

Background: P21-activated kinase (PAK1) is a downstream effector of the GTPase Cdc42.
Results: Inhibition of Cdc42-PAK1 signaling in human islets inhibited insulin secretion. PAK1 knock-out mice showed defects in insulin release and skeletal muscle insulin action, underlying impaired whole body glucose homeostasis.
Conclusion: Attenuated PAK1 abundance/activation may contribute to type 2 diabetes susceptibility.
Significance: Cdc42-PAK1 signaling is crucial for regulating glucose homeostasis in vivo.
Keywords: Cdc42, Glut4, Insulin Resistance, Insulin Secretion, Islet
Abstract
The p21-activated kinase PAK1 is implicated in tumorigenesis, and efforts to inhibit PAK1 signaling as a means to induce tumor cell apoptosis are underway. However, PAK1 has also been implicated as a positive effector of mechanisms in clonal pancreatic beta cells and skeletal myotubes that would be crucial to maintaining glucose homeostasis in vivo. Of relevance, human islets of Type 2 diabetic donors contained ∼80% less PAK1 protein compared with non-diabetics, implicating PAK1 in islet signaling/scaffolding functions. Mimicking this, islets from PAK1−/− knock-out mice exhibited profound defects in the second/sustained-phase of insulin secretion. Reiteration of this specific defect by human islets treated with the PAK1 signaling inhibitor IPA3 revealed PAK1 signaling to be of primary functional importance. Analyses of human and mouse islet beta cell signaling revealed PAK1 activation to be 1) dependent upon Cdc42 abundance, 2) crucial for signaling downstream to activate ERK1/2, but 3) dispensable for cofilin phosphorylation. Importantly, the PAK1−/− knock-out mice were found to exhibit whole body glucose intolerance in vivo. Exacerbating this, the PAK1−/− knock-out mice also exhibited peripheral insulin resistance. Insulin resistance was coupled to ablation of insulin-stimulated GLUT4 translocation in skeletal muscle from PAK1−/− knock-out mice, and in sharp contrast to islet beta cells, skeletal muscle PAK1 loss was underscored by defective cofilin phosphorylation but normal ERK1/2 activation. Taken together, these data provide the first human islet and mammalian in vivo data unveiling the key and crucial roles for differential PAK1 signaling in the multi-tissue regulation of whole body glucose homeostasis.
Introduction
Functional studies have recently implicated the p21-activated kinase (PAK)21 in cell transformation and tumorigenesis and elevated levels of PAK1 are detected in ovarian, breast, and bladder cancers (1, 2). As such, therapeutic targeting of PAK1 in efforts to induce apoptosis of tumor cells has been advocated (3–5). PAK1 is one of six family members, categorized into two groups: PAK1–3 belong to group I and contain a unique autoinhibitory region, distinguishing them from Group II members PAK4–6 (6). PAK1 in particular is a well-established downstream effector of Cdc42 and Rac1 signaling that mediates changes in cytoskeletal functions in a wide array of cell types and processes. Activated-Cdc42 binding to PAK1 promotes PAK1Thr423 phosphorylation, a critical phosphorylation site leading to PAK1 release from autoinhibition to promote its activation (7–9). Activated PAK1 signals downstream as a serine/threonine kinase, utilizing numerous substrates to induce cytoskeletal changes, with PAK1 hyperactivity in signaling being notably exploited in tumorigenesis (10). For these reasons inhibitors of PAK1 signaling are attractive as anti-tumorigenic agents.
Of concern however, is that recent accumulating evidences point to potentially crucial roles for PAK1 in two processes key to the regulation and maintenance of whole body glucose homeostasis: pancreatic beta cell insulin release and skeletal muscle glucose clearance (11, 12). By definition, whole body glucose homeostasis is attained by the coordinated efforts of islet insulin secretion and peripheral insulin action: detection of elevated blood glucose by the beta cell triggers insulin release, and the skeletal muscle responds to the insulin signal by taking up the excess glucose, altogether restoring normal blood glucose levels. Defects in skeletal muscle glucose clearance in vivo are associated with development of peripheral insulin resistance, and the additional defect in beta cell insulin secretion dramatically elevates susceptibility to Type 2 Diabetes (13). Insulin secretion is impaired by PAK1 depletion from clonal MIN6 beta cells, related to a putative essential role for PAK1 as a Cdc42 effector in mediating cytoskeletal remodeling to facilitate insulin granule mobilization to the plasma membrane for insulin release (11). Rac1 activation is dependent upon PAK1 in MIN6 cells (11), but PAK1 involvement in cofilin phosphorylation and/or ERK1/2 activation events, commonly found to be major avenues of downstream PAK1 signaling in other cell types (14–18), remains unknown in beta cells. In clonal L6 skeletal myotubes, PAK1 is implicated as a Rac1 effector in mediating translocation of GLUT4 vesicles to the cell surface to enable glucose uptake into the myocyte (19), based upon evidence showing that insulin-stimulates activation of PAK1, as well as a reduction in phosphorylated cofilin, an event commonly triggered by PAK1 (20–22). Despite the implications of positive roles for PAK1 in cellular mechanisms critically important to regulation of glucose homeostasis, no in vivo evidence, nor data gained from primary islets or skeletal muscle tissues, exists to confirm a physiologically relevant role or requirement for PAK1.
In this report, we provide the first evidence that treatment of human islets with the PAK1 inhibitor IPA3 impairs glucose-stimulated insulin secretion. Further evidence for a physiologically relevant role for PAK1 signaling was gained using Cdc42 depletion to attenuate PAK1 activation in human islets. PAK1 abundance was ∼80% lower in islets from type 2 diabetic humans. Consistent with this, PAK1+/− and PAK1−/− knock-out mice recapitulated defects in insulin secretion, correlating with a clear impairment of whole body glucose intolerance. Compounding this intolerance, PAK1−/− mice also exhibited impaired insulin sensitivity, coordinate with defects in insulin-stimulated GLUT4 vesicle translocation. Surprisingly, PAK1 deficiency led to differential signaling defects in islets versus skeletal muscle. Taken together, these data suggest that deficiency of PAK1 or defects in PAK1 signaling may be linked to type 2 diabetes susceptibility, and that more selective delivery of PAK1 inhibitor to tumor cells may be advised to avert potential diabetogenic complications.
EXPERIMENTAL PROCEDURES
Materials
The mouse anti-Cdc42, phospho-specific anti-PAK1T423, rabbit anti-actin, goat anti-GLUT4, mouse anti-insulin, total ERK1/2, phospho-cofilinS3, and rabbit anti-RhoGDI antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The mouse anti-PAK1, rabbit anti-phospho-AktS473, total-Akt, phospho-ERK1/2T202/Y204, and total cofilin antibodies were purchased from Cell Signaling. Anti-Syntaxin 4, clathrin, and VAMP2 antibodies were obtained from Chemicon, BD Biosciences, and Synaptic Systems, respectively. Donkey anti-goat horseradish peroxidase secondary antibody was purchased from Santa-Cruz. Goat anti-rabbit horseradish peroxidase and anti-mouse horseradish peroxidase secondary antibodies were acquired from Bio-Rad. Enhanced chemiluminescence (ECL) reagent was obtained from Amersham Biosciences. The RIA grade bovine serum albumin, PAK inhibitor IPA3, donkey serum and D-glucose were obtained from Sigma. The sensitive rat insulin and human ultra-sensitive RIA kits and glucagon RIA kit were purchased from Millipore (Billerica, MA).
