

Background: The cartilage superficial zone is critical for tissue function and contains progenitor cells.
Results: Chromatin protein HMGB2 is expressed in stem cells and inhibits differentiation.
Conclusion: HMGB2 is a key regulator of the stem cell pool in mature articular cartilage.
Significance: Understanding age-related changes in HMGB2 expression is crucial to advancing concepts of age-related diseases such as osteoarthritis.
Keywords: Cell Differentiation, Chromatin, Stem Cells, Transcription Regulation, Wnt Pathway
Abstract
The superficial zone (SZ) of articular cartilage is critical in maintaining tissue function and homeostasis and represents the site of the earliest changes in osteoarthritis (OA). The expression of chromatin protein HMGB2 is restricted to the SZ, which contains cells expressing mesenchymal stem cell (MSC) markers. Age-related loss of HMGB2 and gene deletion are associated with reduced SZ cellularity and early onset OA. This study addressed HMGB2 expression patterns in MSC and its role during differentiation. HMGB2 was detected at higher levels in human MSC as compared with human articular chondrocytes, and its expression declined during chondrogenic differentiation of MSC. Lentiviral HMGB2 transduction of MSC suppressed chondrogenesis as reflected by an inhibition of Col2a1 and Col10a1 expression. Conversely, in bone marrow MSC from Hmgb2−/− mice, Col10a1 was more strongly expressed than in wild-type MSC. This is consistent with in vivo results from mouse growth plates showing that Hmgb2 is expressed in proliferating and prehypertrophic zones but not in hypertrophic cartilage where Col10a1 is strongly expressed. Osteogenesis was also accelerated in Hmgb2−/− MSC. The expression of Runx2, which plays a major role in late stage chondrocyte differentiation, was enhanced in Hmgb2−/− MSC, and HMGB2 negatively regulated the stimulatory effect of Wnt/β-catenin signaling on the Runx2 proximal promoter. These results demonstrate that HMGB2 expression is inversely correlated with the differentiation status of MSC and that HMGB2 suppresses chondrogenic differentiation. The age-related loss of HMGB2 in articular cartilage may represent a mechanism responsible for the decline in adult cartilage stem cell populations.
Introduction
The superficial zone (SZ)2 of articular cartilage is critical in maintaining tissue function and homeostasis and represents the site of the earliest changes in osteoarthritis (OA). The SZ contains cells that express mesenchymal stem cell (MSC) markers and multilineage differentiation potential (1–4), and this adult stem cell population may be important in cartilage homeostasis. Differentiation of stem cells or progenitor cells into tissue-specific lineages is associated with global changes in gene expression patterns that are in part controlled by epigenetic mechanisms (5). For instance, gene expression profiling of undifferentiated human ES cell lines showed that genes encoding regulatory proteins such as transcription factors and transcriptional co-regulators are up- or down-regulated during differentiation. Microarray data shows that Hmgb2, which encodes a non-histone chromatin protein, is one of those genes highly up-regulated in human ES cells (6, 7).
HMGB2 is a member of the HMGB protein family, which includes the ubiquitous HMGB1 and the embryo-specific HMGB3. The three proteins are >80% identical at the amino acid sequence level and characterized by two basic HMG box domains followed by a long acidic tail (8). As nuclear proteins, HMGB1 and HMGB2 are known to regulate various cellular activities, including transcription, DNA replication, and repair (9); they both bind to transcription factors such as HOX proteins (10), steroid hormone receptors (11), and p53 and p73 (12) and enhance the transcription and recombination activities of their partner proteins (9). Despite the high degree of amino acid sequence similarity between HMGB1 and HMGB2, studies have shown that they have independent functions. Contrary to the ubiquitous expression of HMGB1, HMGB2 is restricted mainly to lymphoid organs and testes, although it is widely expressed during embryogenesis (13). Hmgb1−/− mice die shortly after birth because of hypoglycemia (14), whereas Hmgb2−/− mice are viable and only show a marked reduction in spermatogenesis (13). We demonstrated that expression of HMGB2 is restricted to the SZ in adult articular cartilage and that expression declines with age in murine and human cartilage (15). In Hmgb2−/− mice, we observed reduced SZ cellularity and accelerated development of OA. These studies demonstrated a correlation between HMGB2 expression and progenitor maintenance in the SZ.
