

Background: TGF-β plays an important role in kidney fibrogenesis, but the downstream target genes remain largely unknown.
Results: Response gene to complement 32 (RGC-32), a TGF-β target, is involved in myofibroblast activation in renal fibrosis.
Conclusion: RGC-32 is a fibrogenic factor contributing to the kidney fibrogenesis.
Significance: Targeting RGC-32 may be a novel strategy to repair kidney injury.
Keywords: Fibrinogenesis, Fibroblast, Kidney, SMAD Transcription Factor, Transforming Growth Factor Beta (TGF-β), Response Gene to Complement 32
Abstract
Response gene to complement 32 (RGC-32) is a downstream target of transforming growth factor-β (TGF-β). TGF-β is known to play a pathogenic role in renal fibrosis. In this study, we investigated RGC-32 function in renal fibrosis following unilateral ureteral obstruction (UUO) in mice, a model of progressive tubulointerstitial fibrosis. RGC-32 is normally expressed only in blood vessels of mouse kidney. However, UUO induces RGC-32 expression in renal interstitial cells at the early stage of kidney injury, suggesting that RGC-32 is involved in interstitial fibroblast activation. Indeed, expression of smooth muscle α-actin (α-SMA), an indicator of fibroblast activation, is limited to the interstitial cells at the early stage, and became apparent later in both interstitial and tubular cells. RGC-32 knockdown by shRNA significantly inhibits UUO-induced renal structural damage, α-SMA expression and collagen deposition, suggesting that RGC-32 is essential for the onset of renal interstitial fibrosis. In vitro studies indicate that RGC-32 mediates TGF-β-induced fibroblast activation. Mechanistically, RGC-32 interacts with Smad3 and enhances Smad3 binding to the Smad binding element in α-SMA promoter as demonstrated by DNA affinity assay. In the chromatin setting, Smad3, but not Smad2, binds to α-SMA promoter in fibroblasts. RGC-32 appears to be essential for Smad3 interaction with the promoters of fibroblast activation-related genes in vivo. Functionally, RGC-32 is crucial for Smad3-mediated α-SMA promoter activity. Taken together, we identify RGC-32 as a novel fibrogenic factor contributing to the pathogenesis of renal fibrosis through fibroblast activation.
Introduction
Fibrotic kidney disease is a major unresolved problem in clinical medicine because of incomplete understanding of its pathophysiology and the lack of effective therapeutic strategies. The progression of renal fibrosis is considered to be a common process that eventually leads to end-stage renal disease, necessitating dialysis or renal transplantation (1–3). Irrespective of the initial cause(s), the striking feature of tubulointerstitial fibrosis is the activation of smooth muscle α-actin (α-SMA)3-positive myofibroblasts, which are thought to be the central effectors responsible for extracellular matrix (ECM) deposition and scar tissue formation in renal disease (4–6). Although multiple sources are proposed (7–9), lineage analyses show that resident mesenchymal fibroblasts are the myofibroblast progenitor pool during fibrosis (10). Therefore, interstitial fibroblast activation remains a very important pathway leading to the generation of ECM-producing myofibroblasts.
Transforming growth factor-β (TGF-β) and its intermediate Smad signaling proteins are viewed as the premier fibrosis-promoting molecules and play major roles in the pathogenesis of renal fibrosis (1, 11–17). However, the downstream targets mediating the fibrogenic effect of TGF-β remain largely unknown. Response gene to complement 32 (RGC-32) is a TGF-β downstream target (18, 19). The present study was designed to investigate the potential role of RGC-32 in renal fibrosis following unilateral ureteral obstruction (UUO), a model of kidney disease with progressive tubulointerstitial fibrosis. Our results show that UUO causes structural damage and collagen deposition along with increased expression of RGC-32 in kidney interstitial cells at the early stage of the injury. Blockade of RGC-32 expression significantly attenuates UUO-induced renal damage, ECM production, and expression of myofibroblast marker. Studies using mesenchymal and kidney fibroblasts indicate that RGC-32 induces fibroblast activation in vitro. Additional studies demonstrate that RGC-32 interacts with Smad3 to enhance its binding to α-SMA promoter, thus regulate Smad3 induction of myofibroblast marker transcription. Together, our results have identified RGC-32 as a novel fibrogenic factor contributing to the onset of renal tubulointerstitial fibrosis.
EXPERIMENTAL PROCEDURES
Experimental Animals and Design
All surgeries were performed according to protocols overseen by the Institutional Animal Care and Use Committee (IACUC) of Georgetown University. Male C57BL6/J mice, weighing 20–25 g at the start of the experiment, were anesthetized with pentobarbital and tracheotomized. UUO was achieved by double-ligating the left ureter with 3–0 silk through a left lateral incision. Sham-operated animals (n = 5) were used as controls. Mice were sacrificed at 3, 5, 7, 10, or 15 days (for each group, n = 5) after the UUO, and the obstructed kidneys were harvested and subjected to the studies described below.
In Vivo Delivery of shRNA and Expression Plasmid
shRNA is constructed in pGeneClip vector (Promega). The delivery was performed by injection of scrambled or RGC-32 shRNA plasmids through the renal artery using the In Vivo Gene Delivery System (Mirus). The mice were anesthetized with pentobarbital (50 mg/kg body wt). The femoral and abdominal areas of the mice were shaved and treated with chlorhexidine as described in UUO model. The abdominal aorta was exposed via a midline abdominal incision. The left femoral artery was exposed by an inguinal incision on the ventral side of the left thigh. Using a stereo-microscope, a PE-10 catheter, heat-stretched to 180 micra, was inserted into the femoral artery (through an appropriately sized incision) and pushed inside the abdominal aorta to a position just above the left renal artery but below the right renal artery. This position was verified by gently lifting the vessel using a small forceps. This procedure allowed the selective and exclusive delivery of reagents into the left kidney. The abdominal aorta was clamped below the right renal artery and below the left renal artery, to ensure delivery of the reagents only into the left kidney. In our preliminary studies, the clamping procedure did not affect renal function or histology. After the injection of the reagents in a 200 μl solution (with lissamine green) in 20 s using a 1 ml tuberculin syringe, the catheter was carefully retracted. The left ureter was then sham- or double-ligated to generate UUO. The incision on the femoral artery was repaired using 11–0 silk sutures and the skin incisions closed.