Plasmids and Adenovirus
The pcDNA3-myc-Cdc42 cDNA (human) plasmid was obtained from Dr. Richard A. Cerione (Cornell University, New York). The plasmid-based siRNA construct pSilencer1.0-Cdc42 (siCdc42) was generated as previously described (11, 23); targeting sequence encoding 19 nucleotides (nt) of human Cdc42 is 5′-CTAACCACTGTCCAAAGAC-3′. Adenoviruses were packaged with green fluorescent protein (GFP) to facilitate identification of transduced cells.
RNA Isolation and RT-PCR
Total RNA from isolated mouse pancreatic islets was obtained using the RNeasy Mini kit (Qiagen, Valencia, CA). RNA (1 μg) was reverse-transcribed with TaqMan (Applied Biosystems, Foster City, CA), and 1% of the product was used for RT-PCR. The primers used for detection of PAK1 (forward: 5′-tgtctgagaccccagcagta; Reverse: 5′-cccgagttggagtaacagga), for PAK2: forward 5′-aacaccagcactgaacacca, reverse 5′-cttggcaccactgtcaacat; for PAK3: forward 5′-gcagcacatcagtcgaatacca, reverse 5′-tttatttggtgcagctggt) were obtained from IDT (San Jose, CA). RT-PCR was performed with BioMix Red (Bioline, Taunton, MA) for 30 cycles: 94 °C for 1 min, 56 °C for 1 min, 71 °C for 1 min with a final 10 min elongation at 71 °C. PCR products were visualized on 2% agarose gels.
Cell Culture, Transient Transfection, and Immunoblotting
To assess the efficiency of siRNA-depleted human Cdc42, CHO-K1 cells were electroporated with 40 μg of DNA as previously described (24). MIN6 cells were cultured and transiently transfected with PAK1 siRNA oligonucleotides as previously reported (11). After 48 h of incubation, cells were harvested in 1% Nonidet P-40 lysis buffer (24) and lysates cleared by centrifugation at 14,000 × g for 10 min at 4 °C. Proteins present in lysates were resolved by 12% SDS-PAGE and depletion detected by immunoblotting. Membranes were incubated with primary antibody at 4 °C overnight, with secondary antibodies conjugated to horseradish peroxidase used for 1 h at room temperature. Bands were visualized by enhanced chemiluminescence.
Human Islet Perifusion
Pancreatic human islets (obtained through the Integrated Islet Distribution Program, IIDP, donor information listed in supplemental Table S3) were used for perifusion in a strategy similar to that used for mouse islet perifusion, as previously described (11, 23). Criteria for human donor islet acceptance: receipt within 36 h of isolation, and of at least 80% purity and 75% viability. Upon receipt, human islets were first allowed to recover in CMRL medium for 2 h, and then were handpicked using a green gelatin filter to eliminate residual non-islet material. Islets of non-diabetic donors were immediately transduced at MOI of 100 with either siCdc42-Ad or siCon-Ad CsCl-purified particles for 1 h at 37 °C. Transduced islets were then washed twice and incubated for 48 h in RPMI 1640 at 37 °C, 5% CO2. Fifty transduced (GFP-positive, GFP+) islets were handpicked onto a column for perifusion analysis (11). Control (siCon or DMSO) islets were run in parallel columns with experimental (siCdc42 or IPA3-treated) islets. Islets were then perifused at a flow rate of 0.3 ml/min, and insulin secreted into eluted fractions was quantitated by the ultrasensitive human insulin RIA kit (Millipore). Human islets treated with and without the PAK1 inhibitor IPA3 were pre-incubated with DMSO or 7.5 μm IPA3 for 16 h in RPMI1640 prior to perifusion.
Mouse Islet Isolation, Perifusion, and Morphometric Analyses
The PAK1 KO mouse is a classic whole-body gene-ablation model on the C57Bl6J strain background, generated as previously described (25). All mice for studies here were obtained by heterozygous crossing and paired littermates used as controls. Mouse pancreatic islets were isolated and used for perifusion, as previously described (11, 23). Briefly, pancreata pooled from five ∼12-week-old male mice were batch-digested with collagenase, purified using a Ficoll density gradient, and incubated overnight in RPMI 1640 at 37 °C, 5% CO2. Fifty islets each of KO and WT were handpicked onto parallel columns for dual perifusion as described (11, 26), and insulin secreted into eluted fractions quantitated using the sensitive rat insulin RIA immunoassay kit from Millipore (Billerica, MA). Mouse islet morphometry was evaluated using anti-insulin immunohistochemical staining of pancreatic sections as described (27). Briefly, pancreata from WT or PAK1−/− KO 5-month-old male mice were fixed with 4% paraformaldehyde, paraffin embedded, and longitudinally sectioned at 5-μm thickness and 100-μm intervals. The sectioned tissues were deparafinized, rehydrated, and blocked in 5% horse serum, and incubated overnight at 4 °C with rabbit anti-insulin antibody (Santa Cruz Biotechnology). Following PBS washes and incubation with HRP-conjugated secondary antibody, the sections were incubated in peroxidase substrates (Vector Labs) and counterstained with hematoxylin. Digital images were acquired on an Axio-Observer Z1 microscope (Zeiss) fitted with an AxioCam high resolution color camera. Percentage of β cell area was calculated using Axio-Vision Software. Data shown are representative of 4 sections per pancreas and 3 pancreata from each group.
Glucagon Secretion Assays
Ten mouse islets were picked into siliconized microcentrifuge tubes, washed three times with KRBH, and incubated in 0.5 ml KRBH at 37 °C, 5% CO2 for 1 h. Supernatants were then collected for analysis and replaced with KRBH containing 16.7 mm glucose and incubated for an additional hour. All supernatants were centrifuged to remove cellular content and subsequently analyzed using the glucagon RIA immunoassay kit from Millipore.
Intraperitoneal Glucose Tolerance and Insulin Tolerance Tests
Male PAK1−/− KO and wild-type (WT) mice (age 4–6 months) were fasted for 18 h prior to the intraperitoneal glucose tolerance test (IPGTT), as described previously (23). One week following the same mice were assessed for insulin sensitivity using the insulin tolerance test (ITT), wherein mice were fasted for 6 h prior to intraperitoneal injection with Humulin R (0.75 units/kg body weight).