Recent studies show that articular chondrocyte hypertrophy is an early event in OA pathogenesis (16). This abnormal chondrocyte differentiation process partially recapitulates the normal endochondral development process in that the degradation and remodeling of the extracellular matrix promotes pathologic articular cartilage calcification (17). OA articular chondrocytes express markers of hypertrophic growth plate chondrocytes such as COL10A1, MMP-13, and RUNX2 (18). RUNX2 plays a major role in late stage chondrocyte differentiation (19). In Runx2−/− mice, chondrocyte maturation is disturbed (20, 21), and overexpression of a dominant-negative form of RUNX2 in chondrocytes severely delays endochondral ossification and suppresses chondrocyte maturation (22, 23). RUNX2 also contributes to the pathogenesis of experimental OA by stimulating chondrocyte hypertrophy and matrix breakdown (18) and directly regulates the Col10a1 promoter (24). Recently, we defined a pathway where protein interactions of HMGB2 and LEF1 enhance Wnt/β-catenin signaling and promote SZ chondrocyte survival (25). As Wnt signaling mediates chondrocyte hypertrophy through activation of Runx2 (26), we hypothesized that HMGB2 might be involved in hypertrophic cartilage differentiation through Runx2 transactivation. In this study, we examined the expression pattern and function of HMGB2 during MSC differentiation and reveal a mechanism by which HMGB2 regulates chondrocyte hypertrophy via interaction with Wnt/β-catenin signaling and the Runx2 promoter.
EXPERIMENTAL PROCEDURES
Human MSC Differentiation Cultures
Human MSC were purchased from Bio-Whittaker. MSC pellets (3 × 105 cells per pellet) were prepared by centrifuging the cells at 500 × g in 15 ml polypropylene conical tubes. Pellets were cultured in 0.5 ml of chondrogenic medium (Lonza), supplemented with TGF-β3 (10 ng/ml) and BMP-2 (100 ng/ml) (27, 28). Medium was changed every 2–3 days. Pellets were collected at the time points indicated for analysis of protein or gene expression and Safranin O staining. Human MSC were also induced toward adipogenic and osteogenic lineages using Poietics Mesenchymal Stem Cell Differentiation Systems according to the manufacturer's instructions (Lonza).
Immunohistochemistry and in Situ Hybridization
Immunohistochemisty was performed with rabbit anti-HMGB2 antibody (Abcam), and RNA in situ hybridization was performed using the Col2a1 and Col10a1 probes as described (29, 30).
Western Blotting
Articular chondrocytes were isolated from normal human knee cartilage and cultured as described (31). After passaging twice, cells were harvested, and a total 20 μg of each cell lysate was separated on 12% (w/v) Tris-glycine SDS-PAGE gels (Invitrogen). Western blotting was performed as described with rabbit anti-HMGB2 antibody (PharMingen), rabbit anti-RUNX2 antibody (IMGENEX), and mouse anti-GAPDH antibody (Ambion) (15).
Quantitative PCR
Total RNA was extracted from monolayer or pellet cultures and oligo(dT)-primed cDNA was prepared using the SuperScript III first strand kit (Invitrogen). The levels of Col2a1, Col10a1, Agc1, CD105, alkaline phosphatase, bone sialoprotein (BSP, encoded by Ibsp), osteocalcin (encoded by Bglap), Runx2, and GAPDH gene expression were assessed by using Taqman primers and probes (Applied Biosystems). Murine Hmgb2 expression was analyzed by SYBR Green system using primers, which were designed not to react with Hmgb1 and Hmgb3 (5′-TCCTGGTAGGCCAACAGGCT; sense and 5′-AGCTAATGTTGAGCTGCACTTG; antisense) and normalized to GAPDH (30). All reactions were performed using the iCycler thermocycler (Bio-Rad).
Construction and Preparation of Lentiviral Vectors
The cDNAs encoding FLAG and full open reading frames of murine Hmgb2 were amplified from a pcDNA3-FLAG-murine HMGB2 vector (25) and subcloned into entry clones (pENTR/SD/D-TOPO) by using the pENTR Directional TOPO Cloning Kit (Invitrogen). Lentiviral vectors were prepared with the ViraPower Lentiviral Expression System (Invitrogen) according to the manufacturer's instructions. Briefly, in vitro recombination was promoted between the pENTR and pLenti4/V5-DEST (Invitrogen) to generate the pLenti4/FLAG-murine HMGB2. The control lentivirus, which expresses GFP under a constitutive PGK promoter, was purchased from Sanford-Burnham Medical Research Institute (La Jolla, CA).