Histopathological and Immunohistochemical Analyses
Kidney tissues were fixed in 4% paraformaldehyde in 1× PBS and embedded in paraffin. Tissue sections (5 μm thick) were stained with H&E or Masson's trichrome staining for histopathological analysis. Immunoreactivity for RGC-32, α-SMA, and collagen I was determined by using standard immunohistochemistry staining. The primary antibodies were rabbit anti-RGC-32 (19), monoclonal mouse anti-α-SMA (Sigma), and mouse anti-collagen I (Abcam). The secondary antibody was affinity-purified goat anti-rabbit or goat anti-mouse IgG (Cell Signaling). Nuclei were counterstained lightly with hematoxylin.
Masson's Trichrome Staining
Masson's staining was performed using a Masson's trichrome staining kit (DAKO), following standard procedures.
Cell Culture, Transfection, and Luciferase Assay
C3H10T1/2 and NRK-49F cells were cultured as previously described (20–22). Cells were transiently transfected (in triplicate) with Lipofectamine LTX plus reagents (Invitrogen), and luciferase assay was performed as described (20, 21).
Reverse Transcription-PCR (RT-PCR) and Quantitative PCR (qPCR)
Total RNA was extracted using TRIzol reagent (Invitrogen) following the manufacturer's instruction. cDNA was synthesized using an iScript cDNA synthesis kit (Bio-Rad). RT-PCR was performed using Bio-Rad C1000 thermal cycler. qPCR was performed in MX3000P qPCR machine using SYBR Green qPCR Mastermix (Agilent). The primers used in PCR were described previously (19).
Western Blotting
Fibroblasts or kidney tissues were lysed or homogenized in RIPA lysis buffer (1% Nonidet P-40, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate, 1 mm sodium orthovanadate, and protease inhibitors in PBS). Samples were separated on SDS-polyacrylamide gels, and electrotransferred onto nitrocellulose membranes (Amersham Biosciences). The membranes were incubated for 16 h at 4 °C with various primary antibodies in blocking buffer containing 5% milk followed by incubation with HRP-conjugated secondary antibody (Bio-Rad). The co-immunoprecipitation (co-IP) assay was performed as described (23, 24).
DNA Affinity Assay
DNA protein binding assay was performed as described (25, 26). Briefly, 5 μg of biotin-labeled double-strand oligonucleotides, and 500 ng of Smad3, Smad4 (SignalChem), and/or RGC-32 proteins were incubated in a binding buffer (0.1% Triton X-100, 4% glycerol, 1 mm EDTA, 10 mm DTT, 10 mm Tris) at 37 °C for 1 h. Streptavidin-coated agarose beads (Sigma) were then added followed by end-over-end rotation at 4 °C overnight. The agarose beads were pelleted and washed with cold 1× PBS three times, and proteins bound to the beads were eluted and immunoblotted with Smad3, Smad4, or RGC-32 antibodies. RGC-32 proteins were obtained by expression of pETDuet-1-RGC32 in E. coli and purified by His-Spin Protein Miniprep kit (Zymo Research). pETDuet-1-RGC32 was cloned by insertion of full-length mouse RGC-32 cDNA into pETDuet-1 expression vector at BamHI/SalI sites. The cDNA insertion was verified by sequencing and the purity of RGC-32 protein was verified by Coomassie Blue staining and Western blot using RGC-32 antibody. The biotin-labeled oligonucleotides used in the DNA affinity assay were from the rat α-SMA promoter region with SBE (26). The sense strand sequences for wild type and mutant oligonucelotides were: SBE: 5′-ACA GAC TTC ATT GAT ACT ACA CAC AGA CTC CAG ACT AC-3′, SBE mutant: 5′-TAC AGA CTT CAT TGA TAC TAC ACA aAG ctT CCA GAC-3′.
Chromatin Immunoprecipitation Assay (ChIP)
ChIP assays were performed as described previously (24, 27). NRK-49F cells were treated with vehicle or TGF-β for 24 h followed by additional 1 h of treatment with new TGF-β to induce Smad nuclear translocation. Chromatin complexes were immunoprecipitated with 3 μg of Smad2 or Smad3 antibody or IgG (negative control). Semi-quantitative PCR and/or qPCR were performed to amplify the α-SMA promoter region containing the functional Smad binding site using the following primer set: 5′-CAT GCA CGT GGA CTG TAC CT-3′ (forward) and 5′-AAA GAT GCT TGG GTC ACC TG-3′ (reverse) (26). The primers for amplifying the SBE region for the PAI-1 promoter were: 5′-CTC TGT GAT GGC TGT CTC CA-3′ (forward) and 5′-CTT CCC TCC CTC CCA GTA AC-3′ (reverse) (28).
Statistical Analysis
All values are expressed as mean ± S.E. Data were analyzed using ANOVA with pairwise comparisons between groups. A level of p < 0.05 was considered statistically significant.
