

Background: Solution structure of the cytoplasmic domain of a podocyte protein Neph1 will provide functional insight into the Neph1 molecule.
Results: A structural model of the Neph1-CD and its complex with ZO-1-PDZ1 was generated, and the interacting sites were mapped.
Conclusion: Neph1-CD adopts a global shape in solution, and its interaction with ZO-1 involves multiple sites.
Significance: This study advances our understanding of the molecular network of podocyte proteins in three dimensions.
Keywords: Cell Biology; Epithelial Cell; Kidney; Membrane Proteins; Molecular Docking; Molecular Modeling; Nephrology; Protein Dynamics; Protein-Protein Interactions; Structural Biology; Glomerulus, SAXS
Abstract
Neph1 is present in podocytes, where it plays a critical role in maintaining the filtration function of the glomerulus, in part through signaling events mediated by its cytoplasmic domain that are involved in actin cytoskeleton organization. To understand the function of this protein, a detailed knowledge of the structure of the Neph1 cytoplasmic domain (Neph1-CD) is required. In this study, the solution structure of this domain was determined by small/wide angle x-ray scattering (SWAXS). Analysis of Neph1-CD by SWAXS suggested that this protein adopts a global shape with a radius of gyration and a maximum linear dimension of 21.3 and 70 Å, respectively. These parameters and the data from circular dichroism experiments were used to construct a structural model of this protein. The His-ZO-1-PDZ1 (first PDZ domain of zonula occludens) domain that binds Neph1-CD was also analyzed by SWAXS, to confirm that it adopts a global structure similar to its crystal structure. We used the SWAXS intensity profile, the structural model of Neph1-CD, and the crystal structure of ZO-1-PDZ1 to construct a structural model of the Neph1-CD·ZO-1-PDZ1 complex. Mapping of the intermolecular interactions suggested that in addition to the C-terminal residues Thr-His-Val, residues Lys-761 and Tyr-762 in Neph1 are also critical for stabilizing the complex. Estimated intensity values from the SWAXS data and in vivo and in vitro pull-down experiments demonstrated loss of binding to ZO-1 when these residues were individually mutated to alanines. Our findings present a structural model that provides novel insights into the molecular structure and function of Neph1-CD.
Introduction
The filtration assembly in glomerulus comprises three major layers: a fenestrated endothelium, the glomerular basement membrane, and podocytes. Podocytes are specialized cells with a unique morphology, which consists of numerous interdigitating processes (foot processes) that arise from their cell bodies and surround the glomerular basement membrane. These foot processes are connected through a thin membranous (40-nm-wide) structure commonly termed the “slit diaphragm,” which serves as a filtration barrier preventing the passage of macromolecules from the capillary lumen (1–3). Injury to podocytes is commonly observed in various glomerular disorders and is associated with loss of filtration function and progression of end stage renal disease. Numerous studies indicate that the podocyte proteins Neph1 and nephrin are critical for maintaining the structural integrity of the slit diaphragm and therefore the filtration function of the glomerulus (4–7). Neph1 is a membrane protein that is structurally related to nephrin and consists of five extracellular immunoglobulin-like repeats, followed by a transmembrane domain and a short cytoplasmic domain (8). Similar to the nephrin gene, patients with mutations in the neph1 gene also develop nephrotic syndrome, and genetic deletion of neph1 in mice results in massive proteinuria with early postnatal death (9). Unlike nephrin, which is primarily a podocyte protein, Neph1 is widely distributed in tissues, including brain, heart, and liver. In podocytes, Neph1 co-localizes with nephrin, and evidence suggests that it forms a complex with nephrin both extracellularly and intracellularly (10, 11). Whereas the extracellular interaction of Neph1 and nephrin determines the structural framework of the slit diaphragm, their intracellular engagement through their cytoplasmic domains plays a key role in signaling events that regulate podocyte actin organization (5, 10, 12).
The primary structural analysis of the Neph1 cytoplasmic domain suggests that it is composed of a SH2-binding domain and a C-terminal PDZ-binding domain (8). In addition to its interaction with nephrin, the cytoplasmic domain of Neph1 interacts with various actin-associated proteins, including podocin, CD2AP, ZO-1 (zonula occludens), CASK, IQGAP1, β-arrestin, Nck, Grb2, α-actinin 4, synaptopodin, and the motor protein Myo1c (1, 2, 13). Although the role of each interaction is not entirely understood, it has been suggested that these interactions participate in transducing signals involved in actin cytoskeleton organization (10, 14). Recent studies indicate that the interactions mediated by the PDZ-binding domain of Neph1 are critical for Neph1 function (12). Currently, ZO-1 and Par proteins have been shown to interact with this domain, and our recent study demonstrates that Neph1 interacts with ZO-1 in a dynamic fashion that is regulated by Neph1 tyrosine phosphorylation (15, 16). Further, Fyn-mediated tyrosine phosphorylation of Neph1 has been shown as a key signaling event that induces actin polymerization through recruitment of the adapter protein Grb2 (10). These studies collectively highlight the dynamic nature of the cytoplasmic domain of Neph1, suggesting that the structural changes induced by binding to other proteins and/or phosphorylation play a role in Neph1 function.
In podocytes, ZO-1 co-localizes with Neph1 at the cell membrane and cell-cell junctions (17). The structural analysis of ZO-1 shows the presence of three PDZ domains, namely, PDZ1, PDZ2, and PDZ3, of which only PDZ1 interacts with Neph1. This interaction has been mapped to the PDZ-binding motif (THV) at the C terminus of Neph1 (17). Although structural analysis of the ZO-1 PDZ1 domain with and without peptide ligands has been performed by x-ray crystallography, no structural insight was available on Neph1. Because the cytoplasmic domain of Neph1 is unique and displays multiple dynamic properties, determining the structure of this domain in solution will significantly enhance our understanding of the biology of this protein. Moreover, this protein is devoid of any disulfide bonds and therefore is fully functional when expressed as a recombinant protein in bacteria (10, 16). To visualize the global structure of Neph1 and its complex with ZO-1, protein samples of tagless, GST-tagged, and His-tagged Neph1, His-ZO-1-PDZ1, and their equimolar mixture(s) were analyzed by SWAXS.7 Dummy residues for the unliganded proteins were used to calculate the ab initio structural models. Placement of the crystal structure of the ZO-1 PDZ1 domain inside the envelope of the scattering data-based model displayed significant similarity between the two structures. The structural features from SWAXS data and circular dichroism spectra from protein samples of the His-Neph1-CD were instrumental in selecting a structural model of this protein from prediction servers. A model representing the Neph1-CD·ZO-1-PDZ1 protein complex was computationally solved by placing the structural model of Neph1-CD and the crystal structure of ZO-1-PDZ1 such that their theoretical SAXS profiles matched with the experimental data. In the model, the Neph1 residues that are predicted to play a significant role in interaction with the ZO-1-PDZ1 were identified, and their point mutants were generated. These mutants were used in the binding assays to experimentally validate their significance in binding of Neph1 with ZO-1. This analysis led to the identification of additional residues in Neph1 that are critical for binding between Neph1 and ZO-1. In the future, this study can be extended to gain novel insight into the structures of other podocyte proteins and their complexes.