Mouse MSC Isolation and Culture
MSC were generated as described previously (32). Hmgb2−/− mice were provided by M. E. Bianchi (San Raffaele University, Milan, Italy) (13). Bone marrow cells from Hmgb2−/− mice and C57Bl/6J wild-type mice were plated at a density of 5 × 106 cells/ml (3.5 ml per well of six-well tissue culture dishes) in Dulbecco's modified Eagle's medium (low glucose) containing 10% fetal bovine serum (HyClone), 3.7 g/liter sodium bicarbonate, 10 mm N-2-hydroxyethylpiper azine-N′-2-ethanesulfonic acid, and 100 units/ml penicillin/streptomycin (Invitrogen). Cultures were kept at 37 °C 5% CO2. The medium was changed 72 h after plating and then every 3 days. At confluence, cultures were split at a 1:2 ratio. At each passage, cells were stained with phycoerythrin-conjugated antibodies to CD44, CD45 (BD PharMingen), CD34 (Biolegend), SCA-1, CD29, and CD31 (eBioscience) and analyzed by flow cytometry to confirm MSC phenotype.
Luciferase Reporter Assay
The 0.6-kb rat proximal Runx2 promoter vector or the mutant TCF1 promoter (mTCF) (gift from G. Stein and J. B. Lian, Massachusetts Medical School) (33), HA-tagged LEF1 expression vector, mutant β-catenin expression vector (25), and human sense and antisense HMGB2 vector (HMGB2-pcDNA3, asHMGB2-pcDNA3; gift from M. Stros, Institute of Biophysics, Brno, Czech Republic) (12) were used as reporter constructs. Chicken antisense HMGB2 was prepared by PCR from the corresponding chicken cDNAs. The amplified DNA samples were gel-purified and cloned in-frame into the mammalian expression vector pcDNA3 (Invitrogen). WNT3A vector was also constructed by subcloning the entire open reading frame of mouse Wnt3a and fused into pCAGGS. Upper sternal chondrocytes were isolated from the cephalic core region of 15-day-old chicken embryo sterna as described previously (26). The cells were seeded in 24-well plates and transfected with expression vectors and reporter constructs using GeneJammer transfection reagent (Stratagene). The cytomegalovirus promoter-driven β-galactosidase reporter gene (pCMVβ) was cotransfected for normalization. Luciferase and β-galactosidase activity were measured using a luciferase assay kit and a β-galactosidase enzyme kit, respectively (Promega). In each experiment, these assays were performed in triplicate.
GST Pulldown Assay
Using FLAG-tagged RUNX2 vector (gift from T. Furumatsu, Okayama University) and LEF1 expression vector (34), RUNX2 and LEF1 were in vitro-transcribed/translated with the TnT reticulocyte lysate kit (Promega) in the presence of [35S]methionine. GST-null and GST-HMGB2 proteins were produced in Escherichia coli and purified and then incubated overnight at 4 °C with the 35S-Met-labeled proteins as described (25). As a positive control, the amount of [35S]methionine-labeled protein loaded was 20% of the input.
ChIP Assay
ChIP assays were performed using the EZ ChIP Assay Kit (Millipore) as described by the manufacturer. ATDC5 cells were incubated in 150-mm dishes overnight and then the cells were cross-linked with 1% formaldehyde for 10 min at 37 °C. The cells were lysed in 1000 μl of SDS lysis buffer for 15 min on ice, followed by nuclear lysis buffer, and sonicated eight times for 15 s. The sonicated samples were incubated with 5 μg of rabbit anti-LEF1 antibody (Cell Signaling), mouse anti-RUNX2 antibody (MBL International), and rabbit anti-HMGB2 antibody (Abcam) or normal mouse IgG or normal rabbit IgG (Santa Cruz Biotechnology) containing protein G-agarose beads for 8 h at 4 °C. Immune complexes were washed following the manufacturer's instructions. Immunoprecipitated DNA was eluted and reverse cross-linked in elution buffer, followed by RNase A and proteinase K treatment. An aliquot of the immunoprecipitated DNA and chromatin input control (1% input) was used for PCR (33 cycles). All reactions were done under an annealing temperature of 58 °C. Two primers for amplifying LEF1 binding sites in the murine Runx2 promoter were described previously (33). As negative controls, primers were set outside of the Runx2 proximal promoter (GGCTCCCTCTCTCCATCTCT and GCTGAACGTGGCCTTTATGT, at −2.5 kb upstream of the Runx2 transcription start site) (35). All PCR products were evaluated on 2% agarose gels for appropriate size (245 bp for Runx2 proximal promoter, 224 bp for −2.5 kb region of the Runx2 gene, 225 bp for the p19ARF promoter).