RESULTS
RGC-32 Is Induced Along with the Progression of Renal Fibrosis in Kidneys with UUO
To determine if RGC-32 is involved in the pathogenesis of renal fibrosis, we first determined if RGC-32 expression in kidney is altered by UUO. RGC-32 was found to be expressed in mouse kidney (29), but precisely which cells express RGC-32 has not been determined. We found that RGC-32 was expressed only in smooth muscle cells (SMC) of blood vessels in the normal mouse kidney as indicated by α-SMA staining (Fig. 1A, panel IV). 5 days after the UUO, RGC-32 was strongly induced in the tubulointerstitium (Fig. 1A, panel V). Importantly, α-SMA expression was also limited to the interstitial cells of obstructed kidney at this early stage (Fig. 1A, panel VIII), suggesting that RGC-32 plays a role in the activation of myofibroblasts from resident fibroblasts. By 15 days after the UUO, RGC-32, and α-SMA were extensively expressed in both kidney interstitium and tubules (Fig. 1A, panels VI and IX). At this time point, severe kidney structural damages including glomeruli atrophy, tubule dilation, and atrophy, inflammatory cell infiltration, and extensive interstitial fibrosis were observed (Fig. 1A, panel III). These results indicate that RGC-32 is induced along with myofibroblast activation by UUO. To examine the dynamics of RGC-32 expression induced by progressive injury due to UUO, kidney proteins were extracted 5, 10, and 15 days after the UUO and blotted with RGC-32 and α-SMA antibodies. As shown in Fig. 1B, both RGC-32 and α-SMA were significantly up-regulated 5 days after UUO. The expression increased with the duration of UUO. It appeared that RGC-32 expression reached the highest level at 10 days after UUO and remains unchanged at 15 days after UUO. Interestingly, the expression of α-SMA correlated with the activation of RGC-32 (Fig. 1C).
FIGURE 1.

RGC-32 is induced, along with myofibroblast activation, in the interstitium of the kidneys with UUO. A, RGC-32 expression in the interstitial cells. Kidney sections of sham-operated mice or mice with UUO for 5 days or 15 days were stained by H&E or immunostained with RGC-32 or α-SMA antibody as indicated. RGC-32 and α-SMA are present only in smooth muscles of blood vessels (V in panels IV and VII) in normal kidneys (Sham). However, RGC-32 and α-SMA are expressed in interstitial cells (Int in A- panels V and VIII) after 5 days of UUO. B and C, quantitative analysis of RGC-32 expression in mouse kidneys with UUO. Protein extracts from sham-operated or mouse kidneys with UUO were prepared as described under “Experimental Procedures.” Western blot was performed to detect RGC-32 and α-SMA protein expression (B). The expression levels were normalized to tubulin (C). * and **, p < 0.01 compared with sham-operated groups for RGC-32 (*) and α-SMA (**), respectively.
RGC-32 Knockdown Attenuates the Fibroblast Activation in Vivo
To test if RGC-32 plays a role in the pathogenesis of renal fibrosis, we developed a method to infuse RGC-32 shRNA into mouse kidney through renal artery using an In Vivo Gene Delivery System. Using this method, we infused control or RGC-32 shRNA into mouse left kidneys followed by UUO for 10 days. As shown in Fig. 2A, shRNA knockdown of UUO-induced RGC-32 expression blocked α-SMA expression in the interstitial cells (Fig. 2A, panels II and IV). RGC-32 shRNA did not block RGC-32 or α-SMA expression in SMC of the blood vessels (Fig. 2A, panels II and IV) because SMC in the blood vessels normally expresses a significant amount of RGC-32 protein before UUO operation (Fig. 1A, panel IV). It is known that shRNA only degrades the newly synthesized mRNA but does not affect pre-existing proteins. Quantitative analyses show that UUO kidneys with control shRNA strongly expressed RGC-32 and α-SMA proteins (Fig. 2, B and C). However, RGC-32 shRNA, which blocked the expression of RGC-32 with an 80% knockdown efficiency, caused a 50% reduction of α-SMA expression induced by UUO (Fig. 2C). These data demonstrate that RGC-32 is essential for myofibroblast activation in kidney interstitium in the onset or progression of UUO-induced renal fibrosis.
FIGURE 2.

RGC-32 knockdown blocks α-SMA expression. Scrambled (shCtrl) or RGC-32 shRNA (shRGC) was infused into the mouse left kidney followed by UUO for 10 days. Kidney sections were stained with RGC-32 or α-SMA antibody as indicated. A, RGC-32 shRNA blocked α-SMA expression in interstitial cells but not in the blood vessels (A- panels II and IV). Western blot (B) and quantitative analysis of RGC-32 and α-SMA expression (C) showed that RGC-32 knockdown significantly inhibited α-SMA expression in kidney interstitium. * and **, p < 0.01 compared with shCtrl groups for RGC-32 (*) or a-SMA (**).
Blockade of RGC-32 Inhibits ECM Deposition in Mouse Kidneys with UUO
Excessive synthesis and accumulation of ECM proteins in kidney interstitium is a hallmark of renal tubulointerstitial fibrosis, as seen in obstructive nephropathy. ECM deposition results in kidney structural damage and functional impairment. To determine if RGC-32 plays a role in ECM deposition as well as structural damage induced by UUO, we observed kidney structural alteration by H&E staining and examined the expression of collagen I by Masson's trichrome staining and Western blot in kidneys infused with control or RGC-32 shRNA. Structurally, UUO caused dilatation of renal tubules, denudation of basement membranes, and interstitial expansion, consistent with previous reports (30) (Fig. 3A, panel II). RGC-32 knockdown by shRNA, however, significantly reduced UUO-induced damage. RGC-32 shRNA restored the normal structure of proximal tubules and eliminated the interstitial expansion (Fig. 3A, panel III). These data suggest that RGC-32 contributed, at least in part, to the structural damage in the progressive nephropathy.
FIGURE 3.

RGC-32 knockdown ameliorates UUO-induced structural damage in kidneys and reduces ECM accumulation. Scrambled (shCtrl) or RGC-32 shRNA (shRGC) was infused into mouse left kidney followed by UUO for 10 days. H&E or Masson's trichrome (Masson's) staining was performed (A) in kidney sections to observe the structural alteration (panels I-III) and collagen deposition (panels IV-VI). Collagen I expression was examined by Western blot (B) and quantified via normalization to tubulin (C). *, p < 0.01 compared with Sham group; **, p < 0.01 compared with shCtrl group.