EXPERIMENTAL PROCEDURES
Expression and Purification of Proteins
DNA encoding the 104 residues representing the first PDZ domain of ZO-1 with an N-terminal His tag (cloned in pET22b(+) expression vector using NdeI/BamHI sites) was obtained as a gift from Dr. Yingnan Zhang (18). Previously described expression and purification protocols were followed (18). Briefly, Escherichia coli BL21 (DE3) strain harboring the desired plasmid was grown at 37 °C to an absorbance of 0.8 at 600 nm in LB (Luria-Bertani) medium (Merck). Expression of His-ZO-1-PDZ1 protein was induced by the addition of 0.4 mm isopropyl β-d-thiogalactopyranoside, and the induced culture was further incubated at 27 °C for 16 h. Bacteria were harvested by centrifugation (8000 rpm for 15 min), and the resulting pellets were washed with 50 mm Tris-HCl buffer (pH 8.0) and stored at −80 °C. The pellets were resuspended in 50 mm Tris-HCl buffer (pH 8.0) containing 500 mm NaCl and protease inhibitor mixture (Merck). To obtain soluble protein, the cells were lysed by sonication followed by centrifugation at 8000 rpm for 20 min. The lysate was added to a Ni2+-NTA column (Merck) and washed with 50 mm Tris-HCl buffer containing 50 mm imidazole, and the bound His-ZO-1-PDZ1 protein was eluted with the wash buffer supplemented with 250 mm imidazole. The eluted fractions containing protein were pooled and dialyzed overnight in buffer containing 50 mm Tris-HCl (pH 8.0), 300 mm NaCl, and 5 mm β-mercaptoethanol. A buffer with a similar composition was used to purify His-ZO-1-PDZ1 protein from minor impurities using a Superdex 200 10/300 GL size exclusion column attached to an ÄKTA Explorer FPLC (GE Biosciences). The purity and identity of the purified protein was confirmed by a single band on SDS-PAGE (refer to Fig. 1B) and a peak at ∼12.1 kDa in a MALDI-TOF mass spectrometer (Voyager 4402, Applied Biosystems). The purified protein was concentrated using membrane concentrators (3 kDa cut-off; Millipore), and the protein concentration was determined by absorbance at 280 nm (A280(1 mg/ml) ∼0.45).
FIGURE 1.

A, protein sequences for the cytoplasmic domain of Neph1 and the first PDZ domain of ZO-1 (ZO-1-PDZ1) used in SWAXS experiments are shown. The sites of ZO-1 interaction discovered from this study and from a previous report are presented in boldface type (positions Arg-750, Lys-761, Tyr-762, and THV (residues 787–789)). B, different Neph1 proteins, including tagless and His- and GST-tagged, and His-tagged ZO-1-PDZ1 were recombinantly expressed and purified prior to SWAXS analysis. The purified proteins were analyzed by SDS-PAGE and staining with Coomassie Blue and Western blotting using Neph1 or His antibodies.
The His-Neph1-CD protein was obtained by cloning the intracellular region of neph1 (238 C-terminal amino acid residues of Neph1) in the pRSETA expression vector (Invitrogen) and then transforming it in the BL21 (DE3) pLysS bacterial strain. To improve the expression and the solubility of His-Neph1-CD protein, the bacteria were cultured in minimal medium containing 4% sucrose. Bacteria grown to an optical density of 0.68 at 37 °C were induced with 0.2 mm isopropyl β-d-thiogalactopyranoside and incubated further for 4 h at 37 °C. The cells were harvested, washed with 20 mm phosphate buffer (pH 7.8), and then resuspended in buffer containing 1% Sarkosyl, 1 mm DTT, and a protease inhibitor mixture (Merck). The cells were lysed by sonication, and the presence of His-Neph1-CD protein in the soluble fraction was confirmed by SDS-PAGE. The supernatant was first centrifuged at 40,000 × g for 30 min and then loaded on a Ni2+-NTA column (Merck). To remove nonspecific weak binders, the column was washed with 5 column volumes of 20 mm phosphate buffer (pH 7.8) containing 0.2% Sarkosyl and 20 mm imidazole. The bound His-Neph1-CD was eluted by increasing the imidazole concentration in the buffer to 300 mm. The protein was concentrated using membrane concentrators (10,000 molecular weight cut-off; Amicon) and purified further by gel filtration chromatography using 20 mm potassium phosphate buffer, pH 7.8, containing 150 mm NaCl on a Superdex 200 10/300 GL column attached to an ÄKTA Explorer (GE Healthcare). The purity and identity of the protein was confirmed by SDS-PAGE and MALDI-TOF (single peak at 35.1 kDa using a Voyager 4402 mass spectrometer, Applied Biosystems) (Fig. 1, A and B).
The GST-Neph1-CD protein was prepared as described previously (10, 16). Further purification of GST-Neph1-CD protein for structural studies was carried out by subjecting the glutathione-eluted GST-Neph1-CD protein preparation to FPLC, and the final product was analyzed by SDS-PAGE and Western blot using Neph1 antibody. The buffer conditions for His-ZO-1-PDZ1, His-Neph1-CD, and their mixtures were optimized by tracking diffusion coefficient values for these proteins by dynamic light scattering (DLS) experiments (DelsaNano C, Beckman Coulter). DLS data were acquired in a microcell at the back scattering angles (165°), and the temperature was maintained by the integrated Peltier set-up. At each temperature, the sample was equilibrated for 3 min before collecting the scattering data for another 3 min. The data were analyzed for diffusion coefficient values using software provided with DelsaNano C.
To obtain the tagless Neph1-CD protein, as a first approach, we digested the GST-Neph1-CD protein (that contains a thrombin cleavage site because it was cloned in the pGEX-4T vector) with thrombin (protease) to remove the GST domain. Although this is a standard approach that is used to remove the GST from a GST-tagged protein, the yield of tagless Neph1-CD using this approach was very poor, and moreover, the protein immediately precipitated upon the removal of GST. While various attempts were made to obtain a soluble form of the tagless Neph-CD protein, a fortuitous discovery was made from our stored His-Neph1-CD protein samples. We observed that the incubation of His-Neph1-CD for extended times at 4 °C resulted in the appearance of a stable 22-kDa protein product in a reproducible fashion. This truncated protein was further purified by FPLC and characterized by N-terminal sequencing, mass spectroscopy (MS) (supplemental Fig. 2), and Western blotting using Neph1 antibody (Fig. 1 and supplemental Fig. 7). The analysis indicated that the cleavage occurred at the Arg-24–Glu-25 peptide bond in the His-Neph1-CD, thus removing the N-terminal His tag and an additional 24 amino acid residues from the N terminus of Neph1-CD. Because this protein product retained all of the potential tyrosine phosphorylation sites in the Neph1-CD molecule (10) and the functionality, as confirmed by its binding to the His-ZO-1-PDZ1 protein (supplemental Fig. 7), we pursued SAXS analysis using this as a tagless Neph1-CD protein.
Separation of the His-Neph1-CD·ZO-1-PDZ1 complex using a single Superdex 200 10/300 GL column (in 20 mm potassium phosphate buffer (pH 7.8) containing 150 mm NaCl) could not be achieved because the elution peaks corresponding to the His-Neph1-CD·ZO-1-PDZ1 complex (∼45 kDa) and His-Neph1-CD (∼35 kDa) overlapped. Therefore, to increase the separation between the peaks, we used two Superdex 200 10/300 GL columns in tandem and attached them to a modified Bio-Rad FPLC system where the flow path between the exit of the second column and the UV detector cell was reduced to below 50 μl. During elution, the proteins were manually collected, and their purity was analyzed by SDS-PAGE.