Skeletal Muscle Subcellular Fractionation
Hindlimb skeletal muscle was subfractionated into plasma membrane and intracellular membrane components, as previously described (28). Male PAK1−/− KO and WT mice (age 4–6 months) were fasted overnight (16 h), injected intraperitoneally with Humulin (21 units/kg body weight) or saline, and sacrificed after 40 min for rapid removal of hindquarter muscles into homogenization buffer (28) for Polytron homogenization. Homogenates were subjected to differential centrifugation and sucrose density gradients to obtain pellets containing plasma membrane fractions apart from fractions containing intracellular vesicles. Pellets were resuspended in lysis buffer for protein resolution by 10% SDS-PAGE and immunodetection of GLUT4, as previously detailed (23).
Statistical Analysis
All data were evaluated for statistical significance using Student's t test. Data are expressed as the average ± S.E.
RESULTS
Cdc42-PAK1 Signaling Is Essential for Second-phase Insulin Release from Human Islets
To address whether Cdc42 is required for human islet glucose-stimulated insulin secretion (GSIS), we generated a human-specific siCdc42 adenovirus. The 40–50% efficiency of targeted knockdown was determined using transfected CHO-K1 cells expressing Myc-tagged human Cdc42 and in human islets transduced with siCdc42 adenovirus (siCdc42-Ad), compared with non-targeting control (siCon-Ad) islets (supplemental Fig. S1 and Fig. 1A). Consistent with the biphasic perifusion pattern observed in human islets (29, 30), human islets transduced with siCon-Ad exhibited a sharp peak indicative of first-phase insulin secretion, followed by a drop and sustainment of a second-phase, upon glucose stimulation (Fig. 1B). In islets transduced with siCdc42-Ad, first-phase secretion was similar to control islets; however, siCdc42-Ad islets had a significant reduction in the second-phase (AUC = 54 ± 20% of siCon-Ad treated islets). Coordinate with impaired secretion, glucose-stimulated PAK1T423 phosphorylation was fully ablated in siCdc42-Ad transduced human islets (Fig. 1C), compared with ∼2-fold increase in siCon-Ad transduced islet lysates. These data are the first to implicate the Cdc42-to-PAK1 signaling axis in insulin release from human islets.
FIGURE 1.
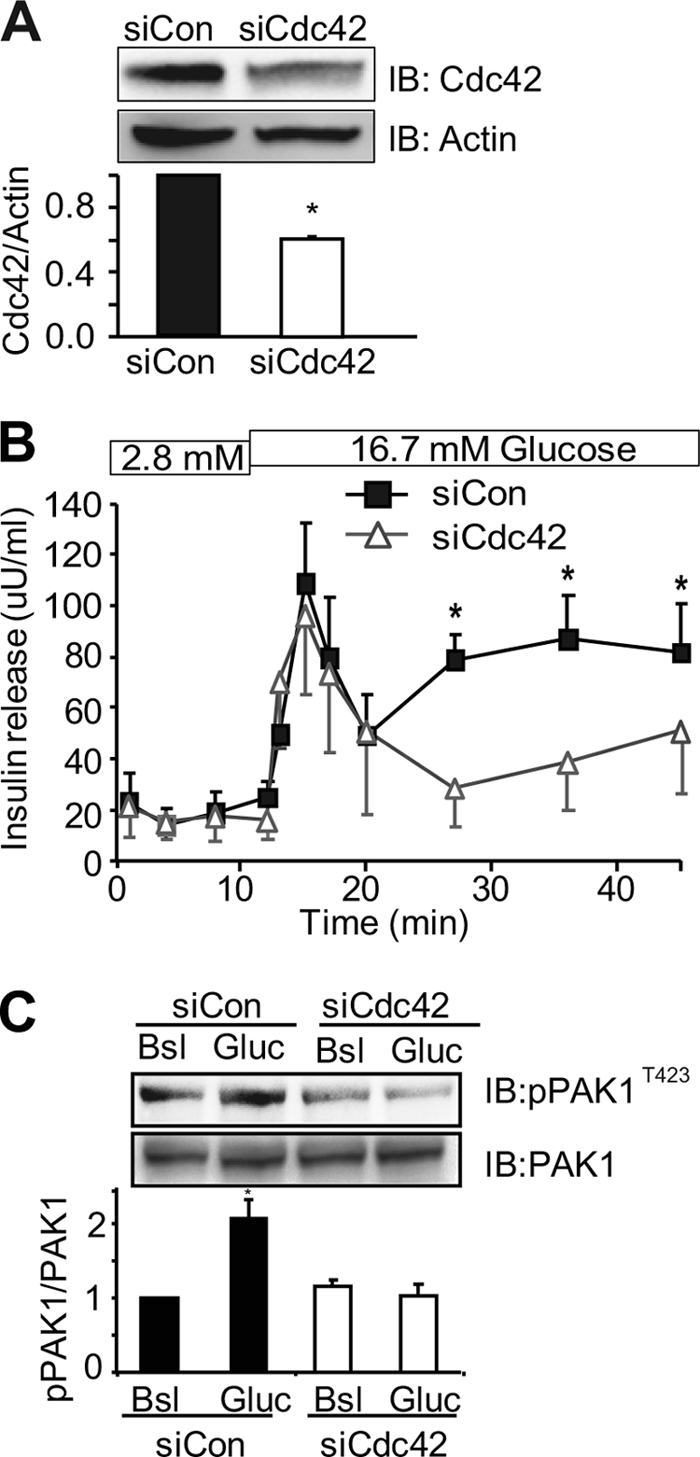
RNAi-mediated depletion of Cdc42 from human islets inhibits glucose-induced second-phase insulin secretion and PAK1 activation. Isolated human islets transduced with control (Con-Ad) or siCdc42 (siCdc42-Ad) adenoviruses for 48 h were: A, solubilized for immunodetection of Cdc42 or actin proteins (*, p < 0.01 versus siCon, n = 3), B, perifused with 2.8 mm and 16.7 mm glucose; curves represent the average ± S.E. of four independent paired experiments, *, p < 0.01 versus siCon; or C, stimulated with 16.7 mm glucose for 10 min for detection of phospho-PAK1T423 by immunoblot. Membranes were subsequently stripped and reblotted for total PAK1 protein. Quantitation of three independent experiments (average ± S.E.) is shown; *, p < 0.01 versus basal siCon.
Next the requirement for downstream PAK1 signaling in human islet function was evaluated. The cell permeable allosteric inhibitor of PAK1 activation, IPA3 (31) was first subjected to dosage- and time-dependence studies in clonal MIN6 beta cells rather than islets due to the limited quantity of human islets, wherein it was determined that GSIS was impaired in a dose-dependent manner with short term (40 min) IPA3 treatment (supplemental Fig. S2); time-dependence studies revealed that a lower dosage of 7.5 μm for 16 h attenuated PAK1T423 phosphorylation (Fig. 2A). At this low dosage in human islets, IPA3 significantly and selectively inhibited the second phase of GSIS (Fig. 2B). A pharmacological approach using the GSIS diazoxide paradigm (discriminates glucose-amplified insulin release independent of elevated [Ca2+]i, analogous to second-phase changes) was used to demonstrate that IPA3 selectively reduced the glucose-amplified secretion in comparison to vehicle-treated cells (Fig. 2C). These data suggest the ability of PAK1 to signal downstream is selective and crucial for sustained insulin release mechanisms.