Statistical Analysis
Results are expressed as mean ± S.D. Means of groups were compared by analysis of variance, and significance of differences was determined by post hoc testing using Bonferroni's method. Statistical comparison between two groups was performed with a two-tailed unpaired t test. p values <0.05 were considered significant.
RESULTS
HMGB2 Expression in MSC and Articular Cartilage
To determine the expression pattern of Hmgb2 in relation to the endogenous chondrocyte differentiation, we examined its expression in mouse embryos by in situ hybridization. Although Hmgb2 was expressed in proliferative and resting zones in the epiphyseal growth plate in the tibia and femur, its expression was significantly lower in prehypertrophic cartilage where Col10a1 was positive at E14.5 (Fig. 1A). At E15.5 and E16.5, Hmgb2 was barely detectable in hypertrophic cartilage in tibia, whereas Hmgb2 was strongly expressed in bone marrow (Fig. 1B). These data suggest a correlation between the decline of Hmgb2 expression and chondrocyte differentiation in developing long bone. Furthermore, in 1-month-old postnatal mouse skeletons, HMGB2 was strongly expressed in the articular cartilage surface as well as bone marrow, whereas it was not detected in hypertrophic zone of a femoral growth plate (Fig. 1C). These observations indicate that HMGB2 expression decreases with chondrocyte differentiation in growth plate but remains at high levels in bone marrow.
FIGURE 1.

HMGB2 expression in developing cartilage. A, Hmgb2 expression is reduced in prehypertrophic cartilage (arrowheads) where Col10a1 is positive in tibia and femur at E14.5. Col2a1 is also reduced in prehypertrophic cartilage in femur (arrow). B, Hmgb2 is eliminated in hypertrophic cartilage in tibia at E15.5 and E16.5. Hmgb2 is highly expressed in bone marrow at E16.5. C, HMGB2 expression in postnatal cartilage. Immunohistochemistry reveals that HMGB2 is expressed in cartilage surface (arrowheads) as well as bone marrow cells (right panel), whereas that expression is not seen in hypertrophic cartilage of femoral growth plate (arrows) at a month old. SO, safranin O staining; ph, prehypertrophic cartilage; h, hypertrophic cartilage; bm, bone marrow; ac, articular cartilage; p, patella. A magnification of ×100 was used.
To further examine the relationship between HMGB2 expression and chondrogenic differentiation, we analyzed human bone marrow-derived MSC and differentiated human articular chondrocytes and found that HMGB2 was highly expressed in MSC compared with human articular chondrocytes (Fig. 2). Interestingly, we detected reduced HMGB2 levels in chondrocytes with aging. HMGB2 levels in young donors were significantly higher than in aged donors, in which HMGB2 was barely detectable. This result suggests a link between the known age-related OA pathogenesis and the observed loss of HMGB2 in humans (15).
FIGURE 2.
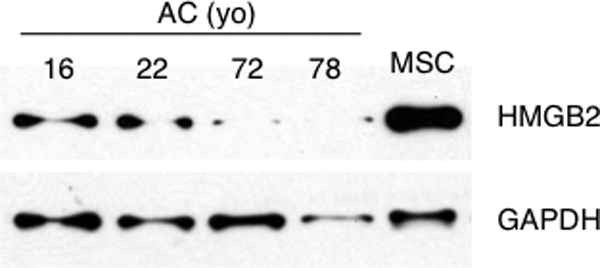
HMGB2 expression in MSC and articular chondrocytes. Human articular chondrocytes (AC) from knee cartilage of donors at the indicated ages, and MSC were cultured in monolayer culture and then used for Western blotting. yo, years old.
HMGB2 Expression during Chondrogenesis of Human MSC
As we found that HMGB2 is highly expressed in undifferentiated cells such as MSC and articular cartilage SZ, which contains a progenitor cell population (1, 4) and is not detectable in differentiated hypertrophic cartilage as shown in Fig. 1, we directly followed changes in HMGB2 expression during chondrogenic differentiation of human bone marrow MSC. Immunohistochemistry showed that HMGB2 was highly expressed at days 1 and 3 but that expression was absent except for the peripheral area at day 7 and further decreased at day 14 (Fig. 3A). This expression pattern was confirmed by Western blotting where a reduction in HMGB2 protein levels was consistently detected by 8 days of pellet culture (Fig. 3B). These results demonstrate that HMGB2 expression pattern is inversely correlated with differentiation status of MSC.