Synchrotron SAXS/WAXS Data Acquisition and Processing
The SWAXS data were collected at the X9 beam line (National Synchrotron Light Source, Brookhaven National Laboratory, Brookhaven, NY). Two charge-coupled detectors (CCDs) simultaneously collected data at small (SAXS) and wide (WAXS) angles. The wavelength of the beam was 0.873 Å, and the ratio of the sample to detector distance to the diameter of the CCD was 20.8. 120 μl of each of the three different concentrations of His-ZO-1-PDZ1, His-Neph1-CD, tagless Neph1-CD, and GST-Neph1-CD proteins were used to collect SWAXS data. To acquire the scattering data, 120 μl of three samples containing different molar mixtures of His-Neph1-CD and His-ZO-1-PDZ1 proteins and their matched buffers were used. Each sample and buffer was exposed to x-rays for 120 s in a quartz flow cell at 15 °C at a flow rate of 50 μl/min. To calibrate the beam intensity at zero angles, SWAXS data sets were collected under identical conditions with five samples of hen egg lysozyme at varying predetermined concentrations using absorption at 280 nm (A280(1 mg/ml) = 2.5). Having determined the actual concentration of samples and the absence of interparticle effect in these proteins at higher concentrations, the SWAXS data on different molar mixtures of His-Neph1-CD·ZO-1-PDZ1 proteins were collected to gain insight into the predominant scattering species in the mixtures. Similar experiments were repeated with various His-Neph1-CD mutant proteins in the presence and absence of the native His-ZO-1-PDZ1.
Using the Python script-based programs written by Dr. Lin Yang (X9 beam line, National Synchrotron Light Source), the images recorded on the two CCDs were scaled, merged, and circularly averaged, and the buffer contribution was subtracted to obtain the scattering intensity (I) as a function of momentum transfer vector, Q (Q = (4πsinθ)/λ), where λ and θ represent the wavelength of x-ray and the scattering angle, respectively. In this study, the SAXS and WAXS intensity profiles were scaled and merged using the Q data between 0.12 and 0.2 Å−1. All of the SAXS experiments described in this study were carried out in duplicate. The protein samples that were subjected to transport to the x-ray facility and exposed to x-rays remained stable and intact, as determined by recharacterization of these samples by SDS-PAGE at the conclusion of the SAXS experiments. The migration profiles of these experimental samples on SDS-PAGE were identical to their parallel aliquots stored in the laboratory at −80 °C.
Circular Dichroism Spectroscopy
The circular dichroism studies were carried out with ∼8 μm protein samples in 20 mm potassium phosphate buffer (pH 7.8) containing 150 mm NaCl on a JASCO 810 spectrometer equipped with temperature control. A quartz cell with 1-mm path length was used for the circular dichroism experiments. Five circular dichroism spectra from 200 to 250 nm were acquired and averaged for each protein solution (His-Neph1-CD, His-Neph1-CD-R750A, His-Neph1-CD-Y762A, His-Neph1-CD-K761A, and His-Neph1-CD-PDZ) and the matching buffer. Each data set (including the buffer) was an average of three scans performed at 200–250 nm. Buffer-subtracted circular dichroism spectra data were used to estimate the secondary structural content in the protein using the K2D2 server (19).
SAXS Data Analyses
Kratky analyses of unliganded proteins and the predominant species present in their mixtures were carried out by interpreting the shape profile of their respective I(Q)Q2 versus Q plot. For globular scattering particles, the Guinier approximation was employed to estimate the Rg of the scattering particle. According to this approximation, for a monodisperse sample of globular protein, a plot of ln(I(Q)) versus Q2, where Q × Rg ≤ 1.3, should be linear and fit into the following equation (Equation 1),
where I0, defined as the intensity of scattering at zero angles, is directly proportional to the product of molar concentration and the molecular mass of the scattering sample and can be approximated by extrapolating the SAXS data to Q ∼0. The Rg is defined as the root mean square of all elemental volumes from the center of mass of the particle, weighted by their scattering densities, and is characteristic of the overall shape of the molecule. For this study, the Guinier analyses were performed using the PRIMUS software package. Using the GNOM45 program, indirect Fourier transformation of the scattering data over the measured Q range computed a pairwise distribution function of interatomic vectors, P(r) (Equation 2),
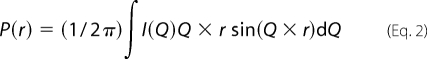 |
where P(r) is a histogram of the frequency of vector lengths connecting small volume elements within the entire volume of the scattering particle. During indirect Fourier transformation, the P(r) was considered as zero for vector lengths equal to 0 and the maximum linear dimension (Dmax). The analyses also provided Rg and I0 from the second moment and the start of P(r), respectively.
Ab Initio Structure Reconstruction and Tertiary Structure Prediction
By employing dummy residues and constraints provided within the measured SAXS I(Q) profile, the three-dimensional shapes of the two proteins were restored using the DAMMINIQ program (20). 10 models were generated without any predefined shape or symmetry bias. An average model that represented all of the individual solutions was generated using the DAMAVER suite of programs (21). Fold recognition-based tertiary structure predictions were performed by submitting the sequence of Neph1-CD to the PHYRE and FUGUE servers. The atomic resolution model of the Neph1-CD·ZO-1-PDZ1 protein complex was generated by steering the structural model of Neph1-CD and the crystal structure of ZO-1-PDZ1 (with engineered His tag) using the SASREF program (22). Open source software programs, including PyMOL, SPDB viewer, and Raswin 2.6UCB, were used for the graphical analysis and figure generation.
Immunoprecipitation and Immunoblotting
The Neph1 mutants, Neph1-R750A, Neph1-K761A, Neph1-Y762A, and Neph1-PDZ, were prepared by a standard PCR technique, using the full-length Neph1 as a template. The detailed procedures of immunoprecipitation and immunoblotting experiments have been described previously (10, 16, 23). Briefly, the wild type Neph1 and its mutants were co-transfected with Myc-ZO-1 (full-length) in COS-7 cells. Forty-eight hours post-transfection, the cells were lysed in radioimmune precipitation buffer (phosphate-buffered 0.9% NaCl (PBS) containing 0.1% SDS, 1% Nonidet P-40, and 0.5% sodium deoxycholate). The lysate was subjected to immunoprecipitation using ZO-1 antibody, and the immunoprecipitated complex was separated by SDS-PAGE and analyzed by Western blotting using Neph1 antibody.
Pull-down Experiments
The recombinant proteins, His-Neph1-CD, the Neph1-CD mutants, including His-Neph1-CD-R750A, His-Neph1-CD-K761A, His-Neph1-CD-Y762A, and His-Neph1-CD-PDZ (prepared by standard PCR technique), and His-ZO-1-PDZ1, were expressed and purified as described above. 5 μg of each Neph1 fusion protein was separately mixed with the purified His-ZO-1-PDZ1 (2 μg), and the His-Neph1-CD·ZO-1-PDZ1 complexes were pulled down using Neph1 antibody bound to protein G-agarose. After washing the agarose beads with PBS containing 0.1% Tween 20, the protein complexes were eluted with SDS-sample buffer and resolved by SDS-PAGE prior to immunoblotting with the histidine antibody.