FIGURE 2.

The PAK inhibitor IPA3 inhibits glucose-induced second-phase insulin secretion in human islets and MIN6 beta cells. A, MIN6 cells incubated with 7.5 μm IPA3 or vehicle (DMSO) for 16 h were glucose stimulated for 10 min for immunoblot confirmation of IPA3-inhibited PAK1T423 phosphorylation; membranes from three independent assays were subsequently stripped and reblotted for total PAK1. B, human islets were cultured in the presence of DMSO or IPA3 (7.5 μm) for 16 h and then perifused; curves represent the average ± S.E. of three independent paired experiments; *, p < 0.01 versus DMSO. C, IPA3 inhibited glucose induced KATP channel-independent insulin secretion as tested by the diazoxide paradigm. MIN6 cells were preincubated in MKRBB for 2 h with 30 μm IPA3 or vehicle control (DMSO) added in the last 10 min of pre-incubation. Diazoxide (250 μm) and KCl (40 mm) were then added, followed by glucose stimulation for an additional 30 min. Insulin release was quantified three independent experiments; data represent the average ± S.E., *, p < 0.05 versus DMSO control. D, quantitation of PAK1 abundance relative to actin in five independent batches of human type 2 diabetic and non-diabetic islets, shown as the average ± S.E.; *, p < 0.05 versus non-diabetic.
Given the observed impairments in biphasic insulin secretion in type 2 diabetic patients and the important role of Cdc42-PAK1 in insulin secretion in human islets, we evaluated the protein expression levels of Cdc42 and PAK1 in islets from type 2 diabetic and non-diabetic donors. Remarkably, PAK1 protein expression levels were substantially reduced in each of five independent batches of type 2 diabetic donor islets (Fig. 2D). In contrast, no significant changes were observed in Cdc42 or RhoGDI (data not shown) protein abundances, such that normalization to either reiterated the nearly 80% loss of PAK1 protein from diabetic human islets. The specific reduction of PAK1 inspired us to explore the role of PAK1 in the development of diabetes using a PAK1 knock-out (KO) mouse model (25).
PAK1-deficient Islets Exhibit Impairments in Second-phase Insulin Secretion and ERK1/2 Activation
Pak1 KO mice have been examined previously (25), but not in tissues associated directly with maintaining glucose homeostasis. Quantitative real-time PCR and immunoblot analyses confirmed the complete absence of PAK1 from PAK1−/− KO islets; mRNA abundances of the remaining Group I isoforms, PAK2 and PAK3, were unaffected (supplemental Fig. S3, A-B). PAK1 protein was confirmed absent from PAK1−/− KO heart, liver, and skeletal muscle extracts when compared with wild-type (WT) littermate tissues (supplemental Fig. S3C), while expression of other key insulin exocytosis factors in the same extracts was unchanged (Cdc42, RhoGDI, the t-SNARE protein Syntaxin 4, and the v-SNARE protein VAMP2). No significant differences in overall body weight or weights of organs/tissues were detected between WT and PAK1−/− KO mice at 6 months of age (supplemental Table S1). At 12 months of age only one difference was noted: PAK1−/− KO mice exhibited increased epididymal fat mass relative to littermate WT mice (6.7 ± 1.5% versus 2.4 ± 0.7%, p < 0.05).
Similar to the profile observed with IPA3 treated human islets, second-phase insulin secretion from PAK1−/− KO islets was selectively and significantly impaired (Fig. 3A). The impairment was not due to defect in islet architecture of the PAK1−/− KO pancreata (Fig. 3B) or a reduction in insulin content (Fig. 3C). No defects in glucose-induced glucagon secretion or in total glucagon content were seen between PAK1−/− KO and WT mouse islets (Fig. 3, D and E), suggesting against an impact of differential glucagon secretion upon insulin secretion and content in the PAK1−/− KO mouse islets. In addition, fasting levels of serum triglycerides, cholesterol, nonesterified fatty acids (NEFAs), IL-6, and TNFα did not differ between WT and PAK1−/− KO mice; glucose and insulin levels from fed mice were not statistically different (supplemental Table S2). Striking decreases were observed however in glucose-induced ERK1/2 activation of PAK1−/− KO islets and PAK1-depleted (by siRNA) MIN6 beta cells (Fig. 3, F–H) in the absence of altered ERK1/2 protein abundances. No differences in basal ERK1/2 phosphorylation were detected in siPAK1-depleted MIN6 cells (relative to siCon = 1.0, siPAK1 = 1.2 ± 0.2). Downstream cofilin phosphorylation was essentially unchanged, suggesting PAK1 signaling in the islet beta cell to be primarily in the direction of ERK1/2 and not cofilin.
FIGURE 3.

PAK1 is required for glucose-induced ERK1/2 activation and second-phase insulin secretion in mouse islet beta cells. A, isolated PAK1−/− KO and WT mouse islets were perifused in 2.8 m glucose and 16.7 mm glucose; curves represent the average ± S.E. of three independent paired experiments, *, p < 0.01 versus WT. B, insulin staining of WT and PAK1−/− KO pancreas sections showed equivalent islet density, size and beta cell mass in 3 pancreata of each genotype. C, total insulin content in PAK1−/− KO and WT islets used for perifusions was equivalent. D, glucagon secretion from basal or glucose-stimulated WT or PAK1−/− KO islets, *, p < 0.01 versus WT basal. E, islet total glucagon content quantification of three independent sets of islets. F, glucose-induced ERK1/2 activation in three independent paired batches of WT and PAK1−/− KO islet lysates (16.7 mm Gluc, 20 min) *, p < 0.05 versus WT pERK normalized to total ERK1/2. G, glucose-induced ERK1/2 and cofilin phosphorylation representative of four sets of MIN6 cells transfected with PAK1 siRNA oligonucleotides (20 mm Gluc, 20 min). H, quantitation of the ratio of pERK normalized to total ERK in each of three independent sample sets of immunoblots represented in panels F and G above; *, p < 0.05 versus WT pERK normalized to total ERK1/2.
Defective Glucose Homeostasis in the PAK1 Knock-out Mouse in Vivo
To determine the consequences of impaired second phase insulin secretion upon whole body glucose homeostasis, PAK1−/− KO and WT mice were subjected to intraperitoneal glucose tolerance tests (IPGTT). While fasting blood glucose levels were similar in WT and KO mice (Fig. 4A), PAK1−/− KO mice showed significantly higher peak blood glucose levels at 30 and 60 min time points after glucose injection, signifying defects in glucose clearance. Both the PAK1−/− KO and WT mice did elicit an acute insulin response to the glucose challenge (15 min after injection; serum insulin content rose from 0.63 ± 0.04 to 0.84 ± 0.05 ng/ml in WT mice, and from 0.48 ± 0.02 to 0.91 ± 0.09 ng/ml in KO mice), corresponding to normal first-phase insulin secretion of the perifused KO islets. Importantly, PAK1+/− heterozygous mice also showed significantly impaired glucose tolerance (AUC: PAK1+/− = 45,981 ± 3,490, versus WT = 36,405 ± 1,285, p < 0.01), indicating that a paucity of PAK1 by only 50% was sufficient to exert this dominant effect. Taken together with the observed ∼80% decrease in PAK1 protein abundance in human type 2 diabetic islets, PAK1 abundance may be an important mediator of whole body glucose homeostasis.