FIGURE 3.

HMGB2 expression during chondrogenesis of human MSC. A, immunohistochemistry shows that HMGB2 is expressed at days 1 and 3 but that expression is reduced at days 7 and 14 upon induction of chondrogenesis. Arrowheads indicate Safranin O (SO)-positive region. Boxes are shown in bottom as enlarged pictures. B, human bone marrow MSC from three different donors were collected from chondrogenesis pellet cultures on days 2, 4, and 8, and Western blotting was performed to detect HMGB2 and GAPDH. A reduction in HMGB2 protein levels is consistently seen by 8 days of pellet culture.
Overexpression of HMGB2 in MSC Inhibits Chondrogenesis
To address the role of HMGB2 during chondrogenesis, MSC were transduced with lentivirus (LV) encoding HMGB2 or control LV expressing GFP. Immunohistochemistry showed that HMGB2 was intensely expressed in LV/HMGB2 pellets at day 7, whereas in LV/GFP pellets, HMGB2-positive cells were barely detectable (Fig. 4A). After 3 weeks of culture, the size of chondrogenic pellets with LV/HMGB2 was significantly smaller than that of control pellets expressing GFP (Fig. 4B), suggesting that extracellular matrix production, which increases with chondrocytic differentiation is inhibited. At day 14, Safranin O staining was positive in the center of LV/GFP pellets but not in LV/HMGB2 pellets (Fig. 4C). Col2a1-positive cells were found extensively in control pellets but not in LV/HMGB2 pellets. At day 21, control pellets showed high Safranin O staining, whereas LV/HMGB2 pellets were still negative. Quantitative PCR showed that extracellular matrix genes Col2a1, Agc1, and Col10a1 were suppressed in LV/HMGB2-transduced pellets compared with control LV/GFP pellets (Fig. 4D). In contrast, CD105, a marker of human MSC that decreases in chondrogenesis culture (36), was higher in LV/HMGB2 control group. These findings suggest that sustained expression of HMGB2 in MSC blocks chondrogenic differentiation and maintains immature status of MSC even after chondrogenesis was induced.
FIGURE 4.

Ectopic expression of HMGB2 by lentivirus transduction in human MSC. A, LV/GFP transduction in MSC of a monolayer culture by immunofluorescence (top left panel). HMGB2-positive cells are found in LV/HMGB2 pellets (bottom right panel) at day 7 (7d). B, macroscopic image of differentiating MSC showing the size of chondrogenic pellet with LV/HMGB2 and LV/GFP control pellet at day 21. GFP is still observed in control pellet with LV/GFP. C, at day 14 upon chondrogenesis induction, safranin O is positive in the center of LV/GFP control pellet (arrowheads) but not in LV/HMGB2 pellet. Col2a1-positive cells are found extensively in control pellet but are absent in LV/HMGB2 pellet. At day 21, GFP pellet shows high Safranin O staining, whereas the LV/HMGB2 pellet remains negative. D, quantitative PCR shows that Col2a1, Col10a1, and Agc1 are suppressed in LV/HMGB2 pellets compared with control LV/GFP pellets, whereas CD105 is higher in the LV/HMGB2 (*, p < 0.01; n = 3).
Chondrogenesis in MSC from Wild-type and Hmgb2−/− Mice
To further address the function of HMGB2 during MSC differentiation, we harvested mouse bone marrow-derived MSC from 6- to 8-week-old wild-type and Hmgb2−/− mice (13). At each passage, cells were analyzed by flow cytometry for CD44, CD29, SCA-1, CD45, CD31, and CD34 to monitor and confirm the MSC phenotype (supplemental Fig. 1) (37) and then induced chondrogenesis, osteogenesis, and adipogenesis to confirm their multipotency (Fig. 5A). In chondrogenesis cultures, Hmgb2 levels were gradually reduced in wild-type MSC pellets (Fig. 5B), which was similar to the observations with human MSC as shown in Fig. 3. Western blotting confirmed that HMGB2 was expressed in wild-type MSC but not in Hmgb2−/− MSC (Fig. 5C). In situ hybridization demonstrated extensive Col2a1 expression in pellets with both types of MSC (Fig. 5D). Col10a1 was expressed only in the peripheral area of pellets with wild-type MSC, whereas pellets with Hmgb2−/− MSC showed increased Col10a1 expression. Quantitative PCR showed that Col2a1 and Agc1, representative chondrogenic markers, were comparable between the two groups. In contrast, the chondrocyte hypertrophy marker Col10a1 was more strongly expressed in Hmgb2−/− MSC compared with wild-type MSC in pellet culture at day 14 (Fig. 5E). These data demonstrate that chondrogenesis starts in the absence of Hmgb2, and chondrogenic differentiation toward hypertrophic status was significantly up-regulated. Overall, these data strongly suggest that HMGB2 is not required for initial chondrogenic differentiation, but it negatively regulates expression of the hypertrophy marker Col10a1 during chondrocyte differentiation of bone marrow MSC.