RESULTS AND DISCUSSION
Unliganded ZO-1-PDZ1
Because none of the podocyte proteins have previously been analyzed by SWAXS, it was necessary to validate our approach by determining the solution structure of a protein whose structure has been determined by alternate biophysical methods. Therefore, we first deciphered the predominant solution shape of the ZO-1-PDZ1 domain (Fig. 1, A and B) that binds Neph1-CD and its crystal structure was resolved using x-ray diffraction analysis (18). SWAXS data were acquired using four samples of His-ZO-1-PDZ1, varying in protein concentration from 1.7 to 9.5 mg/ml (Fig. 2A, left). The peaklike profile of the Kratky plots confirmed the globular nature of the ZO-1 molecules in solution (Fig. 2A, middle). The Guinier approximation of the globular nature over a Q range of 0.009–0.08 Å−1 suggested that the Rg of His-ZO-1 is about 15.5 Å (Fig. 2A, left inset, and Table 1). Indirect Fourier transformations of the data sets over a much wider angular range (Q range 0.009–1 Å−1) provided a good solution at Dmax ∼50 Å, which computed an Rg of 15.6 ± 0.01 Å (Fig. 2A, right, and Table 1). When compared with the I0 value of 14 arbitrary units extrapolated for a sample of 1 mg/ml lysozyme, estimated I0 values of 66 and 12 arbitrary units showed that the concentrations of the His-ZO-1-PDZ1 samples were in the range of 1.7–9.5 mg/ml (Table 1). Lack of variation in the Rg values, estimated using Guinier analyses and P(r) analyses as a function of protein concentration, affirmed the absence of an “interparticle effect” in this protein at higher concentrations (24).
FIGURE 2.

SWAXS data analyses from the samples of His-ZO-1-PDZ1 (A), His-Neph1-CD (B), and their 1:1.2 molar mixtures (C). Left, SWAXS intensity profiles are plotted versus Q for the samples and mixtures. The linear Guinier region for each data set is shown in the inset. Middle, the Kratky analyses of the samples highlight the global nature of molecules in the solution. Right, P(r) curves computed from the SWAXS data highlight the frequency distribution of interatomic vectors in the predominant scattering species.
TABLE 1.
Structural parameters deduced by performing indirect Fourier transformation on the SWAXS data obtained from samples of His-ZO-1-PDZ1 and three different constructs of His-Neph1-CD
| Unliganded proteins | Indirect Fourier transformation |
Molecular mass | Concentration |
|||
|---|---|---|---|---|---|---|
| Dmax | Rg | I0 | mg/ml | μm | ||
| Å | Å | kDa | ||||
| Hen egg white lysozyme | 44 | 14.2 ± 0.01 | 14 | 14.2 | 1a | |
| His-ZO-1-PDZ1 | 12.1 | |||||
| Sample 1 | 50 | 15.6 ± 0.01 | 66 | 9.5 | 460 | |
| Sample 2 | 50 | 15.7 ± 0.01 | 49 | 7 | 339 | |
| Sample 3 | 50 | 15.6 ± 0.02 | 24 | 3.4 | 165 | |
| Sample 4 | 50 | 15.7 ± 0.07 | 12 | 1.7 | 82 | |
| His-Neph1-CD | 35 | |||||
| Sample 1 | 70 | 21.3 ± 0.03 | 152 | 4.4 | 125 | |
| Sample 2 | 70 | 21.4 ± 0.05 | 93 | 2.7 | 77 | |
| Sample 3 | 70 | 21.4 ± 0.07 | 41 | 1.2 | 34 | |
| Neph1-CD | 22 | |||||
| Sample 1 | 70 | 18.2 ± 0.3 | 11 | 0.5 | 22 | |
| Sample 2 | 70 | 18.3 ± 0.6 | 6.5 | 0.3 | 13.6 | |
| GST-Neph1-CD | 53 | |||||
| Sample 1 | 110 | 24.1 ± 0.2 | 15.7 | 0.3 | 5.6 | |
a Extrapolated values from a dilution series of lysozyme in the concentration range of 6.2–1.7 mg/ml.
The scattering shape of His-ZO-1-PDZ1 was generated by averaging 10 individual solution shapes, which in turn were calculated using the SWAXS data acquired from sample with the highest concentration (i.e. 9.5 mg/ml) (Fig. 3A). The structure showed a globular domain with a small projection originating from one side of the molecule. To compare the overall shape profile, we placed the crystal structure of the ZO-1-PDZ1 domain (Protein Data Bank code 2H3M) inside the volume of our model by manually aligning the surface topology of the two structures. Interestingly, the crystal structure fitted well within the volume of our model, except for the projected area that was left unoccupied. It is likely that this extra volume represents the His tag present in our protein (Fig. 3A). In parallel, we also collected the circular dichroism spectrum for His-ZO-1 with a sample concentration of ∼0.1 mg/ml (Fig. 3C). Deconvolution of acquired data suggested that the major part of this protein adopts a β-sheet structure in solution (∼40% β-sheets and 15% α-helices). These values are comparable with the secondary structural content seen in the crystal structure of this protein (37% β-sheets and 9% α-helices). Overall, our results conclude that the ZO-1-PDZ1 adopts a solution structure similar to its structure resolved using x-ray crystallography.
FIGURE 3.

A, two rotated views (upper and lower panels) compare the predominant scattering shape of His-ZO-1-PDZ1 in solution with its tagless crystal structure. Left, average scattering shape of the His-ZO-1-PDZ1 molecule calculated within the shape constraints encoded in the SWAXS intensity profile (gray, CPK model). Middle, the crystal structure of ZO-1-PDZ1 protein lacking the N-terminal His tag (Protein Data Bank entry 2H3M; black, CPK model). Right, manual placement of the crystal structure inside the shape profile of the scattering data-based model brought forth an unoccupied volume in our SWAXS-based model, which may represent the His tag present in the N terminus of our construct. B, two rotated views (top and bottom) compare the scattering data-based global structure of the His tagged Neph1 with its in silico model that best fits the experimental data. Left, average scattering shape of the His-Neph1 molecule computed using dummy residues within the shape profile of the acquired scattering intensity data (gray, CPK model). Middle, predicted structural model of the His-Neph1-CD that best fits the structural parameters deduced from the scattering data (black, CPK model). Right, automated superimposition of the SWAXS data-based model and the predicted structure by aligning their inertial axes. C, the circular dichroism spectral profiles of the unliganded His-ZO-1-PDZ1 (△), unliganded His-Neph1-CD (▿), and their 1:1 equimolar mixture (solid line). The data represent mean residual ellipticity (MRE) as a function of wavelength.
Unliganded His-Neph1-CD
To decipher the solution structure-function relationship of Neph1-CD, we primarily used the Neph1-CD construct with a His tag at its N terminus (Fig. 1, A and B). SWAXS data sets were acquired from three samples of this protein varying in concentration from 1.2 to 4.4 mg/ml (Fig. 2B, left). The peaklike behavior of the Kratky plots supported a globular nature for this protein in solution (Fig. 2B, middle). The Guinier approximation for globular scattering particles in the Q range 0.005–0.06 Å−1 suggested an Rg of ∼21.0 Å for the unliganded His-Neph1-CD (Fig. 2B, left inset). Considering a much wider Q range (0.005–1 Å−1) and the monodisperse globular nature of the His-Neph1-CD molecules, indirect Fourier transformation provided a good solution for all of the samples at Dmax ∼70 Å with an Rg close to 21.3 Å (Fig. 2B, right, and Table 1). Estimated I0 values of 41–1 arbitrary units indicated that the concentration of His-Neph1-CD (∼35 kDa) protein was in the range of 1.2–4.4 mg/ml. Comparable dimensions for the protein molecules in three samples supported lack of interparticle effects for His-Neph1-CD under the concentration range analyzed. The predominant solution scattering shape of this protein was restored by averaging 10 individual uniform density models solved using the measured SWAXS data for the sample with His-Neph1-CD protein concentration of ∼4.4 mg/ml (Fig. 3B). The global shape appeared as a single domain with a maximum dimension of ∼70 Å. To supplement the global shape information from the scattering data, we acquired the circular dichroism spectrum of this protein at a protein concentration of ∼0.3 mg/ml (Fig. 3C). Using the measured circular dichroism data, the K2D2 server predicted that this construct contains about 25% α-helices and 20% β-sheets.