RGC-32 appears to be important for ECM production in UUO-induced nephropathy. Compared with sham-operated mice, UUO induced a significant amount of collagen accumulation in kidney tubulointerstitium (Fig. 3A, panel V). RGC-32 shRNA, however, significantly inhibited the accumulation of collagen (Fig. 3A, panel VI) in kidney interstitium. Western blot showed that UUO induced a significant increase in collagen I expression in the kidney 10 days after the UUO. RGC-32 knockdown, however, significantly blocked collagen I expression (Fig. 3, B and C). These data demonstrate that RGC-32 is required for collagen I synthesis and accumulation in the renal interstitium in obstructive nephropathy.
RGC-32 Is Important for Fibroblast Activation
Although myofibroblasts may originate from several sources including resident mesenchymal fibroblasts, epithelial cells via EMT, endothelial cells via endothelial-mesenchymal transition, and circulating fibroblast-like cells called fibrocytes derived from bone-marrow stem cells (7–9), interstitial resident mesenchymal fibroblast activation remains one of the important pathways leading to the generation of ECM-producing myofibroblasts. UUO induced interstitial expression of both RGC-32 and α-SMA in mouse kidneys, especially in the early stages of obstructive nephropathy (Fig. 1A, panels V, VIII), suggesting that the α-SMA-producing myofibroblasts were derived from interstitial fibroblasts.
TGF-β, which is activated in kidneys 3 days after UUO, plays a central role in activating fibroblasts in renal fibrosis (2, 31, 32). Because RGC-32 is a TGF-β downstream target (18, 19) and is important for the generation of myofibroblasts in the kidney with UUO (Figs. 2 and 3), we hypothesize that RGC-32 is induced by TGF-β during myofibroblast activation from mesenchymal fibroblasts. To test this hypothesis, we used C3H10T1/2 (10T1/2) mesenchymal progenitor cells (Fig. 4A, B, and D)) and kidney fibroblast NRK-49F cells (Fig. 4C) as models to study the induction of myofibroblasts by TGF-β. As shown in Fig. 4, A–C, TGF-β induced myofibroblast differentiation of 10T1/2 and NRK-49F cells as indicated by the expression of myofibroblast marker genes α-SMA and collagen I. TGF-β also induced RGC-32 expression during the fibroblast activation (Fig. 4, A--C). The induction of RGC-32 occurred as early as 2 h after TGF-β induction (Fig. 4A), suggesting that RGC-32 plays a role in TGF-β-induced fibroblast activation.
FIGURE 4.

RGC-32 induces activation of mesenchymal fibroblasts. A, TGF-β induces RGC-32 and myofibroblast markers in 10T1/2 cells. 10T1/2 cells were treated with vehicle (0) or TGF-β for the times indicated. Western blot was performed using RGC-32, α-SMA, collagen I, or fibronectin antibody. B and C, RGC-32 knockdown blocks TGF-β-induced fibroblast activation. Control (shCtrl/Ad-GFP) or RGC-32 shRNA (shRGC32/Ad-shRGC32) was expressed in 10T1/2 cells (B) or NRK-49F (C) followed by vehicle or TGF-β treatment as indicated. RT-PCR (B) or Western blot (C) was performed to detect α-SMA, collagen, and fibronectin expression. D, RGC-32 induces fibroblast activation. RGC-32 cDNA was overexpressed in 10T1/2 cells. Myofibroblast markers α-SMA, collagen, and fibronectin expression were examined by RT-PCR.
To test if RGC-32 is important for TGF-β-induced fibroblast activation, we expressed control or RGC-32 shRNA in 10T1/2 (Fig. 4B) and NRK-49F cells (Fig. 4C) and then treated the cells with TGF-β for 24 h and studied the effect of RGC-32 knockdown on myofibroblast markers. Control shRNA did not affect the expression of RGC-32 and myofibroblast markers in TGF-β-treated cells. RGC-32 shRNA, however, significantly inhibited TGF-β-induced expression of RGC-32 as well as α-SMA and collagen I (Fig. 4, B and C). These results indicate that RGC-32 is essential for TGF-β-induced fibroblast activation. To determine if RGC-32 alone induces fibroblast activation, RGC-32 cDNA was transfected in 10T1/2 cells; and myofibroblast marker gene expression was examined. Interestingly, RGC-32 by itself was sufficient to induce the expression of myofibroblast markers (Fig. 4D).
RGC-32 Physically Interacts with Both Smad3 and Smad2
TGF-β function is mediated by both Smad2 and Smad3. In fibroblasts, it appears that Smad3, but not Smad2, is important for the activation of α-SMA promoter activity (20, 33). TGF-β-induced collagen type I and III expression is ablated in Smad3 knock-out (KO) mouse embryo fibroblasts (MEF), but not in the Smad2 KO MEF (15). Although Smad2 is also involved, Smad3 appears to play a major role in activating fibroblasts and fibrosis (15–17, 20, 33). Because RGC-32 and Smad3 are both involved in regulating the promoter activity of myofibroblast marker α-SMA in different cells (18, 34–36), we hypothesized that RGC-32 interacts with Smad3 to induce fibroblast activation. To test if RGC-32 and Smad3 physically interacts, Co-IP was performed by overexpressing RGC-32, Smad2 and Smad3 individually or in combination in 10T1/2 cells. As shown in Fig. 5A, RGC-32 strongly interacted with Smad3, but slightly with Smad2 in 10T1/2 cells. To confirm these interactions, Co-IP was performed using endogenous proteins extracted from 10T1/2 cells. Interestingly, as shown in Fig. 5, B and C, RGC-32 appeared to interact with both Smad2 and Smad3 (left panels of Fig. 5, B and C). These interactions were specific because shRNA knockdown of Smad2 or Smad3 diminished the Co-IP between RGC-32 and Smad2 or Smad3 (Fig. 5, B and C, right panels).
FIGURE 5.

