6D the PHF6 region adopts a strand that is again part of a β-hairpin. Note that in both structures, residues 291 and 322 are too far apart to form a disulfide bond; i.e. their Cα interatomic distances are 71.6 Å and 68.1 Å, respectively. Hence, although the ensemble does contain conformers that place aggregation-prone subsequences in conformations that can readily form cross-β-structure, structures that can form intramolecular disulfide bonds do not. Moreover, although it has been argued that intramolecular disulfide bonds prevent aggregation simply by forming relatively compact states that cannot form extended structure (19, 28), our data suggest that the explanation is more complex. Indeed, the radii of gyration of the four structures in Fig. 5 are 28.7, 26.8, 47.0, and 32.1 Å for structures A, B, C, and D, respectively. Interestingly, the radius of gyration of structures 6, A and D, only differ by 3.4 Å. Whereas the radius of gyration of the structures shown in Fig. 6, A and D, are similar, the conformers differ in other ways. The structures that can form disulfide bonds do not place aggregation-prone sequences in a conformation that is conducive to the formation of cross-β-structure and structures that do not form disulfide bonds have aggregation-prone features (Fig. 6, C and D).
DISCUSSION
Understanding the structural determinants of tau self-association is of the utmost importance as tau aggregation has been implicated in a number of neurodegenerative disorders (4–7). A number of environmental factors such as the oxidation state and the presence of polyanions and fatty acids have been shown to have a significant effect on the aggregation kinetics of tau protein in vitro (19).
In this study we demonstrate that fully oxidized species form more fibrils relative to the WT (Fig. 2) but oxidized forms of K18 that contain an intramolecular disulfide bond are aggregation-resistant. The greater propensity to form fibrillar aggregates by the fully oxidized state is explained by the fact that the oxidized protein contains higher order structures that contain intermolecular disulfide bonds, thereby facilitating tau self-association. By contrast, explaining the aggregation-resistant properties of the oxidized monomeric protein is less straightforward. Previous studies on tau mutants suggest that monomeric forms of tau protein, which cannot form intermolecular disulfide bonds, can still aggregate (24). Consequently, the role that intramolecular disulfide bonds play in preventing tau aggregation is more complex than simply stabilizing the monomeric protein. We therefore hypothesized that intramolecular disulfide bonds introduce conformational preferences that retard tau aggregation. Hence, although the importance of the tau oxidation state has yet to be clarified in vivo, the existence of an aggregation-resistant form of tau (i.e. the oxidized monomeric protein) provides a unique opportunity to deduce structural features of the unfolded ensemble that retard tau aggregation.
Although previous studies have argued that oxidized tau monomers are aggregation-resistant precisely because intramolecular disulfide bonds lead to the formation of compact monomers that cannot form extended structure (19, 25, 26, 28–31), our data argue that the effects of intramolecular disulfide bond formation are more subtle. By analyzing an atomistic ensemble for K18, we are able to correlate data from the disulfide trapping experiments with structural preferences in the unfolded state. Our method for generating the atomistic structural ensemble is based on a BW formalism (21). The result is a Bayes' ensemble for K18 that consists of a set of structures and a set of population weights. In Fig. 6, A and B, we show the two structures in the ensemble that can potentially form disulfide bonds. Although only two structures are shown, their associated population weights allow us to quantify how much of the ensemble resembles these structures; i.e. ∼2.1% of the structures in the unfolded ensemble resemble the structures shown in Fig. 6, A and B. Moreover, an added advantage of the BW method is that we can add error bounds to this estimate. In the present case we can say with 90% confidence that the percentage of structures in the K18 unfolded ensemble that resemble the structures shown in Fig. 6, A and B, is between 0.2 and 2.9%.
Structures in the ensemble that can potentially form disulfide bonds have similar radii of gyration to structures that have aggregation-prone features. More precisely, our observations argue that the structures having intramolecular disulfide bonds have distinct conformational preferences in that they place the aggregation-prone sequences, PHF6* and PHF6, in conformations that cannot readily form cross-β-structures. Hence, it is the precise conformational preferences in aggregation-prone subsequences within tau that may explain the aggregation properties of disulfide-bonded K18 monomers and not simply their degree of compaction. These data highlight the need to interpret experimental observations on IDPs in terms of precise atomistic models that capture important features of the unfolded ensemble. Moreover, studies that further our understanding of the molecular features that prevent tau aggregation can serve as a springboard for the design of therapies that prevent tau aggregation. For example, therapies that stabilize conformations that are relatively aggregation-resistant (e.g. the structures shown in Fig. 6, A and B) may form a viable means of preventing tau aggregation in vivo.