Structural analysis of the Neph1-CD using sequence similarity-based searches suggested that it is not homologous to any existing protein structure in the database. Because no templates were found, we tried fold recognition-based prediction protocols from the PHYRE and FUGUE servers to obtain structural models for the His-Neph1-CD construct. Whereas the PHYRE server provided a set of three discontinuous structures, the FUGUE server predicted two models for the entire sequence (supplemental Fig. 1). Interestingly, the structural parameters of the model of His-Neph1-CD based on Protein Data Bank entry 2V3Q (earlier submitted as 2CAP) had a theoretical Rg and Dmax of 21 and 71 Å, respectively. Moreover, this model had a secondary structural content of 25% α-helices and 15% β-sheets, similar to the results from measured circular dichroism spectrum of His-Neph1-CD protein. In addition, the CRYSOL program computed a χ2 value of only 1.7 over the Q range 0.005–0.25 Å−1, exhibiting good similarity between the theoretical SAXS profile of this model with the measured SAXS data for His-Neph1-CD protein. Next, this predicted model was superimposed on the SWAXS data-based model by aligning their inertial axes (Fig. 3B). As expected from the analysis of the SAXS data, the predicted model fit well within the envelope volume of our SAXS-based model. The predicted model of His-Neph1-CD was based on the crystal structure of human phosphate-binding apolipoprotein (Protein Data Bank entry 2V3Q) (Fig. 4A). Recent reports suggest that the human phosphate-binding apolipoproteins play important roles as phosphate scavengers and are involved in protection against phosphate-related disorders like atherosclerosis, diabetes, and kidney stones. This model of the His-Neph1-CD suggested that the linker region at its N terminus (including the His tag) folds along the length of Neph1-CD molecule, whereas the functionally important C-terminal tail (containing the THV motif that interacts with ZO-1 and Par proteins) remains exposed at the surface of this molecule (Fig. 4A).
FIGURE 4.

A, two rotated views of the structural model of His-Neph1-CD predicted by the FUGUE server, which best compares with the structural parameters Rg and Dmax deduced from analyzing the scattering data, highlight the position of the functionally critical C-terminal THV residues (blue, sticks), secondary structural content in the protein (red, α-helix; yellow, β-sheet; green, loops), and the N-terminal tag (black). The space-filled representations are shown on the respective right panels. B, these plots show the relative decrease in the diffusion coefficient values of three different forms of Neph1-CD as a function of temperature (left) and storage time at 4 °C (right). C (left), the two rotated views show the average scattering shape of tagless Neph1-CD with its structural model aligned inside the volume. D (right), the two rotated views present the average scattering shape of GST-Neph1-CD. The crystal structure of GST and model of Neph1-CD have been aligned inside the volume of the SAXS data-based model.
It is possible that the tag at the N terminus of Neph1-CD induces structural changes in this domain that are different from the untagged protein. To rule out or explore this as a possibility, two additional Neph1-CD constructs were prepared: tagless Neph1-CD and GST-Neph1-CD. Because the C terminus of Neph1 is required for binding to ZO-1, the tag could not be placed at the C terminus of this protein. The 22-kDa tagless Neph1-CD protein was obtained by the proteolytic processing of His-Neph1-CD, as described under “Experimental Procedures.” The processed sample contained a 22-kDa protein as a major component (refer to supplemental Fig. 2) that was further purified by FPLC, concentrated, and characterized by mass spectrometry (single peak at 22.79 kDa) and N-terminal sequencing (Fig. 1B). Although this protein did not bind the Ni2+-NTA-agarose due to the lack of a His affinity tag, this cleavage did not affect its three-dimensional structure and function, which was confirmed by its ability to interact with the His-ZO-1-PDZ1 protein (supplemental Fig. 7). GST-Neph1-CD protein was also expressed and purified by FPLC, and its identity was confirmed by mass spectrometry. GST-Neph1-CD was found to interact with His-ZO-1-PDZ1 with the same efficiency as His-Neph1-CD and tagless Neph1-CD proteins (supplemental Fig. 7). However, both the tagless and the GST-Neph1-CD purified proteins displayed a strong tendency to aggregate and precipitate out of the solution during storage. This observation was confirmed by the relative decrease of the diffusion coefficient values of the purified protein samples in DLS experiments upon varying temperature and by analyzing the fractions of samples stored at 4 °C (Fig. 4B). We further observed that the 50% soluble to aggregated state was achieved at ∼45 and ∼50 °C for fresh FPLC-purified tagless Neph1-CD and GST-Neph1-CD proteins. A similar concentration of His-Neph1-CD protein (0.5–1 mg/ml) was relatively more stable and aggregated in a three-staged manner with a central stage from 60–68 °C. Importantly, a comparative DLS analysis of these purified proteins stored in the same buffer at 4 °C showed that although aggregation occurred in the tagless and GST-tagged proteins (at concentrations of ∼1 mg/ml), His-Neph1-CD protein was remarkably more stable for longer durations. An earlier report also indicated that Neph1-CD protein was unstable in the solution and prone to aggregation (15). Overall, our results indicate that the His tag provides increased stability to Neph1-CD; however, the mechanism for this stability is uncertain.
Because aggregation occurred during the storage of proteins, we acquired SWAXS data from the freshly purified and diluted samples of tagless and the GST-Neph1-CD proteins. Indirect Fourier transformation of the measured data sets suggested that the tagless 22-kDa and the GST-tagged Neph1-CD proteins adopt predominant solution shapes with Dmax of about 70 and 110 Å, respectively (Table 1). Structure reconstruction of the predominant scattering species in the solution indicated a single and a bilobal shape for Neph1-CD and the GST-tagged protein, respectively (Fig. 4, C and D). The superimposition highlights that the modeled structure of the Neph1-CD (tagless) fits inside the shape profile of the scattering data-based model of the Neph1-CD (Fig. 4C). The automated superimposition also highlights the shape similarity and the “cleft” formed due to the absence of the folded tag (Fig. 4C). Importantly, the CRYSOL program computed a χ2 value of 1.3 between the theoretical SAXS profile of the modeled structure (placed inside) and the measured SAXS profile for this protein, supporting that the modeled structure compared well with the shape preferred by this protein in solution. In the other analysis, the crystal structure of glutathione (Protein Data Bank entry 1GTA) and the model of tagless Neph1-CD could be easily placed inside the bilobal shape of GST-Neph1 (Fig. 4D). A χ2 value of 1.5 computed by the CRYSOL program between the theoretical scattering profile of the relatively placed structures and the experimental data supported the spatial positioning of the GST and Neph1-CD in the SAXS data-based model. The model shows that the GST tag on the N-terminal of Neph1-CD folds in a manner that is different from the His tag. Our SAXS data based models indicate that whereas the His tag folds along the length of Neph1-CD, the GST domain is oriented away from the parent molecule (Fig. 4, A and D). In addition, the averaged shape of GST-Neph1-CD shows unoccupied volume in comparison with the structural model of Neph1-CD and the crystal structure of GST. Importantly, comparing the shape models of the tagless and GST-Neph1-CD proteins with the model of His-Neph1-CD suggests that the ZO-1 binding region in Neph1-CD is exposed in all three constructs. This further suggests that the presence or absence of a tag will have little or no effect on the binding ability of Neph1 with ZO-1. Accordingly, the pull-down experiment performed with the GST- and His-tagged and tagless Neph1-CD proteins shows that the binding between Neph1-CD and His-ZO-1-PDZ1 is not affected by the presence and nature of a tag (supplemental Fig. 7). Considering the poor solubility of the tagless and the GST-tagged Neph1 proteins, further structural and pull-down studies (including the identification of additional residues critical for the Neph1/ZO-1 binding) were performed using the His-Neph1-CD protein and mutants derived from this protein.