RGC-32 physically interacts with Smad3 in mesenchymal fibroblasts. A, RGC-32 physical interaction with Smad3. Flag-tagged Smad2, Smad3, and T7-tagged RGC-32 plasmids were transfected into 10T1/2 cells as indicated. Co-IP was performed. RGC-32-interacting proteins were pulled down with T7 antibody (IP:T7), and blotted (IB) with Flag antibody. T7 antibody blotting was a Co-IP positive control. Smad2, Smad3, or RGC-32 expression was monitored in whole cell lysates (WCL IB) with Flag or T7 antibodies as indicated. B and C, co-IP using endogenous proteins. TGF-β-treated 10T1/2 cells were lysed with Co-IP buffer (Pierce). RGC-32 interacting proteins were pulled down using RGC-32 (RGC32) antibody and blotted with Smad2 (S2, B-left panel) or Smad3 (S3, C-left panel). IgG was a negative control. The specificity of RGC-32 interaction with Smad2/3 was verified by Smad2 (shS2, B-right panel) or Smad3 (shS3, C-right panel) shRNA. Control shRNA (shCtrl) was used as a negative control for shRNA function. Smad2 (S2) and Smad3 (S3) protein bands are indicated.
RGC-32 Enhances Smad3 Binding to Smad Binding Element (SBE) in the Promoters of Fibroblast Activation-related Genes
Smad3 regulates α-SMA gene transcription through binding to SBE in the α-SMA promoter (35). To test if Smad2 or Smad3 interacts with SBE in α-SMA promoter in a chromatin setting in kidney fibroblasts, CHIP assays were performed. We found that Smad3, but not Smad2, bound the functional SBE in α-SMA promoter in vivo in kidney fibroblasts (Fig. 6A). Because RGC-32 interacts with Smad3, we sought to determine if RGC-32 plays a role in Smad3 interaction with α-SMA promoter in vivo. Adenoviral vector expressing GFP or RGC-32 shRNA was transduced into NRK-49F cells followed by vehicle or TGF-β treatment. Endogenous Smad3 binding to SBE in α-SMA promoter was assessed by CHIP assay. We found that TGF-β induced an enhanced binding of Smad3 to α-SMA promoter (Fig. 6, B and C). RGC-32 knockdown, However, significantly blocked TGF-β-enhanced Smad3 interaction with the promoter (Fig. 6, B and C), suggesting that RGC-32 is essential for Smad3 binding to SBE in α-SMA promoter in kidney fibroblasts, thus regulating Smad3-mediated α-SMA promoter activity.
FIGURE 6.

RGC-32 enhances Smad3 binding to the promoters of fibroblast activation-related genes in vivo. A, Smad3, but not Smad2, binds to SBE in α-SMA promoter in a chromatin setting. CHIP assays were performed using TGF-β-treated NRK-49F cells with control IgG, Smad2, or Smad3 antibody. B—E, RGC-32 knockdown blocks TGF-β-enhanced Smad3 binding to SBEs of α-SMA (B, C) and PAI-1 gene promoters (D, E). Ad-GFP or Ad-shRGC-32 was transduced into NRK-49F cells followed by vehicle (−) or TGF-β induction for 24 h. Cells were then treated with TGF-β for additional 1 h to induce Smad3 nuclear translocation. CHIP assays were performed using control IgG or Smad3 antibody. RT-PCR (B, D) and qPCR (C, E) were performed to show the enrichment of Smad3 binding to SBEs in the α-SMA (B, C) and PAI-1 (D, E) promoters. Smad3 binding in vehicle-treated group was set as 1. *, p < 0.01 compared with vehicle-treated group (−); #, p < 0.01 compared with Ad-GFP group. TGF-β enhanced Smad3 binding to SBE. RGC-32 knockdown, however, diminished TGF-β-induced effect.
To determine if RGC-32 enhances Smad3 interaction with promoters of other fibroblast activation-related genes, we tested Smad3 binding to the SBE in the promoter of plasminogen activator inhibitor-1 (PAI-1). PAI-1 plays an important role in fibroblast activation and renal fibrosis (37). As shown in Fig. 6, D and E, similar to α-SMA promoter, TGF-β increased the Smad3 binding to SBE of PAI-1 promoter, shRNA knockdown of RGC-32, however, significantly inhibited Smad3 binding. These results suggest that RGC-32 is crucial for Smad3 interaction with promoters of multiple fibroblast activation genes.
RGC-32 Increases the Affinity of Smad3 Binding to α-SMA Promoter
To further determine the role of RGC-32 in Smad3 interaction with α-SMA promoter, we performed DNA affinity assay with purified Smad3, Smad4, and RGC-32 proteins and biotin-labeled DNA oligonucleotides containing wild type or mutant SBE. Smad3 bound to α-SMA in a dose-dependent manner; SBE mutation diminished the binding (Fig. 7A). Both Smad4 and RGC-32 enhanced the Smad3 binding to the SBE site (Fig. 7, B and C). Importantly, combination of RGC-32 and Smad4 further increased the Smad3 binding (Fig. 7, B and C), demonstrating that RGC-32 is able to increase the Smad3 binding affinity to α-SMA promoter with or without the presence of Smad4.
FIGURE 7.

RGC-32 increases Smad3 binding affinity to α-SMA promoter. A, dose-dependent binding of Smad3 to the SBE region of α-SMA promoter. Mutation at the SBE site (SBEmt) blocked the binding. B, RGC-32 enhances Smad3 binding affinity. Smad3, Smad4, and RGC-32 (RGC32) were incubated with biotin-labeled oligonucleotides as indicated. DNA binding assay was performed as described under “Experimental Procedures.” C, quantitative analysis of Smad3 binding to SBE. Smad3 alone binding was set as 1. *, p < 0.01 compared with Smad3 alone group. #,p < 0.01 compared with Smad3 with Smad4 or Smad3 with the RGC-32 group.