FIGURE 5.

Chondrogenesis of MSC established from 6- to 8-week-old wild-type and Hmgb2−/− mice. A, multipotency of bone marrow MSC established from C57Bl/6 wild-type mice. MSC were cultured under conditions to induce chondrogenesis, osteogenesis and adipogenesis as determined by Safranin O (SO), alizarin red S, and Oil Red staining, respectively. B, in chondrogenic pellet cultures, Hmgb2 levels are gradually reduced in wild-type MSC pellets, whereas Col2a1 and Col10a1 are increased. mono, monolayer MSC. C, HMGB2 expression in WT and Hmgb2−/− (KO) MSC by Western blotting. D, in situ hybridization shows that Col2a1 expression is found extensively in both types of pellets. Col10a1 is expressed exclusively in peripheral areas of wild-type MSC, although Hmgb2−/− MSC shows high Col10a1 expression throughout. SO, safranin O straining. E, quantitative PCR for Col2a1, Agc1, and Col10a1 expression in wild-type and Hmgb2−/− MSC at 14 days (14d) pellet culture (*, p < 0.05, n = 4).
Osteogenesis in MSC from Wild-type and Hmgb2−/− Mice
Given that the loss of HMGB2 is correlated with accelerated chondrocyte differentiation, we assumed that HMGB2 might also modulate osteogenesis. Thus, we investigated the role of HMGB2 in osteogenesis using wild-type and Hmgb2−/− MSC. Similar to the reduction of HMGB2 expression during chondrogenesis, there was a time-dependent reduction of Hmgb2 levels during osteogenic differentiation in wild-type MSC (Fig. 6A). Using Hmgb2−/− MSC under osteogenesis conditions, alizarin red S staining was significantly stronger as compared with wild-type MSC (Fig. 6B). In addition, the levels of alkaline phosphatase and Ibsp, an early marker of osteoblastic differentiation (38), were elevated in Hmgb2−/− MSC. Bglap, a late differentiation marker (38), was also expressed robustly at day 7 in Hmgb2−/− MSC but was barely detectable in wild-type MSC (Fig. 6C).
FIGURE 6.

Osteogenesis of MSC from wild-type and Hmgb2−/− mice. A, a time-dependent reduction of Hmgb2 level in wild-type MSC (*, p < 0.05, n = 3). mono, monolayer MSC. B, after 8 days of culture under osteogenesis conditions, alizarin red S staining is much stronger in Hmgb2−/− MSC compared with WT MSC. C, quantitative PCR shows that alkaline phosphatase (Alpl), Ibsp, Bglap, and Runx2 are significantly increased in Hmgb2−/− MSC at day 7 (7d) upon osteogenesis induction (*, p < 0.05, n = 4). D, Western blotting shows that RUNX2 is strongly expressed in Hmgb2−/− (KO) MSC at day 7 during osteogenesis.
Importantly, we found that Runx2, a transcriptional regulator of Ibsp and Bglap (39), showed a significantly greater elevation during osteogenesis in Hmgb2−/− MSC compared with wild-type MSC (Fig. 6C). RUNX2 protein was also more strongly expressed in Hmgb2−/− MSC as compared with wild-type at day 7 upon osteogenesis induction (Fig. 6D). These findings indicate that osteogenesis was enhanced in Hmgb2−/− MSC, and this enhancement was related to increased RUNX2 expression.
HMGB2 Negatively Regulates Runx2
As RUNX2 not only regulates osteogenesis but also promotes hypertrophic differentiation in growth plate and OA cartilage (24, 40), we hypothesized that RUNX2 expression is up-regulated during chondrogenesis in the absence of Hmgb2 and examined Runx2 expression in wild-type and Hmgb2−/− MSC during chondrocyte differentiation. In wild-type MSC, Runx2 expression showed a time-dependent decrease. Runx2 expression was significantly higher in Hmgb2−/− MSC at each time point during chondrogenesis culture (Fig. 7A), demonstrating an up-regulation of Runx2 in the absence of Hmgb2. To directly determine whether HMGB2 regulates RUNX2 expression, we employed siRNA and demonstrated that loss of Hmgb2 potentiated Runx2 expression in murine articular chondrocytes (Fig. 7B). MSC were transduced with LV/HMGB2 to sustain HMGB2 expression, and this functional manipulation suppressed Runx2 mRNA levels (Fig. 7C).