1:1 Complex of His-Neph1-CD·ZO-1-PDZ1
The gel filtration profiles of different molar mixtures of the His-Neph1-CD and ZO-1-PDZ1 proteins showed that preincubation for ∼30 min at 4 °C was sufficient to induce 90–95% binding between these proteins (supplemental Fig. 3). The 1:1 His-Neph1-CD·ZO-1-PDZ1 complex (∼47 kDa) eluted close to the volume where ovalbumin (44 kDa) eluted from the two tandem Superdex 200 10/300 GL columns. In these experiments, a running buffer composed of 20 mm potassium phosphate buffer (pH 7.8) and 150 mm NaCl was used. The peak corresponding to the 1:1 His-Neph1-CD·ZO-1-PDZ1 protein complex was confirmed by varying the molar ratios of the His-Neph1-CD and ZO-1-PDZ1 proteins in the incubated mixture. The stability of the binary complex was ascertained by reinjecting the purified binary complex to the tandem Superdex 200 10/300 GL columns (supplemental Fig. 4). Importantly, the FPLC profile of the 1:1 complex reinjected after 2 h of purification showed no sign of dissociation into constituent proteins. Based on these gel filtration results, three different molar mixtures of His-Neph1-CD and ZO-1-PDZ1 proteins were prepared from the purified proteins about 1 h prior to SAXS data acquisition.
To further validate the structural model of His-Neph1-CD and to gain insight into the structural features of the His-Neph1-CD·ZO-1-PDZ1 protein complex, these proteins were mixed at the indicated molar ratios (concentrations of the unliganded proteins were estimated based on I0 values; Table 1), and the SWAXS data were acquired on these mixtures (Table 2). The Kratky analyses confirmed that the predominant scattering species were globular in shape in all of the three mixtures and slightly bigger in dimensions than the His-Neph1-CD (Fig. 2C, right). The Guinier approximation suggested an Rg value of ∼23.7 Å for the predominant scattering species in the His-Neph1-CD·ZO-1-PDZ1 protein mixtures (Fig. 2C, left inset). This was in close agreement with the indirect Fourier transformation analysis, which suggested the Dmax and Rg to be close to 80 and 23.1–24.2 Å, respectively, for the primary entity in the mixtures. An increase in the dimensions of the primary scattering species in solution when compared with that of the unliganded protein was the first indicator of binding between these two proteins. To estimate the extent of binding, we calculated the I0 of a solution assuming 100% binding between the two available proteins and remaining unliganded protein (Table 2). A comparison of the observed versus expected I0 values indicated that 90–95% of the 47-kDa complex was present in the mixture during SWAXS data collection. Ab initio structure reconstruction using the data obtained after analyzing the 1:1.2 molar mixture of His-Neph1-CD·ZO-1-PDZ1 proteins resulted in a structural model of this complex (Fig. 5A). To incorporate the residue level information in the envelope model obtained from the SAXS data, we used the SASREF program to orient the predicted model of tagged Neph1-CD and the crystal structure of ZO-1-PDZ1 until the theoretical SAXS profile of the complex matched with the experimental data. Four unique structural models of the complex with low χ2 values (recalculated using the CRYSOL program over the Q range of 0.005–0.25 Å−1) were generated (supplemental Fig. 5). The orientation with a minimum calculated χ2 value (1.7) between the calculated SAXS profile of the docking-based model and the experimental scattering intensity profile is presented in Fig. 5A (right). In this model, the ZO-1-PDZ1 docked close to the C terminus of Neph1, which has been demonstrated previously to be the binding region between these two proteins (17). Because these two proteins bind at almost 95% of their capacity (Table 2), the data obtained from circular dichroism analyses of Neph1-ZO-1 protein mixtures suggested that the secondary structural content of their 1:1 complex is about 25% α-helices and 25% β-sheets (Fig. 3C). There was a slight increase in the β-sheet content for the complex when compared with the unliganded Neph1-CD, possibly due to contribution from the β-sheet-rich ZO-1-PDZ1.
TABLE 2.
Structural parameters of the predominant scattering entity (1:1 complex of His-Neph1-CD·His-ZO-1-PDZ1) from different molar ratios of these proteins
| Molar ratio of His-ZO-1/His-Neph1 | Indirect Fourier transformation |
Expected I0 | Percentage of 1:1 binding | ||
|---|---|---|---|---|---|
| Dmax | Rg | I0 | |||
| Å | Å | % | |||
| 0.8 | 80 | 23.9 ± 0.05 | 187 | 205a | 90 |
| 1.0 | 80 | 24.0 ± 0.07 | 190 | 212b | 90 |
| 1.2 | 80 | 24.2 ± 0.15 | 197 | 207c | 95 |
a Expected I0 value was calculated based on 4.0 mg/ml 47-kDa complex and 0.5 mg/ml unliganded His-Neph1-CD.
b Expected I0 value was calculated based on 4.5 mg/ml 47-kDa complex and 0.07 mg/ml unliganded His-ZO-1.
c Expected I0 value was calculated based on 4.4 mg/ml 47-kDa complex and 0.2 mg/ml unliganded His-ZO-1.
FIGURE 5.

A, the two rotated views (top and bottom) offer a comparison between the scattering data-based global structure of the 1:1 complex between His-Neph1-CD and His-ZO-1-PDZ1 and their theoretical structure obtained from high resolution rigid body docking. Left, the average scattering shape of the His-Neph1-CD·ZO-1-PDZ1 complex calculated within the shape profile of the acquired scattering intensity data (purple, CPK model). Middle, the structural model of the 1:1 complex obtained by modeling the relative placement of the crystal structure of ZO-1-PDZ1 (Protein Data Bank entry 2H3M; cyan, CPK model) and the predicted structure of Neph1-CD (green, CPK model) as per the shape constraints in the SAXS data searched by the SASREF program. Right, the automated superimposition of the SWAXS data-based model of the complex and its in silico model by aligning their inertial axes. B, based on the in silico model of the Neph1-CD·ZO-1-PDZ1 complex, this figure highlights the key residues of Neph1 (green) involved in binding with ZO-1-PDZ1 (cyan). Left, the two proteins in their space-filled mode. The yellow surface represents the THV residues, which are located at the C terminus of Neph1, whereas Arg-750, Lys-761, and Tyr-762 are shown in blue, purple, and red, respectively. Tyr-637 and Tyr-638 residues are shown in brown. Two zoomed-in views on the right highlight how the THV residues play a central role in Neph1-CD/ZO-1-PDZ1 binding.