RGC-32 Plays an Essential Role in Smad3-mediated Activation of Myofibroblast Marker Gene Transcription
To determine if RGC-32-mediated Smad3 binding to α-SMA promoter plays a role in fibroblast activation, we tested if RGC-32 affects Smad3-induced α-SMA promoter activity in both 10T1/2 (Fig. 8A) and NRK-49F cells (Fig. 8B). We found that only Smad3, but not Smad2, significantly increased α-SMA promoter activity in both cells (Fig. 8), consistent with their role in renal fibrosis (15–17, 20, 33). Smad3-induced promoter activity was diminished, however, by RGC-32 shRNA in both mesenchymal (Fig. 8A) and kidney fibroblasts (Fig. 8B), demonstrating that RGC-32 is required for Smad3 function in inducing fibroblast activation. RGC-32 shRNA also reduced α-SMA promoter activity in control (Fig. 8B) or Smad2-transfected cells (Fig. 8, A and B), but this effect is likely due to the interruption of endogenous RGC-32/Smad3 interaction by RGC-32 shRNA. It was not an effect on Smad2 activity because Smad2 did not increase α-SMA promoter activity (Fig. 8). These data demonstrate that RGC-32 enhances Smad3 binding, and thus activates the promoters of myofibroblast marker genes to mediate TGF-β-induced fibroblast activation, which may lead to the generation of myofibroblasts in kidney interstitium when the mouse is subjected to progressive injury as seen in obstructive injury and UUO.
FIGURE 8.
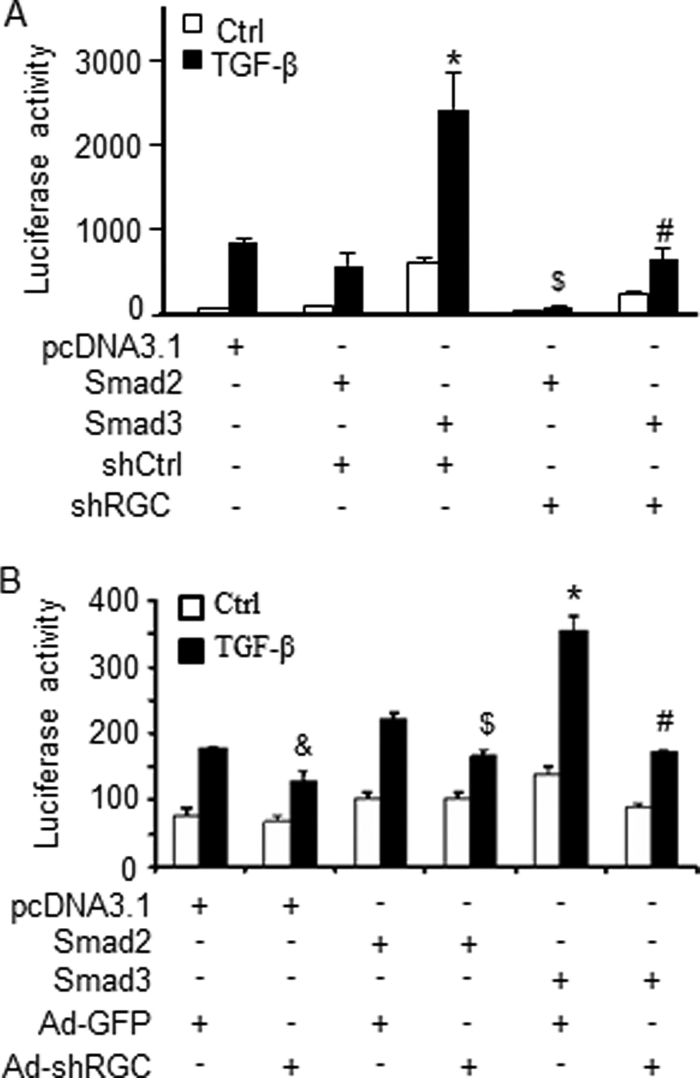
RGC-32 is required for Smad3-mediated α-SMA promoter activity. Control (shCtrl/Ad-GFP) or RGC-32 shRNA (shRGC/Ad-shRGC) was expressed in 10T1/2 (A) or NRK-49F cells (B) for 2 days followed by α-SMA promoter reporter cotransfection with pcDNA3.1, Smad2, or Smad3 expression plasmid as indicated. The cells were then treated with vehicle (Ctrl) or TGF-β for 18 h. Luciferase assay was performed. &, p < 0.05 compared with pcDNA3.1 with Ad-GFP group treated with TGF-β; $, p < 0.05 compared with Smad2 with shCtrl (A) or Ad-GFP group (B) treated with TGF-β; *, p < 0.01 compared with other groups treated with TGF-β; #, p < 0.01 compared with Smad3 with Ad-GFP group treated with TGF-β. RGC-32 shRNA blocked Smad3-induced α-SMA promoter activity.
DISCUSSION
Emerging evidence indicates that TGF-β is a key mediator in the progression of renal fibrosis (38). Tubulointerstitial fibrosis is an inevitable common pathway of chronic kidney diseases (CKD) resulting from many disorders, including hypertension, diabetes, infection, inflammation, kidney stones, and cysts (39). The extent of tubulointerstitial damage correlates with renal prognosis (40). Therefore, elucidation of the pathogenetic mechanisms involved in the progression of tubulointerstitial fibrosis is important for designing a strategy to prevent progressive kidney diseases leading to end-stage renal failure. Many studies have shown that TGF-β signaling intermediate Smad proteins including Smad3, Smad4, and Smad7 play important roles in the progression of renal fibrosis (16, 41, 42). However, the downstream targets essential for the onset of the fibrosis especially fibroblast activation, however, remain largely unknown.
RGC-32, a complement-activated gene, was identified as a TGF-β downstream target, which plays an important role in smooth muscle differentiation from neural crest cells (18). RGC-32 is also involved in the EMT of renal epithelial cells in vitro (19). Because the relative contribution of EMT to renal fibrosis is still being debated (43), RGC-32-induced EMT may not contribute to the development of renal fibrosis in vivo. Indeed, our data show that RGC-32 is mainly induced in interstitial cells in mouse kidneys with UUO, especially in the early stage of renal fibrosis. The induction correlates with the expression of myofibroblast marker α-SMA (Fig. 1), suggesting a role of RGC-32 in fibroblast activation in the kidney interstitium. Knockdown of RGC-32 by shRNA significantly attenuates the expression of both RGC-32 and α-SMA after 10 days of UUO (Fig. 2), demonstrating that RGC-32 is essential for fibroblast activation. Knockdown of RGC-32 also ameliorates kidney structural damage and ECM deposition in the kidney interstitium caused by UUO, suggesting that RGC-32 may be an ideal target for reversing the progression of renal fibrosis. Although RGC-32 is also expressed in renal tubules at the later stages of fibrosis, e.g. 15 days after UUO, the EMT events of the tubule cells such as migration and invasion are not evident in kidneys with UUO (10). Therefore, the role of RGC-32 in EMT of renal tubule cells in vivo can only be determined after the EMT mechanism in the obstructive nephropathy is well-defined.