This work was supported, in whole or in part, by National Institutes of Health Grant 5R21NS063185-02.
- IDP
- intrinsically disordered protein
- BW
- Bayesian weighting
- FTDP-17
- frontotemporal dementia with parkinsonism linked to chromosome 17
- 4R
- four microtubule-binding repeats
- ThT
- thioflavin T
- RDC
- residual dipolar coupling.
REFERENCES
- 1. Buée L., Bussière T., Buée-Scherrer V., Delacourte A., Hof P. R. (2000) Tau protein isoforms, phosphorylation, and role in neurodegenerative disorders. Brain Res. Rev. 33, 95–130 [DOI] [PubMed] [Google Scholar]
- 2. Paglini G., Peris L., Mascotti F., Quiroga S., Caceres A. (2000) Tau protein function in axonal formation. Neurochem. Res. 25, 37–42 [DOI] [PubMed] [Google Scholar]
- 3. Grundke-Iqbal I., Iqbal K., Tung Y. C., Quinlan M., Wisniewski H. M., Binder L. I. (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Terry R. D. (1996) The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J. Neuropathol. Exp. Neurol. 55, 1023–1025 [PubMed] [Google Scholar]
- 5. Delaère P., Duyckaerts C., Brion J. P., Poulain V., Hauw J. J. (1989) Tau, paired helical filaments, and amyloid in the neocortex: a morphometric study of 15 cases with graded intellectual status in aging and senile dementia of Alzheimer type. Acta Neuropathol. 77, 645–653 [DOI] [PubMed] [Google Scholar]
- 6. Giannakopoulos P., Herrmann F. R., Bussière T., Bouras C., Kövari E., Perl D. P., Morrison J. H., Gold G., Hof P. R. (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology 60, 1495–1500 [DOI] [PubMed] [Google Scholar]
- 7. Gómez-Isla T., Hollister R., West H., Mui S., Growdon J. H., Petersen R. C., Parisi J. E., Hyman B. T. (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann. Neurol. 41, 17–24 [DOI] [PubMed] [Google Scholar]
- 8. Hutton M., Lendon C. L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A., Hackett J., Adamson J., Lincoln S., Dickson D., Davies P., Petersen R. C., Stevens M., de Graaff E., Wauters E., van Baren J., Hillebrand M., Joosse M., Kwon J. M., Nowotny P., Che L. K., Norton J., Morris J. C., Reed L. A., Trojanowski J., Basun H., Lannfelt L., Neystat M., Fahn S., Dark F., Tannenberg T., Dodd P. R., Hayward N., Kwok J. B., Schofield P. R., Andreadis A., Snowden J., Craufurd D., Neary D., Owen F., Oostra B. A., Hardy J., Goate A., van Swieten J., Mann D., Lynch T., Heutink P. (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705 [DOI] [PubMed] [Google Scholar]
- 9. Kim M. L., Zhang B., Mills I. P., Milla M. E., Brunden K. R., Lee V. M. (2008) Effects of TNFα-converting enzyme inhibition on amyloid β production and APP processing in vitro and in vivo. J. Neurosci. 28, 12052–12061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clos A. L., Lasagna-Reeves C. A., Castillo-Carranza D. L., Sengupta U., Jackson G. R., Kelly B., Beachkofsky T. M., Kayed R. (2011) Formation of immunoglobulin light chain amyloid oligomers in primary cutaneous nodular amyloidosis. Br. J. Dermatol. 165, 1349–1354 [DOI] [PubMed] [Google Scholar]
- 11. Lasagna-Reeves C. A., Castillo-Carranza D. L., Sengupta U., Clos A. L., Jackson G. R., Kayed R. (2011) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brunden K. R., Trojanowski J. Q., Lee V. M. (2008) Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J. Alzheimer Dis. 14, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Frishman D., Argos P. (1995) Knowledge-based protein secondary structure assignment. Proteins 23, 566–579 [DOI] [PubMed] [Google Scholar]
- 14. Goedert M., Spillantini M. G., Jakes R., Rutherford D., Crowther R. A. (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3, 519–526 [DOI] [PubMed] [Google Scholar]
- 15. Fisher C. K., Stultz C. M. (2011) Protein structure along the order-disorder continuum. J. Am. Chem. Soc. 133, 10022–10025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dunker A. K., Lawson J. D., Brown C. J., Romero P., Oh J. S., Oldfield C. J., Campen A. M., Ratliff C. M., Hipps K. W., Ausio J., Nissen M. S., Reeves R., Kang C., Kissinger C. R., Bailey R. W., Griswold M. D., Chiu W., Garner E. C., Obradovic Z. (2001) Intrinsically disordered protein. J. Mol. Graph. Model. 19, 26–59 [DOI] [PubMed] [Google Scholar]