Validation of the Computed Model
The resulting His-Neph1-CD·ZO-1-PDZ1 protein complex filtered from docking using structural parameters from the scattering analysis was considered as the working model. Therefore, we analyzed the protein-protein interface to identify the residues critical for stabilizing the interprotein contacts (Fig. 5B). Mapping of the electrostatic interactions reinforced the central role of the THV motif at the C terminus and to a lesser extent the role of the Lys-761 and Tyr-762 residues of Neph1 in binding with ZO-1. The THV motif fitted into the cavity formed by the only helix and the adjacent β-sheet, as seen in the crystal structures of ZO-1-PDZ1 with and without peptides (25). To further validate our working model, we generated various His-Neph1-CD mutants, specifically lacking the THV motif (His-Neph1-CD-PDZ), K761A (His-Neph1-CD-K761A), and Y762A (His-Neph1-CD-Y762A) (supplemental Fig. 6A). In the last two point mutants, the THV motif was retained. All mutants were expressed and purified to homogeneity (supplemental Fig. 6A). The gel filtration profiles of His-Neph1-CD and its mutants showed that the mutant proteins eluted with retention times similar to the native protein (supplemental Fig. 6B). The SWAXS data sets were collected from the mutant His-Neph1-CD proteins under identical conditions in the unliganded and the equimolar mixture with His-ZO-1-PDZ1 (Table 3). The structural parameters deduced by indirect Fourier transformation of the intensity profiles suggested that all of the mutant proteins adopted a shape highly similar to that of the native His-Neph1-CD (Table 3). In addition, close similarities in the structural parameters deduced from SAXS data analyses ruled out any gross misfolding in the shape of the mutants compared with that of the native protein. Further support for the similarity in the structural content in the solution structure of the wild-type His-Neph1-CD and its mutants was supported by the comparable profiles of their circular dichroism spectra (Fig. 6A). Deconvolution of the circular dichroism spectra using the K2D2 server indicated that all of the mutant proteins have ∼25 ± 8% α-helix and 20 ± 12% β-sheet secondary structural content.
TABLE 3.
Structural parameters deduced by performing indirect Fourier transformation on the SWAXS data obtained from samples of His-Neph1-CD mutants and their mixtures with native His-ZO-1-PDZ1

a Estimated for all possible 47-kDa complex and left-over unliganded His-ZO-1-PDZ1.
FIGURE 6.

The amino acids Lys-761 and Tyr-762 in Neph1 are also involved in mediating the binding of Neph1 with ZO-1. A, CD spectra of wild-type His-Neph1-CD and its mutants (-PDZ, R750A, K761A, and Y762A) at ∼8 μm concentration under identical conditions. Spectra were vertically translated for clarity. B and C, COS-7 cells were co-transfected with the plasmids encoding Neph1 or its various indicated mutants and Myc-ZO-1. ZO-1 was immunoprecipitated (IP) from the cell lysates, and immune complexes were evaluated for binding of Neph1 with ZO-1. The experiment was repeated three times with similar results, and quantitative analysis was performed. The bar diagram shows mean pixel intensity of the Neph1 bands in the blots. Error bars, S.D. D, the purified His-Neph1 protein (cytoplasmic domain) or its indicated mutants were incubated with the purified His-ZO-1-PDZ1 protein, and the pull-down was performed using Neph1 antibody. The complex was analyzed for the presence of His-ZO-1-PDZ1 using His antibody.
Based on the estimated protein concentration from the observed I0 values and the volumes mixed, we calculated expected I0 values for the mutant-ZO-1-PDZ1 protein mixtures. The observation of a very low I0 value of 148 arbitrary units for the Neph1-PDZ-ZO-1-PDZ1 protein complex with respect to its expected I0 value of 202 arbitrary units for the formation of a 47-kDa complex confirmed no binding between this mutant and ZO-1. This observation was expected because it has been previously demonstrated that the deletion of the THV motif from the C terminus of Neph1 abolishes its ability to interact with ZO-1 (16, 17). Interestingly, the I0 values observed from the mixtures of Neph1 mutants K761A and Y762A with ZO-1-PDZ1 also suggested that similar to the Neph1-PDZ mutant, these Neph1 mutant proteins failed to interact with the ZO-1-PDZ1 in solution. In contrast, mutation of the Arg-750, which is positioned far away from the ZO-1-binding interface, should have no effect on the Neph1 and ZO-1 binding. As expected, the mutation of this residue to alanine (His-Neph1-CD-R750A) did not affect the binding between His-Neph1-CD and His-ZO-1-PDZ1 proteins (Table 3). These results from SWAXS experiments indicate that the point mutations in His-Neph1-CD do not affect the protein folding but result in almost complete loss of binding to ZO-1-PDZ1. Therefore, using this system, we are now able to demonstrate the significance of two additional sites in the Neph1 that mediate its interaction with the ZO-1.
To further validate our results obtained from SWAXS data analyses, we determined the binding of Neph1 mutants with ZO-1 both in vivo and in vitro. Various Neph1 mutants, including the point mutants, K761A, Y762A, and R750A, and the deletion mutant Neph1-PDZ (in which the last three amino acids, THV, from the C terminus were removed) were co-transfected with the Myc-ZO-1 in COS-7 cells, and Neph1 was immunoprecipitated from the cell lysates. Only the wild-type Neph1 and R750A mutant interacted with ZO-1, whereas the binding of ZO-1 with the Neph1 mutants K761A, Y762A, and Neph1-PDZ was significantly reduced (Fig. 6B). A quantitative analysis from three independent binding experiments suggested a decrease of more than 90% in the binding of Neph1 mutants K761A, Y762A, and Neph1-PDZ with ZO-1 (Fig. 6C). To further confirm these results, a similar binding study was performed in vitro with the same purified proteins used for SWAXS analysis. Purified His-Neph1-CD proteins, including the wild type and indicated mutants, were mixed with the purified His-ZO-1-PDZ1 protein, and Neph1 was immunoprecipitated with the Neph1 antibody. The immunoprecipitated complex was analyzed for the presence of ZO-1 by Western blot analysis with a His antibody. Similar to the in vivo binding experiment, the binding between the wild-type His-Neph1-CD and the His-Neph1-CD-R750A mutant with ZO-1 remained unaltered, whereas the binding between Neph1 mutant proteins, His-Neph1-CD-K761A, His-Neph1-CD-Y762A, and His-Neph1-CD-PDZ, with ZO-1 could not be detected (Fig. 6D). These results are in complete agreement with our SWAXS analysis, which shows that, in addition to the PDZ-binding domain, residues Lys-761 and Tyr-762 in Neph1 are also required for binding with the ZO-1. In the current model, residues Tyr-637/638 (Fig. 5B) are oriented distantly from the Neph1-ZO-1 binding site. In addition to the significance of these residues for Neph1 signaling, the recent results suggest that these residues are also important for mediating the interaction between Neph1 and ZO-1 (10, 16). However, the interaction mediated by these residues was dependent on Fyn-mediated phosphorylation of these tyrosine residues (16). This further suggests a need to understand the significance of phosphorylation-dependent changes in the Neph1 structure. The Neph1-CD is a signaling protein whose function includes interaction with Grb2 and ZO-1, which is significantly influenced by its phosphorylation. There is a need to determine the phosphorylation-dependent structural changes in Neph1, which will be the focus of our future investigation. Nevertheless, this current study presents a basic working structural model of Neph1 that provides greater insight in to the Neph1 function and can now be applied to understanding the phosphorylation-dependent changes in this molecule. This study has paved the way to ask these challenging questions.