RGC-32 is important for fibroblast activation in both mesenchymal and kidney fibroblasts. RGC-32 knockdown by shRNA blocks TGF-β-induced marker gene expression (Fig. 4, B and C). Mechanistically, RGC-32 appears to partner with Smad3 to regulate fibroblast activation. It is well documented that Smad3 is critical for the induction of renal fibrosis (16, 42, 44). RGC-32 shares several common characteristic with Smad3. Both of RGC-32 and Smad3 are TGF-β downstream effectors important for fibroblast activation (Fig. 4) (15–17, 20, 33); both are nuclear proteins regulating the same myofibroblast marker genes such as α-SMA (18, 20, 29, 33); and both play roles in renal fibrosis. These observations suggest that RGC-32 interacts with Smad3 in activating fibroblasts. Indeed, RGC-32 physically interacts with Smad3 (Fig. 5). Although RGC-32 also interacts with Smad2, Smad2 does not bind to α-SMA promoter and is not involved in TGF-β-induced fibroblast activation (Figs. 6A and 8).
RGC-32 appears to induce fibroblast activation through enhancing Smad3 binding to SBE of myofibroblast marker genes. Smad3, but not Smad2, binds to the SBE of α-SMA promoter. TGF-β enhances Smad3 binding. TGF-β-induced Smad3-SBE interaction requires RGC-32 because knockdown of RGC-32 attenuates Smad3 binding to α-SMA promoter in vivo. In addition to α-SMA, RGC-32 also increases Smad3 binding to PAI-1 promoter, suggesting that RGC-32 may regulate several different fibroblast activation-related genes via enhancing Smad3 binding to SBE in their promoters. Importantly, RGC-32 appears to increase Smad3 binding affinity independent of Smad4 because RGC-32 is able to enhance Smad3 binding to SBE of α-SMA promoter in the absence of Smad4. In the presence of Smad4, RGC-32 can further increase Smad4-enhanced Smad3 binding. Functionally, RGC-32 is essential for TGF-β-induced α-SMA promoter activation in fibroblasts. Smad3 is required for TGF-β function in activating the promoter, but Smad3 function is dependent on RGC-32 because RGC-32 knockdown blocks Smad3-mediated promoter activity (Fig. 8), suggesting that RGC-32 functionally interacts with Smad3 in the activation of α-SMA promoter.
In conclusion, we have identified RGC-32 as a novel fibrogenic factor in the progression of renal fibrosis in obstructive nephropathy in UUO model. We have also demonstrated that RGC-32 mediates TGF-β-induced fibroblast activation by interacting with Smad3, leading to an enhanced Smad3 binding to SBE, which activate the transcription programs of myofibroblast marker genes.
This work was supported, in whole or in part, by Grants HL093429 and HL107526 (to S. Y. C.) and DK39308 and HL23081 (to P. A. J.) from the National Institutes of Health.
- α-SMA
- smooth muscle α-actin
- TGF
- transforming growth factor
- UUO
- unilateral ureteral obstruction
- ECM
- extracellular matrix.
REFERENCES
- 1. Eddy A. A. (2000) Pediatr. Nephrol. 15, 290–301 [DOI] [PubMed] [Google Scholar]
- 2. Klahr S. (2003) J. Nephrol. 16, 179–185 [PubMed] [Google Scholar]
- 3. Owen W. F., Jr. (2003) J. Am. Soc. Nephrol 14, S76–80 [DOI] [PubMed] [Google Scholar]
- 4. Dai C., Yang J., Bastacky S., Xia J., Li Y., Liu Y. (2004) J. Am. Soc. Nephrol 15, 2637–2647 [DOI] [PubMed] [Google Scholar]
- 5. Hewitson T. D., Becker G. J. (1995) Am. J. Nephrol 15, 111–117 [DOI] [PubMed] [Google Scholar]
- 6. Essawy M., Soylemezoglu O., Muchaneta-Kubara E. C., Shortland J., Brown C. B., el Nahas A. M. (1997) Nephrol. Dial Transplant. 12, 43–50 [DOI] [PubMed] [Google Scholar]
- 7. Wynn T. A. (2008) J. Pathol. 214, 199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang N., Wu L. L., Nikolic-Paterson D. J., Ng Y. Y., Yang W. C., Mu W., Gilbert R. E., Cooper M. E., Atkins R. C., Lan H. Y. (1998) Nephrol. Dial Transplant. 13, 1967–1974 [DOI] [PubMed] [Google Scholar]
- 9. Masur S. K., Dewal H. S., Dinh T. T., Erenburg I., Petridou S. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 4219–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Humphreys B. D., Lin S. L., Kobayashi A., Hudson T. E., Nowlin B. T., Bonventre J. V., Valerius M. T., McMahon A. P., Duffield J. S. (2010) Am. J. Pathol. 176, 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jernigan S. M. (2000) Experimental Insights into the Mechanisms of Tubulo-interstitial Scarring, Oxford University Press, Oxford [Google Scholar]
- 12. Isaka Y., Fujiwara Y., Ueda N., Kaneda Y., Kamada T., Imai E. (1993) J. Clin. Invest. 92, 2597–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li J. H., Zhu H. J., Huang X. R., Lai K. N., Johnson R. J., Lan H. Y. (2002) J. Am. Soc. Nephrol 13, 1464–1472 [DOI] [PubMed] [Google Scholar]