- 17. Fisher C. K., Stultz C. M. (2011) Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol. 21, 426–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Di Noto L., DeTure M. A., Purich D. L. (1999) Disulfide-cross-linked tau and MAP2 homodimers readily promote microtubule assembly. Mol. Cell Biol. Res. Commun. 2, 71–76 [DOI] [PubMed] [Google Scholar]
- 19. Barghorn S., Mandelkow E. (2002) Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry 41, 14885–14896 [DOI] [PubMed] [Google Scholar]
- 20. Furukawa Y., Kaneko K., Nukina N. (2011) Tau protein assembles into isoform- and disulfide-dependent polymorphic fibrils with distinct structural properties. J. Biol. Chem. 286, 27236–27246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fisher C. K., Huang A., Stultz C. M. (2010) Modeling intrinsically disordered proteins with bayesian statistics. J. Am. Chem. Soc. 132, 14919–14927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mo Z. Y., Zhu Y. Z., Zhu H. L., Fan J. B., Chen J., Liang Y. (2009) Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J. Biol. Chem. 284, 34648–34657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhattacharya K., Rank K. B., Evans D. B., Sharma S. K. (2001) Role of cysteine 291 and cysteine 322 in the polymerization of human tau into Alzheimer-like filaments. Biochem. Biophys. Res. Commun. 285, 20–26 [DOI] [PubMed] [Google Scholar]
- 24. Sahara N., Maeda S., Murayama M., Suzuki T., Dohmae N., Yen S. H., Takashima A. (2007) Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur. J. Neurosci. 25, 3020–3029 [DOI] [PubMed] [Google Scholar]
- 25. Meraz-Ríos M. A., Lira-De León K. I., Campos-Peña V., De Anda-Hernández M. A., Mena-López R. (2010) Tau oligomers and aggregation in Alzheimer's disease. J. Neurochem. 112, 1353–1367 [DOI] [PubMed] [Google Scholar]
- 26. Kuret J., Chirita C. N., Congdon E. E., Kannanayakal T., Li G., Necula M., Yin H., Zhong Q. (2005) Pathways of tau fibrillization. Biochim. Biophys. Acta 1739, 167–178 [DOI] [PubMed] [Google Scholar]
- 27. Sugino E., Nishiura C., Minoura K., In Y., Sumida M., Taniguchi T., Tomoo K., Ishida T. (2009) Three-/four-repeat-dependent aggregation profile of tau microtubule-binding domain clarified by dynamic light scattering analysis. Biochem. Biophys. Res. Commun. 385, 236–240 [DOI] [PubMed] [Google Scholar]
- 28. Schweers O., Mandelkow E. M., Biernat J., Mandelkow E. (1995) Oxidation of cysteine 322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of paired helical filaments. Proc. Natl. Acad. Sci. U.S.A. 92, 8463–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hua Q., He R. Q. (2003) Tau could protect DNA double helix structure. Biochim. Biophys. Acta 1645, 205–211 [DOI] [PubMed] [Google Scholar]
- 30. Hikosou R., Kurabayashi Y., Doumoto M., Hoshitoku K., Mizushima F., Minoura K., Tomoo K., Ishida T. (2007) Effect of DNA on filament formation of tau microtubule-binding domain: structural dependence of DNA. Chem. Pharm. Bull. 55, 1030–1033 [DOI] [PubMed] [Google Scholar]
- 31. Wei Y., Qu M. H., Wang X. S., Chen L., Wang D. L., Liu Y., Hua Q., He R. Q. (2008) Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PloS One 3, e2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barghorn S., Biernat J., Mandelkow E. (2004) in Methods in Molecular Biology (Sigurdsson E. M., ed) pp. 35–51, Humana Press, Totowa, NJ [Google Scholar]
- 33. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 34. Jiang C., Chang J. Y. (2007) Isomers of human α-synuclein stabilized by disulfide bonds exhibit distinct structural and aggregative properties. Biochemistry 46, 602–609 [DOI] [PubMed] [Google Scholar]
- 35. Jha A. K., Colubri A., Freed K. F., Sosnick T. R. (2005) Statistical coil model of the unfolded state: resolving the reconciliation problem. Proc. Natl. Acad. Sci. U.S.A. 102, 13099–13104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fischer D., Mukrasch M. D., von Bergen M., Klos-Witkowska A., Biernat J., Griesinger C., Mandelkow E., Zweckstetter M. (2007) Structural and microtubule binding properties of tau mutants of frontotemporal dementias. Biochemistry 46, 2574–2582 [DOI] [PubMed] [Google Scholar]