In conclusion, this study utilizes fast developing SAXS technology to obtain greater insight into the molecular structure and interactions of Neph1. More importantly, this study describes the solution structures of critical domains from podocyte proteins Neph1 and ZO-1 and their complex, which will be of immense value in providing a structural overview of the molecular network of these proteins. Subsequently, similar studies can now be undertaken with other podocyte proteins to gain insight into their molecular interactions. Because the integrity of the slit diaphragm is highly dependent on these interactions, the structural insight of these interactions will be highly significant in developing tools that regulate the function of podocyte proteins in glomerular disorders.
Supplementary Material
Acknowledgment
We acknowledge the use of the SAXS facility at CSIR-CCMB (Center for Scientific and Industrial Research–Center for Cellular and Molecular Biology), which allocated time for data collection to characterize the protein samples and optimization of buffer conditions prior to experiments at the X9 beamline.
This work was supported, in whole or in part, by National Institutes of Health, NIDDK, Grants K01 DK072047-03, 5K01DK072047-04, and RO1 1R01DK087956 (to D. N.) and National Institutes of Health, NIDDK, Grant DK080751 (to L. B. H.). Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-98CH10886.

This article contains supplemental Figs. 1–7.
- SWAXS
- small/wide angle x-ray scattering
- SAXS
- small angle x-ray scattering
- WAXS
- wide angle x-ray scattering
- CD
- cytoplasmic domain
- DLS
- dynamic light scattering
- CCD
- charge-coupled detector.
REFERENCES
- 1. Patrakka J., Tryggvason K. (2010) Molecular make-up of the glomerular filtration barrier. Biochem. Biophys. Res. Commun. 396, 164–169 [DOI] [PubMed] [Google Scholar]
- 2. Tryggvason K., Pikkarainen T., Patrakka J. (2006) Nck links nephrin to actin in kidney podocytes. Cell 125, 221–224 [DOI] [PubMed] [Google Scholar]
- 3. Moreno J. A., Sanchez-Niño M. D., Sanz A. B., Lassila M., Holthofer H., Blanco-Colio L. M., Egido J., Ruiz-Ortega M., Ortiz A. (2008) A slit in podocyte death. Curr. Med. Chem. 15, 1645–1654 [DOI] [PubMed] [Google Scholar]
- 4. Johnstone D. B., Holzman L. B. (2006) Clinical impact of research on the podocyte slit diaphragm. Nat. Clin. Pract. Nephrol. 2, 271–282 [DOI] [PubMed] [Google Scholar]
- 5. Liu G., Kaw B., Kurfis J., Rahmanuddin S., Kanwar Y. S., Chugh S. S. (2003) Neph1 and nephrin interaction in the slit diaphragm is an important determinant of glomerular permeability. J. Clin. Invest. 112, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Otaki Y., Miyauchi N., Higa M., Takada A., Kuroda T., Gejyo F., Shimizu F., Kawachi H. (2008) Dissociation of NEPH1 from nephrin is involved in development of a rat model of focal segmental glomerulosclerosis. Am. J. Physiol. Renal Physiol. 295, F1376–F1387 [DOI] [PubMed] [Google Scholar]
- 7. Saleem M. A., Ni L., Witherden I., Tryggvason K., Ruotsalainen V., Mundel P., Mathieson P. W. (2002) Co-localization of nephrin, podocin, and the actin cytoskeleton. Evidence for a role in podocyte foot process formation. Am. J. Pathol. 161, 1459–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sellin L., Huber T. B., Gerke P., Quack I., Pavenstädt H., Walz G. (2003) NEPH1 defines a novel family of podocin-interacting proteins. FASEB J. 17, 115–117 [DOI] [PubMed] [Google Scholar]
- 9. Donoviel D. B., Freed D. D., Vogel H., Potter D. G., Hawkins E., Barrish J. P., Mathur B. N., Turner C. A., Geske R., Montgomery C. A., Starbuck M., Brandt M., Gupta A., Ramirez-Solis R., Zambrowicz B. P., Powell D. R. (2001) Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol. Cell Biol. 21, 4829–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garg P., Verma R., Nihalani D., Johnstone D. B., Holzman L. B. (2007) Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol. Cell Biol. 27, 8698–8712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gerke P., Huber T. B., Sellin L., Benzing T., Walz G. (2003) Homodimerization and heterodimerization of the glomerular podocyte proteins nephrin and NEPH1. J. Am. Soc. Nephrol. 14, 918–926 [DOI] [PubMed] [Google Scholar]
- 12. Hartleben B., Schweizer H., Lübben P., Bartram M. P., Möller C. C., Herr R., Wei C., Neumann-Haefelin E., Schermer B., Zentgraf H., Kerjaschki D., Reiser J., Walz G., Benzing T., Huber T. B. (2008) Neph-Nephrin proteins bind the Par3-Par6-atypical protein kinase C (aPKC) complex to regulate podocyte cell polarity. J. Biol. Chem. 283, 23033–23038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arif E., Wagner M. C., Johnstone D. B., Wong H. N., George B., Pruthi P. A., Lazzara M. J., Nihalani D. (2011) Motor protein Myo1c is a podocyte protein that facilitates the transport of slit diaphragm protein Neph1 to the podocyte membrane. Mol. Cell Biol. 31, 2134–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Verma R., Kovari I., Soofi A., Nihalani D., Patrie K., Holzman L. B. (2006) Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J. Clin. Invest. 116, 1346–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harita Y., Kurihara H., Kosako H., Tezuka T., Sekine T., Igarashi T., Hattori S. (2008) Neph1, a component of the kidney slit diaphragm, is tyrosine-phosphorylated by the Src family tyrosine kinase and modulates intracellular signaling by binding to Grb2. J. Biol. Chem. 283, 9177–9186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wagner M. C., Rhodes G., Wang E., Pruthi V., Arif E., Saleem M. A., Wean S. E., Garg P., Verma R., Holzman L. B., Gattone V., Molitoris B. A., Nihalani D. (2008) Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J. Biol. Chem. 283, 35579–35589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huber T. B., Schmidts M., Gerke P., Schermer B., Zahn A., Hartleben B., Sellin L., Walz G., Benzing T. (2003) The carboxyl terminus of Neph family members binds to the PDZ domain protein zonula occludens-1. J. Biol. Chem. 278, 13417–13421 [DOI] [PubMed] [Google Scholar]
- 18. Appleton B. A., Zhang Y., Wu P., Yin J. P., Hunziker W., Skelton N. J., Sidhu S. S., Wiesmann C. (2006) Comparative structural analysis of the Erbin PDZ domain and the first PDZ domain of ZO-1. Insights into determinants of PDZ domain specificity. J. Biol. Chem. 281, 22312–22320 [DOI] [PubMed] [Google Scholar]
- 19. Perez-Iratxeta C., Andrade-Navarro M. A. (2008) K2D2. Estimation of protein secondary structure from circular dichroism spectra. BMC Struct. Biol. 8, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Petoukhov M. V., Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nihalani D., Wong H., Verma R., Holzman L. B. (2007) Src family kinases directly regulate JIP1 module dynamics and activation. Mol. Cell Biol. 27, 2431–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jacques D. A., Trewhella J. (2010) Small-angle scattering for structural biology. Expanding the frontier while avoiding the pitfalls. Protein. Sci. 19, 642–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doyle D. A., Lee A., Lewis J., Kim E., Sheng M., MacKinnon R. (1996) Crystal structures of a complexed and peptide-free membrane protein-binding domain. Molecular basis of peptide recognition by PDZ. Cell 85, 1067–1076 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.