FIGURE 4.

Impaired glucose intolerance and insulin sensitivity in PAK1−/− KO mice. A, IPGTT of PAK1−/− KO and WT mice was performed by intraperitoneal injection of d-glucose (2 g/kg body weight) into 7 pairs of male mice (age 4–6 months) fasted for 18 h; *, p < 0.05 versus WT. B, insulin tolerance testing of PAK1−/− KO (n = 6) and WT male mice (n = 7) was performed by intraperitoneal injection of insulin (0.75 units/kg body weight) into male mice (age 4–6 months) fasted for 6 h. Blood glucose levels were normalized to basal = 100% for each animal for calculation of the mean percent ± S.E.; *, p < 0.05 versus WT.
Cdc42 and F-actin remodeling are known to be important in insulin-stimulated GLUT4 vesicle translocation in skeletal muscle cells as a means to evoke clearance of excess blood glucose (32, 33), although PAK1 has yet to be placed in this process of peripheral insulin action. To determine whether the glucose-intolerant phenotype of the PAK1−/− KO mice might also be impacted by the absence of PAK1 in skeletal muscle, which facilitates ∼80% of glucose clearance, insulin tolerance testing of the PAK1−/− KO mice was performed. Fasting (6 h) glucose levels were similar prior to the intraperitoneal insulin injection (KO = 180 ± 17 mg/dl versus WT = 182 ± 12 mg/dl). However, PAK1−/− KO mice had elevated levels of blood glucose compared with the WT mice at all time points following injection (Fig. 4B), and overall showed significantly elevated area under the curve (AUC: KO = 3,029 ± 146 versus WT = 2,626 ± 120, p < 0.05), indicative of peripheral insulin resistance.
PAK1-deficient Skeletal Muscle Exhibits Impairments in GLUT4 Vesicle Translocation and Cofilin Phosphorylation
The insulin resistance phenotype of the PAK1−/− KO mice could emanate from deficient GLUT4 protein, defective insulin-stimulated GLUT4 vesicle translocation, and/or impaired insulin signaling in peripheral insulin-responsive tissues. To address the first possibility, skeletal muscle and adipose tissue extracts from WT and PAK1−/− KO mice were evaluated for GLUT4 abundance; equivalent GLUT4 levels were found in each tissue type (Fig. 5A). To interrogate a role for PAK1 in GLUT4 vesicle translocation, hindlimb skeletal muscle homogenates prepared from WT and PAK1−/− KO mice injected with insulin or vehicle control (saline) were fractionated to quantify the contingent of plasma membrane localized GLUT4 vesicles, apart from intracellular vesicles, using sucrose density gradients (28). The plasma membrane fraction contains both sarcolemma and transverse tubules, a caveat of this fractionation method. Nevertheless, this method did sufficiently detect a nearly 2-fold increase in GLUT4 protein translocation to the plasma membrane compartments from insulin-stimulated WT mice, consistent with other studies (Fig. 5B). However, in PAK1−/− KO mice, GLUT4 vesicles failed to translocate/accumulate in plasma membrane fractions following insulin stimulation. As shown in Fig. 5C, AKT abundance and insulin-stimulated activation were similar among the same WT and PAK1−/− KO plasma membrane skeletal muscle fractions, and in contrast to islets, insulin-stimulated ERK1/2 activation was normal (pERK:total ERK normalized to WT basal = 1.0, insulin = 3.0 ± 0.6; KO basal = 0.9 ± 0.2, insulin = 3.0 ± 1.1). Also different from islet signaling were changes in cofilin phosphorylation/dephosphorylation. While phospho-cofilinS3 levels were reduced in response to insulin in WT skeletal muscle (relative to WT basal = 1.0, insulin = 0.6 ± 0.06; p < 0.001), PAK1−/− KO muscle was otherwise unresponsive to insulin stimulation (KO basal = 0.8 ± 0.2, insulin = 0.9 ± 0.2). Efforts to determine if the phospho-cofilin defect was related to altered LIMK activity, downstream of PAK1 activation, were thwarted by poor LIMK antibody detection in muscle tissue. Importantly, insulin did stimulate PAK1 phosphorylation/activation in primary skeletal muscle from WT mice (Fig. 5D). Altogether, these data implicate, for the first time, a crucial role for PAK1 in the process of insulin-stimulated GLUT4 vesicle translocation in skeletal muscle, at a step distal to or independent of AKT activation involving cofilin.
FIGURE 5.