FIGURE 7.

HMGB2 negatively regulates RUNX2 expression. A, Runx2 in Hmgb2−/− MSC is significantly higher than in WT MSC throughout chondrogenesis culture (*, p < 0.05, n = 4). mono, monolayer MSC. B, Runx2 is significantly increased by Hmgb2 siRNA in murine articular chondrocytes. N/C, negative control siRNA. C, Runx2 is suppressed in LV/HMGB2 pellets compared with LV/GFP control 3 days after chondrogenesis induction (*, p < 0.05, n = 3). D, a schematic illustration of the Runx2 promoter showing the relative positions of binding sites, including functionally validated RUNX2, HIF-2α, NKX3–2, TCF/LEF, and vitamin D-responsive elements (VDRE). E and F, HMGB2 affects Wnt/β-catenin-dependent transactivation from the Runx2 promoter in chicken upper sternal chondrocytes. The cells were transfected with the Runx2 promoter or mutant Runx2 promoter (mTCF) (100 ng), second reporter plasmid pCMVβ (10 ng), LEF1 expression construct (33 ng), WNT3A, β-catenin, HMGB2, or antisense HMGB2 (100 ng each) plus empty vector (pcDNA3) to compensate for equal DNA amounts. Cells were harvested after 24 (E) or 48 h (F), followed by luciferase assay (*, p < 0.05, n = 3). asHMGB2, antisense HMGB2. N.S., Not significant. G, the relative levels of endogenous RUNX2 expression in chicken chondrocytes transfected with the LEF1 (150 ng), β-catenin (450 ng), HMGB2, or antisense HMGB2 (450 ng) expression construct. Cells were harvested after 48 h for protein isolation and subsequent analysis by Western blotting.
To address the mechanism involved in HMGB2 regulation of RUNX2 in more detail, we focused on Wnt/β-catenin signaling, which mediates chondrocyte hypertrophy through activation of Runx2 (26). As we reported that HMGB2 physically interacts with LEF1 and enhances Wnt signaling according to the result of TOPflash promoter vector and TOP-GFP in zebrafish (25, 41), we examined whether HMGB2 affects Wnt/β-catenin-dependent activation of the Runx2 proximal promoter (0.6 kb), which contains the TCF/LEF consensus site (Fig. 7D) (26, 33). Runx2 and its promoter activity were enhanced by ectopic expression of β-catenin and Wnt in chicken upper sternal chondrocytes as reported (26). Importantly, transfection of antisense HMGB2 significantly enhanced the WNT3A-dependent transcriptional activation from the Runx2 promoter, whereas expression of sense HMGB2 plasmid did not have significant effects. Mutation of the TCF-binding site resulted in loss of activation by WNT3A (Fig. 7E). After transfection with LEF1/β-catenin and antisense HMGB2, Runx2 promoter activity was increased (Fig. 7E), and the relative levels of endogenous protein RUNX2 were elevated (Fig. 7F).
LEF1 physically interacts with RUNX2, and this down-regulates Runx2 promoter activity (42, 43). Interestingly, HMGB2 is also capable of binding RUNX2 as well as LEF1 by GST pulldown assay (Fig. 8A). To detect the interaction of HMGB2 with RUNX2 or LEF1 in the normal genomic context, ChIP assay was performed in chondrogenic ATDC5 cells using primer pairs designed to encompass the TCF/LEF element within the Runx2 promoter (33). The results showed that LEF1 and HMGB2 are associated with the Runx2 promoter (Fig. 8B). RUNX2 has previously been found to bind the promoter region of the Runx2 gene that includes several RUNX2 sites (43, 44). The specificity of the binding of these proteins to the Runx2 proximal promoter was confirmed by the absence of binding outside of this region and to a promoter of an unrelated gene (Fig. 8B). Thus, both LEF1/HMGB2 and RUNX2 occupy the proximal promoter to control levels of Runx2. These results suggest that HMGB2 regulates MSC chondrogenesis in part by modulating the LEF1-dependent transactivation of Runx2.
FIGURE 8.