- 37. Mukrasch M. D., Markwick P., Biernat J., Bergen M., Bernadó P., Griesinger C., Mandelkow E., Zweckstetter M., Blackledge M. (2007) Highly populated turn conformations in natively unfolded tau protein identified from residual dipolar couplings and molecular simulation. J. Am. Chem. Soc. 129, 5235–5243 [DOI] [PubMed] [Google Scholar]
- 38. Mylonas E., Hascher A., Bernadó P., Blackledge M., Mandelkow E., Svergun D. I. (2008) Domain conformation of tau protein studied by solution small angle x-ray scattering. Biochemistry 47, 10345–10353 [DOI] [PubMed] [Google Scholar]
- 39. Kurita J., Shimahara H., Utsunomiya-Tate N., Tate S. (2003) Measurement of 15N chemical shift anisotropy in a protein dissolved in a dilute liquid crystalline medium with the application of magic angle sample spinning. J. Magn. Reson. 163, 163–173 [DOI] [PubMed] [Google Scholar]
- 40. Bernadó P., Bertoncini C. W., Griesinger C., Zweckstetter M., Blackledge M. (2005) Defining long-range order and local disorder in native α-synuclein using residual dipolar couplings. J. Am. Chem. Soc. 127, 17968–17969 [DOI] [PubMed] [Google Scholar]
- 41. Neal S., Nip A. M., Zhang H., Wishart D. S. (2003) Rapid and accurate calculation of protein 1H, 13C and 15N chemical shifts. J. Biomol. NMR 26, 215–240 [DOI] [PubMed] [Google Scholar]
- 42. Sowdhamini R., Srinivasan N., Shoichet B., Santi D. V., Ramakrishnan C., Balaram P. (1989) Stereochemical modeling of disulfide bridges: criteria for introduction into proteins by site-directed mutagenesis. Protein Eng. 3, 95–103 [DOI] [PubMed] [Google Scholar]
- 43. Neve R. L., Harris P., Kosik K. S., Kurnit D. M., Donlon T. A. (1986) Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 387, 271–280 [DOI] [PubMed] [Google Scholar]
- 44. Lee G., Rook S. L. (1992) Expression of tau protein in non-neuronal cells: microtubule binding and stabilization. J. Cell Sci. 102, 227–237 [DOI] [PubMed] [Google Scholar]
- 45. Butner K. A., Kirschner M. W. (1991) Tau protein binds to microtubules through a flexible array of distributed weak sites. J. Cell. Biol. 115, 717–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goedert M., Jakes R. (1990) Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 9, 4225–4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gustke N., Trinczek B., Biernat J., Mandelkow E. M., Mandelkow E. (1994) Domains of tau protein and interactions with microtubules. Biochemistry 33, 9511–9522 [DOI] [PubMed] [Google Scholar]
- 48. Mukrasch M. D., Biernat J., von Bergen M., Griesinger C., Mandelkow E., Zweckstetter M. (2005) Sites of tau important for aggregation populate β-structure and bind to microtubules and polyanions. J. Biol. Chem. 280, 24978–24986 [DOI] [PubMed] [Google Scholar]
- 49. von Bergen M., Barghorn S., Li L., Marx A., Biernat J., Mandelkow E. M., Mandelkow E. (2001) Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local β-structure. J. Biol. Chem. 276, 48165–48174 [DOI] [PubMed] [Google Scholar]
- 50. von Bergen M., Friedhoff P., Biernat J., Heberle J., Mandelkow E. M., Mandelkow E. (2000) Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β-structure. Proc. Natl. Acad. Sci. U.S.A. 97, 5129–5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tang H. Y., Speicher D. W. (2004) Enzymatic digestion of proteins in solution. Curr. Protoc. Protein Sci. 11.1, 1–12 [DOI] [PubMed] [Google Scholar]
- 52. Khurana R., Coleman C., Ionescu-Zanetti C., Carter S. A., Krishna V., Grover R. K., Roy R., Singh S. (2005) Mechanism of thioflavin T binding to amyloid fibrils. J. Struct. Biol. 151, 229–238 [DOI] [PubMed] [Google Scholar]
- 53. Saeed S. M., Fine G. (1967) Thioflavin-T for amyloid detection. Am. J. Clin. Pathol. 47, 588–593 [DOI] [PubMed] [Google Scholar]
- 54. Huang A., Stultz C. M. (2008) The effect of a ΔK280 mutation on the unfolded state of a microtubule-binding repeat in Tau. PLoS Comput. Biol. 4, e1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]