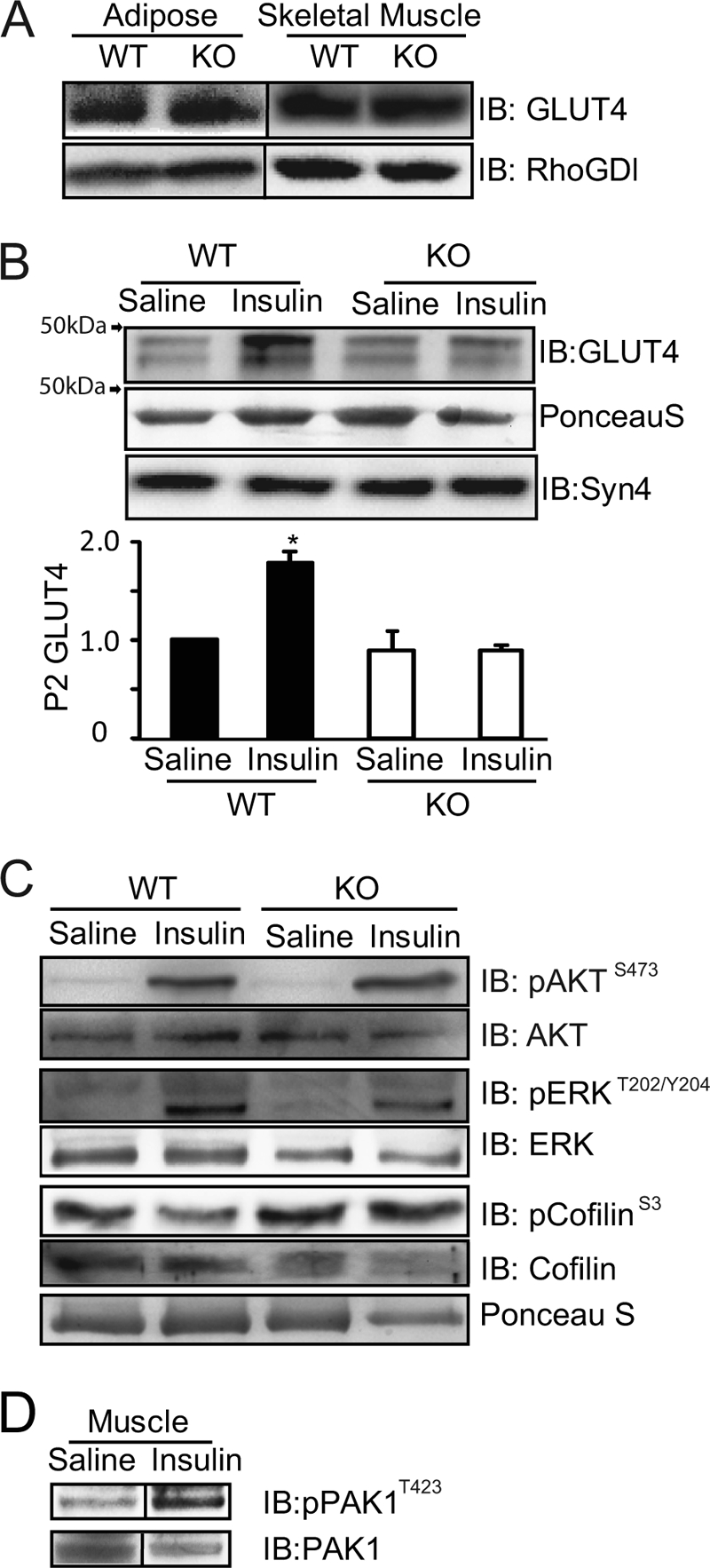
Insulin-stimulated GLUT4 translocation is impaired in skeletal muscle from PAK1−/− KO mice. A, GLUT4 protein expression in adipose (epididymal) and hind limb skeletal muscle tissue cleared detergent homogenates, representative of lysates from three pair PAK1−/− KO and WT littermate mice. B, GLUT4 protein accumulated in sarcolemma/transverse tubule plasma membrane fractions (P2) under basal (16 h fasted, saline-injected) or insulin-stimulated conditions (40 min after injection with 21 units/kg body weight); quantified from three independent assays, with each normalized to WT basal = 1; *, p < 0.05 versus basal. C, P2 fraction proteins described in panel B above were immunoblotted for phospho-AKTS473, phospho-ERK1/2T202/Y204, and phospho-cofilinS3. Total protein abundances of AKT, ERK1/2 and cofilin were obtained by strip/reblot (AKT) or from parallel identical lanes of the same gel. Data shown are representative of one of four sets of mouse experiments. D, skeletal muscle lysates from WT mice left unstimulated or insulin-stimulated for 10 min were resolved on SDS-PAGE for immunoblot detection of phospho-PAK1T423. The same membrane was stripped and reprobed for total PAK1 protein. Immunoblots are representative of skeletal muscle lysates from three pair of WT mice.
DISCUSSION
In the current study, we demonstrate for the first time that Cdc42-PAK1 signaling is essential for glucose-induced second-phase insulin secretion in human islets. Strikingly, human islets from type 2 diabetic donors lacked ∼80% of the observed normal component of PAK1 protein, suggesting the possibility of linkage between PAK1 abundance and disease susceptibility. Supporting this was our finding of significant glucose intolerance in the PAK1+/− heterozygous mice. PAK1−/− KO mice showed similar glucose intolerance, and this was linked to impaired second-phase insulin secretion. Surprisingly, the PAK1−/− KO mice were also found to have peripheral insulin resistance, likely emanating from defective insulin-stimulated GLUT4 vesicle translocation in the skeletal muscle of these mice. Although PAK1−/− KO mice maintained normal fasted and fed glucose and insulin levels (supplemental Table S2), suggestive of compensation at the 4–6 month age assessed, further aging and/or dietary stress could expose the PAK1−/− KO mouse as a new model of study for diabetes susceptibility.
Although roles for PAK1 in smooth (34) and cardiac (35) muscle contraction have been widely addressed, findings here that PAK1−/− KO mice display severe peripheral insulin resistance and defects in skeletal muscle GLUT4 translocation following insulin stimulation are novel. Interestingly, PAK1 has been shown to function downstream of insulin in multiple cell types (36, 37), one of which being L6 myotubes (21). Consistent with our data showing normal AKT activation in insulin-stimulated PAK1 KO skeletal muscle extracts, Klip and colleagues showed PAK1 (termed PAK65) to be downstream of PI-3 kinase in the insulin signaling cascade (21). More recent data extend this by showing PAK1 to be downstream of Rac1, and a Rac1 knock-out mouse exhibits defective insulin stimulated GLUT4 translocation (22). Because there is no evidence for the participation of Cdc42 in insulin action in skeletal muscle, it is very likely that Rac1 is the Rho family GTPase that signals to PAK1 in this tissue (20, 22). Insulin was recently shown to reduce the amount of phosphocofilin in L6 rat skeletal myoblasts (19). Our data confirm this finding in mouse skeletal muscle, and further show that ablation of PAK1 deregulates this alteration in cofilin phosphorylation. Not previously recognized however was our finding that in skeletal muscle, PAK1 is dispensable for ERK1/2. Since PAK1 preferentially signals through cofilin and is downstream of Rac1, this implicates a role for PAK1 in F-actin reorganization in the process of GLUT4 vesicle translocation in skeletal muscle cells. Interestingly, mRNA levels of PAK1 but not of Rac1, are significantly reduced in soleus and gastrocnemius skeletal muscle from obese diabetes-susceptible BTBR mice, at both 4 and 10 weeks of age (38). Since the BTBR ob mouse is not yet diabetic at 4 weeks, changes in gene expression at this time are proposed to be potential causes rather than consequences of hyperglycemia. Consistent with a special requirement for PAK1, human type 2 diabetic islets lacked ∼80% of PAK1 but had normal levels of Cdc42 protein. Taken together, this implicates PAK1 as a novel key factor in impaired peripheral insulin sensitivity and diabetes development.
Having placed PAK1 downstream of Cdc42 in a new signaling axis that is specifically required for only second-phase insulin release from both human and mouse islets, the question remains as to why PAK1 is required and how it is linked to insulin granule exocytosis. Pertinent to this, previous studies of the bone marrow-derived mast cells of PAK1 KO mice demonstrated reduced stimuli-induced degranulation and altered depolymerization of cortical F-actin (25). Relatedly, in MIN6 beta cells, PAK1 activation is detected at a time concurrent with that of F-actin reorganization, ∼5 min postglucose stimulation. While PAK1 is required for downstream activation of Rac1 in the MIN6 cells (11), this latter event is not detectable until after the visualization of F-actin reorganization and well into the second-phase of insulin secretion. Given this disconnect, it seems plausible that PAK1 signals downstream bifurcate; one arm to trigger Rac1, and a second as of yet unidentified signaling arm to evoke F-actin reorganization and sustain insulin release in the second phase. Our data secure the placement of PAK1 upstream of ERK1/2 activation, which fits well with prior studies that have implicated ERK1/2 in the later phase of insulin release (39–42). In addition, Raf kinase may be a missing link between PAK1 and ERK1/2 activations, since Raf kinases (B-Raf in human islets and Raf-1 in mouse islets) are implicated in insulin release (40, 43, 44). The PAK1 KO islets will be invaluable to test this putative step in the pathway.