- 14. Dooley S., Hamzavi J., Breitkopf K., Wiercinska E., Said H. M., Lorenzen J., Ten Dijke P., Gressner A. M. (2003) Gastroenterology 125, 178–191 [DOI] [PubMed] [Google Scholar]
- 15. Wang W., Koka V., Lan H. Y. (2005) Nephrology 10, 48–56 [DOI] [PubMed] [Google Scholar]
- 16. Sato M., Muragaki Y., Saika S., Roberts A. B., Ooshima A. (2003) J. Clin. Invest. 112, 1486–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fujimoto M., Maezawa Y., Yokote K., Joh K., Kobayashi K., Kawamura H., Nishimura M., Roberts A. B., Saito Y., Mori S. (2003) Biochem. Biophys. Res. Commun. 305, 1002–1007 [DOI] [PubMed] [Google Scholar]
- 18. Li F., Luo Z., Huang W., Lu Q., Wilcox C. S., Jose P. A., Chen S. (2007) J. Biol. Chem. 282, 10133–10137 [DOI] [PubMed] [Google Scholar]
- 19. Huang W. Y., Li Z. G., Rus H., Wang X., Jose P. A., Chen S. Y. (2009) J. Biol. Chem. 284, 9426–9432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen S., Kulik M., Lechleider R. J. (2003) Nucleic Acids Res. 31, 1302–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen S., Lechleider R. J. (2004) Circ. Res. 94, 1195–1202 [DOI] [PubMed] [Google Scholar]
- 22. Hao S., Shen H., Hou Y., Mars W. M., Liu Y. (2010) Am. J. Pathol. 177, 1164–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo X., Jose P. A., Chen S. Y. (2011) Am. J. Physiol. Cell Physiol. 300, C1415–C1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xie W. B., Li Z., Miano J. M., Long X., Chen S. Y. (2011) J. Biol. Chem. 286, 15050–15057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu Y., Saunders M. A., Yeh H., Deng W. G., Wu K. K. (2002) J. Biol. Chem. 277, 6923–6928 [DOI] [PubMed] [Google Scholar]
- 26. Hu B., Wu Z., Phan S. H. (2003) Am. J. Respir. Cell Mol. Biol. 29, 397–404 [DOI] [PubMed] [Google Scholar]
- 27. Kawai-Kowase K., Kumar M. S., Hoofnagle M. H., Yoshida T., Owens G. K. (2005) Mol. Cell Biol. 25, 8009–8023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo B., Inoki K., Isono M., Mori H., Kanasaki K., Sugimoto T., Akiba S., Sato T., Yang B., Kikkawa R., Kashiwagi A., Haneda M., Koya D. (2005) Kidney Int. 68, 972–984 [DOI] [PubMed] [Google Scholar]
- 29. Badea T., Niculescu F., Soane L., Fosbrink M., Sorana H., Rus V., Shin M. L., Rus H. (2002) J. Biol. Chem. 277, 502–508 [DOI] [PubMed] [Google Scholar]
- 30. Cochrane A. L., Kett M. M., Samuel C. S., Campanale N. V., Anderson W. P., Hume D. A., Little M. H., Bertram J. F., Ricardo S. D. (2005) J. Am. Soc. Nephrol 16, 3623–3630 [DOI] [PubMed] [Google Scholar]
- 31. Klahr S., Morrissey J. (2002) Am. J. Physiol. Renal Physiol. 283, F861–F875 [DOI] [PubMed] [Google Scholar]
- 32. Diamond J. R. (1995) Am. J. Kidney Dis. 26, 133–140 [DOI] [PubMed] [Google Scholar]
- 33. Qiu P., Feng X. H., Li L. (2003) J. Mol. Cell Cardiol. 35, 1407–1420 [DOI] [PubMed] [Google Scholar]
- 34. Sinha S., Hoofnagle M. H., Kingston P. A., McCanna M. E., Owens G. K. (2004) Am. J. Physiol. Cell Physiol. 287, C1560–C1568 [DOI] [PubMed] [Google Scholar]
- 35. Kennard S., Liu H., Lilly B. (2008) J. Biol. Chem. 283, 1324–1333 [DOI] [PubMed] [Google Scholar]
- 36. Hu B., Wu Z., Liu T., Ullenbruch M. R., Jin H., Phan S. H. (2007) Am. J. Respir. Cell Mol. Biol. 36, 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eddy A. A., Fogo A. B. (2006) J. Am. Soc. Nephrol. 17, 2999–3012 [DOI] [PubMed] [Google Scholar]
- 38. Border W. A., Noble N. A. (1994) N. Engl. J. Med. 331, 1286–1292 [DOI] [PubMed] [Google Scholar]
- 39. Chatziantoniou C., Dussaule J. C. (2005) Am. J. Physiol. Renal Physiol. 289, F227–234 [DOI] [PubMed] [Google Scholar]
- 40. Eddy A. A. (1996) J. Am. Soc. Nephrol. 7, 2495–2508 [DOI] [PubMed] [Google Scholar]
- 41. Fukasawa H., Yamamoto T., Togawa A., Ohashi N., Fujigaki Y., Oda T., Uchida C., Kitagawa K., Hattori T., Suzuki S., Kitagawa M., Hishida A. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 8687–8692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Inazaki K., Kanamaru Y., Kojima Y., Sueyoshi N., Okumura K., Kaneko K., Yamashiro Y., Ogawa H., Nakao A. (2004) Kidney Int. 66, 597–604 [DOI] [PubMed] [Google Scholar]
- 43. Schnaper H. W., Kopp J. B. (2006) J. Am. Soc. Nephrol. 17, 1777–1781 [DOI] [PubMed] [Google Scholar]
- 44. Lan H. Y., Mu W., Tomita N., Huang X. R., Li J. H., Zhu H. J., Morishita R., Johnson R. J. (2003) J. Am. Soc. Nephrol 14, 1535–1548 [DOI] [PubMed] [Google Scholar]