Since PAK1 ablation failed to alter cofilin phosphorylation, it seems plausible that PAK1-ERK-actin signaling may underlie beta cell F-actin reorganization, as has been shown in macrophages (45). This is somewhat remarkable given that islet beta cells, being neuroendocrine cells, retain numerous stimulus-secretion coupling mechanisms of their neuronal cousins, yet as seen in hippocampal neurons of an independent PAK1 KO mouse model (46), the LIMK-Cofilin pathway was impacted. Perhaps more likely, ERK1/2 activation may phosphorylate synapsin I to liberate granules from the actin cytoskeleton to contribute to second phase insulin release (41). Since it remains a possibility that the lack of effects upon cofilin in beta cells or ERK1/2 in skeletal muscle could be due to compensation from remaining Group I members PAK2 or PAK3, future studies will require double and triple knock-out/knockdown formats to comprehensively delineate these pathway requirements.
In addition to these classic signaling avenues however, it seems likely that PAK1 signaling to evoke F-actin reorganization in the beta cell will involve a beta cell-specific factor. This prediction is based upon contradictory findings between beta cells and myocytes: both require PAK1 for exocytosis, but pharmacological depolymerization of F-actin by latrunculin potentiates stimulus-induced insulin release (47) while it inhibits insulin-stimulated GLUT4 translocation (48). It is interesting that we were unable to detect defects in glucagon secretion from the PAK1−/− KO islets. This could however be related to inherent differences in exocytosis mechanisms, such that α-cells require the deactivation of exocytosis, whereas β-cells need the rapid acceleration of exocytosis, and thus insulin secretion, following a glucose challenge. Third, although Cdc42, PAK1 and Rac1 are known participants in numerous F-actin remodeling and secretory events, their ordered use in these events can vary substantially; i.e. Rac1 signals downstream to PAK1 in platelets (49), yet PAK1 signals downstream to Rac1 in fibroblasts (50). Altogether, it would not be surprising to discover that highly specialized secretory cells have co-evolved with specific factors to tightly regulate specialized exocytotic and secretory processes, particularly for a cell such as the islet beta cell that is uniquely designed to make and secrete insulin in a highly regulated and selective manner. Given that our data demonstrated here that PAK1 plays an important role in both pancreatic β-cells and skeletal muscle, discovery and cell-specific targeting of the PAK1-signaling cascade has tremendous potential as a putative therapy for multifactorial diseases such as type 2 diabetes.
Acknowledgments
We thank Drs. Stephanie Yoder and Bernhard Maier (Indiana University School of Medicine) for critical reading of this manuscript and Dr. Amira Klip (University of Toronto) and Dr. Owen McGuinness (Vanderbilt University) for helpful discussions. We thank Latha Ramalingam for assistance with the skeletal muscle PAK1 activation assessments. Pancreatic human islets were obtained through the Integrated Islet Distribution Program, IIDP.
This study was supported, in whole or in part, by Grants DK067912 and DK076614 (to D. C. T.), CTSI-KL2 RR025760 (to Z. W.), and CA058836 (to J. C.) from the National Institutes of Health and from the Showalter Trust of Indiana University School of Medicine (to E. O.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3 and Tables S1–S3.
- PAK
- p21-activated kinase
- GDI
- guanosine diphosphate-dissociation inhibitor
- GLUT4
- glucose transporter type 4
- G-protein (GTPase)
- guanosine triphosphate (GTP)-binding protein
- GSIS
- glucose-stimulated insulin secretion
- KRBB
- Krebs-Ringer bicarbonate buffer
- MOI
- multiplicity of infection
- PVDF
- polyvinylidene fluoride
- RIA
- radioimmunoassay
- siRNA
- small interfering RNA. SNARE: soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein
- Syn4
- syntaxin 4
- VAMP2
- vesicle-associated membrane protein 2
- T2D
- Type 2 Diabetes.
REFERENCES
- 1. Vadlamudi R. K., Adam L., Wang R. A., Mandal M., Nguyen D., Sahin A., Chernoff J., Hung M. C., Kumar R. (2000) J. Biol. Chem. 275, 36238–36244 [DOI] [PubMed] [Google Scholar]
- 2. Ong C. C., Jubb A. M., Haverty P. M., Zhou W., Tran V., Truong T., Turley H., O'Brien T., Vucic D., Harris A. L., Belvin M., Friedman L. S., Blackwood E. M., Koeppen H., Hoeflich K. P. (2011) Proc. Natl. Acad. Sci. U.S.A. 108, 7177–7182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ong C. C., Jubb A. M., Zhou W., Haverty P. M., Harris A. L., Belvin M., Friedman L. S., Koeppen H., Hoeflich K. P. (2011) Oncotarget 2, 491–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arias-Romero L. E., Chernoff J. (2010) Small Gtpases 1, 124–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murray B. W., Guo C., Piraino J., Westwick J. K., Zhang C., Lamerdin J., Dagostino E., Knighton D., Loi C. M., Zager M., Kraynov E., Popoff I., Christensen J. G., Martinez R., Kephart S. E., Marakovits J., Karlicek S., Bergqvist S., Smeal T. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 9446–9451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arias-Romero L. E., Chernoff J. (2008) Biol. Cell 100, 97–108 [DOI] [PubMed] [Google Scholar]
- 7. Yu J. S., Chen W. J., Ni M. H., Chan W. H., Yang S. D. (1998) Biochem. J. 334, 121–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zenke F. T., King C. C., Bohl B. P., Bokoch G. M. (1999) J. Biol. Chem. 274, 32565–32573 [DOI] [PubMed] [Google Scholar]
- 9. Lei M., Lu W., Meng W., Parrini M. C., Eck M. J., Mayer B. J., Harrison S. C. (2000) Cell 102, 387–397 [DOI] [PubMed] [Google Scholar]
- 10. Wang R. A., Zhang H., Balasenthil S., Medina D., Kumar R. (2006) Oncogene 25, 2931–2936 [DOI] [PubMed] [Google Scholar]
- 11. Wang Z., Oh E., Thurmond D. C. (2007) J. Biol. Chem. 282, 9536–9546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chiu T. T., Jensen T. E., Sylow L., Richter E. A., Klip A. (2011) Cell Signal. 23, 1546–1554 [DOI] [PubMed] [Google Scholar]
- 13. Prentki M., Nolan C. J. (2006) J. Clin. Invest. 116, 1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Edwards D. C., Sanders L. C., Bokoch G. M., Gill G. N. (1999) Nat Cell Biol. 1, 253–259 [DOI] [PubMed] [Google Scholar]
- 15. Coniglio S. J., Zavarella S., Symons M. H. (2008) Mol. Cell. Biol. 28, 4162–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beeser A., Jaffer Z. M., Hofmann C., Chernoff J. (2005) J. Biol. Chem. 280, 36609–36615 [DOI] [PubMed] [Google Scholar]
- 17. Delorme V., Machacek M., DerMardirossian C., Anderson K. L., Wittmann T., Hanein D., Waterman-Storer C., Danuser G., Bokoch G. M. (2007) Dev Cell 13, 646–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stockton R., Reutershan J., Scott D., Sanders J., Ley K., Schwartz M. A. (2007) Mol. Biol. Cell 18, 2346–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chiu T. T., Patel N., Shaw A. E., Bamburg J. R., Klip A. (2010) Mol. Biol. Cell 21, 3529–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. JeBailey L., Wanono O., Niu W., Roessler J., Rudich A., Klip A. (2007) Diabetes 56, 394–403 [DOI] [PubMed] [Google Scholar]
- 21. Tsakiridis T., Taha C., Grinstein S., Klip A. (1996) J. Biol. Chem. 271, 19664–19667 [DOI] [PubMed] [Google Scholar]
- 22. Ueda S., Kitazawa S., Ishida K., Nishikawa Y., Matsui M., Matsumoto H., Aoki T., Nozaki S., Takeda T., Tamori Y., Aiba A., Kahn C. R., Kataoka T., Satoh T. (2010) FASEB J. 24, 2254–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spurlin B. A., Thomas R. M., Nevins A. K., Kim H. J., Kim Y. J., Noh H. L., Shulman G. I., Kim J. K., Thurmond D. C. (2003) Diabetes 52, 1910–1917 [DOI] [PubMed] [Google Scholar]
- 24. Thurmond D. C., Ceresa B. P., Okada S., Elmendorf J. S., Coker K., Pessin J. E. (1998) J. Biol. Chem. 273, 33876–33883 [DOI] [PubMed] [Google Scholar]
- 25. Allen J. D., Jaffer Z. M., Park S. J., Burgin S., Hofmann C., Sells M. A., Chen S., Derr-Yellin E., Michels E. G., McDaniel A., Bessler W. K., Ingram D. A., Atkinson S. J., Travers J. B., Chernoff J., Clapp D. W. (2009) Blood 113, 2695–2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oh E., Thurmond D. C. (2009) Diabetes 58, 1165–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maier B., Ogihara T., Trace A. P., Tersey S. A., Robbins R. D., Chakrabarti S. K., Nunemaker C. S., Stull N. D., Taylor C. A., Thompson J. E., Dondero R. S., Lewis E. C., Dinarello C. A., Nadler J. L., Mirmira R. G. (2010) J. Clin. Invest. 120, 2156–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou M., Sevilla L., Vallega G., Chen P., Palacin M., Zorzano A., Pilch P. F., Kandror K. V. (1998) Am. J. Physiol. 275, E187–E196 [DOI] [PubMed] [Google Scholar]
- 29. Henquin J. C., Dufrane D., Nenquin M. (2006) Diabetes 55, 3470–3477 [DOI] [PubMed] [Google Scholar]
- 30. Henquin J. C. (2009) Diabetologia 52, 739–751 [DOI] [PubMed] [Google Scholar]
- 31. Deacon S. W., Beeser A., Fukui J. A., Rennefahrt U. E., Myers C., Chernoff J., Peterson J. R. (2008) Chem. Biol. 15, 322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Usui I., Imamura T., Huang J., Satoh H., Olefsky J. M. (2003) J. Biol. Chem. 278, 13765–13774 [DOI] [PubMed] [Google Scholar]
- 33. Kanzaki M., Pessin J. E. (2001) J. Biol. Chem. 276, 42436–42444 [DOI] [PubMed] [Google Scholar]
- 34. Gerthoffer W. T. (2007) Circ. Res. 100, 607–621 [DOI] [PubMed] [Google Scholar]
- 35. Ke Y., Lei M., Solaro R. J. (2008) Prog. Biophys. Mol. Biol. 98, 238–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sun J., Khalid S., Rozakis-Adcock M., Fantus I. G., Jin T. (2009) Oncogene 28, 3132–3144 [DOI] [PubMed] [Google Scholar]
- 37. Stanley F. M. (2007) Endocrinology 148, 5874–5883 [DOI] [PubMed] [Google Scholar]
- 38. Keller M. P., Choi Y., Wang P., Davis D. B., Rabaglia M. E., Oler A. T., Stapleton D. S., Argmann C., Schueler K. L., Edwards S., Steinberg H. A., Chaibub Neto E., Kleinhanz R., Turner S., Hellerstein M. K., Schadt E. E., Yandell B. S., Kendziorski C., Attie A. D. (2008) Genome Res. 18, 706–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rondas D., Tomas A., Halban P. A. (2011) Diabetes 60, 1146–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kowluru A., Veluthakal R., Rhodes C. J., Kamath V., Syed I., Koch B. J. (2010) Diabetes 59, 967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Longuet C., Broca C., Costes S., Hani E. H., Bataille D., Dalle S. (2005) Endocrinology 146, 643–654 [DOI] [PubMed] [Google Scholar]
- 42. Tomas A., Yermen B., Min L., Pessin J. E., Halban P. A. (2006) J. Cell Sci. 119, 2156–2167 [DOI] [PubMed] [Google Scholar]
- 43. Alejandro E. U., Lim G. E., Mehran A. E., Hu X., Taghizadeh F., Pelipeychenko D., Baccarini M., Johnson J. D. (2011) FASEB J. in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Trümper J., Ross D., Jahr H., Brendel M. D., Göke R., Hörsch D. (2005) Diabetologia 48, 1534–1540 [DOI] [PubMed] [Google Scholar]
- 45. Smith S. D., Jaffer Z. M., Chernoff J., Ridley A. J. (2008) J. Cell Sci. 121, 3729–3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Asrar S., Meng Y., Zhou Z., Todorovski Z., Huang W. W., Jia Z. (2009) Neuropharmacology 56, 73–80 [DOI] [PubMed] [Google Scholar]
- 47. Thurmond D. C., Gonelle-Gispert C., Furukawa M., Halban P. A., Pessin J. E. (2003) Mol. Endocrinol. 17, 732–742 [DOI] [PubMed] [Google Scholar]
- 48. Khayat Z. A., Tong P., Yaworsky K., Bloch R. J., Klip A. (2000) J. Cell Sci. 113, 279–290 [DOI] [PubMed] [Google Scholar]
- 49. Vidal C., Geny B., Melle J., Jandrot-Perrus M., Fontenay-Roupie M. (2002) Blood 100, 4462–4469 [DOI] [PubMed] [Google Scholar]
- 50. Abramovici H., Mojtabaie P., Parks R. J., Zhong X. P., Koretzky G. A., Topham M. K., Gee S. H. (2009) Mol. Biol. Cell 20, 2049–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]