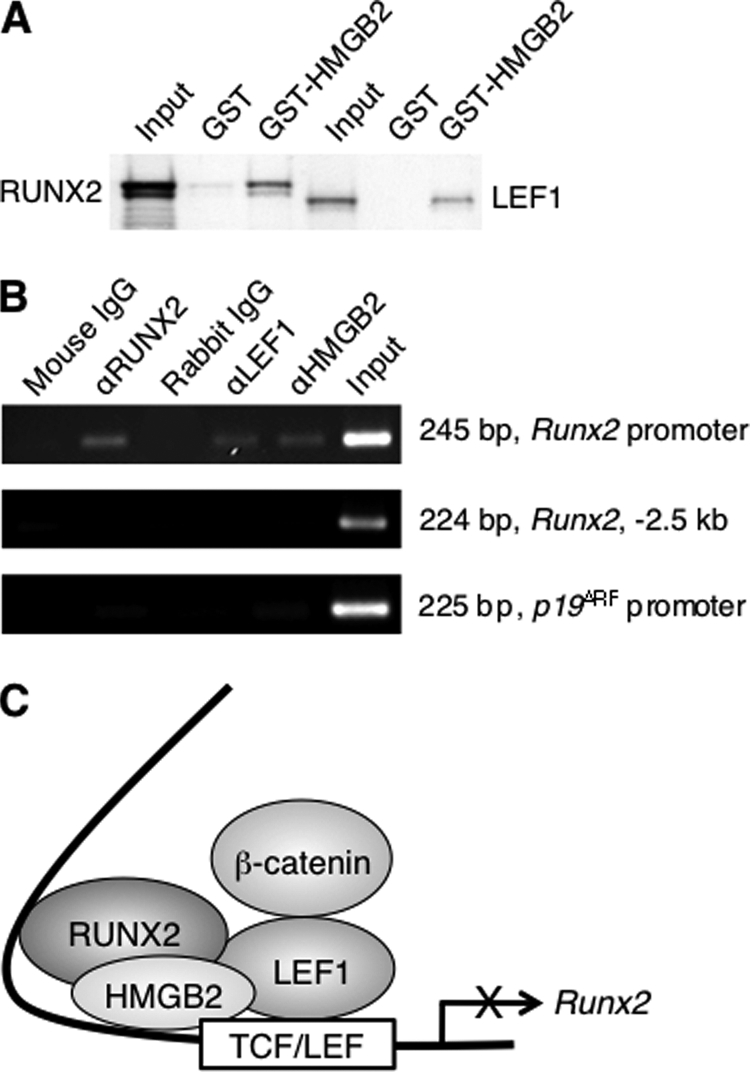
Physical interaction of HMGB2 in the Runx2 promoter. A, interaction of in vitro-translated RUNX2 with GST-HMGB2. In vitro-translated LEF1 was used as positive control. B, ChIP assay was performed in chondrogenic ATDC5 cells using primer pairs designed to encompass the TCF/LEF element within the Runx2 promoter. LEF1, RUNX2, and HMGB2 are associated with the promoter region of the Runx2 gene but not outside this region of the Runx2 gene (Runx2, −2.5kb) and to a promoter of an unrelated gene, p19ARF. C, summarizing schema showing functional and physical interactions between HMGB2, RUNX2, and LEF1 on the Runx2 proximal promoter containing TCF/LEF motif. HMGB2 can bind with LEF1 as well as RUNX2, and this down-regulates Runx2 promoter activity. When HMGB2 is decreased during MSC differentiation, Wnt/β-catenin-dependent activation of Runx2 promoter is enhanced, and this accelerates chondrogenesis and osteogenesis.
DISCUSSION
In this study, we demonstrate that the chromatin protein HMGB2 is highly expressed in undifferentiated MSC, and its expression is down-regulated during chondrogenesis and osteogenesis. HMGB2 is identified as a novel regulator of MSC differentiation as it inhibits chondrogenesis, and this is related to binding with LEF1 and suppression of RUNX2.
As expression of HMGB2 is restricted to the SZ of articular cartilage that contains progenitor cells (1, 25), we analyzed HMGB2 expression in undifferentiated human bone marrow MSC and differentiated articular chondrocytes and found that HMGB2 is more highly expressed in MSC. In vitro chondrogenesis studies revealed that Hmgb2 transcript levels are reduced with progressive differentiation of MSC. To directly investigate the role of HMGB2 in regulating chondrogenic differentiation we used lentiviral transduction for ectopic expression of HMGB2 and found that LV/HMGB2 suppressed glycosaminoglycan production, chondrogenic markers Col2a1, Agc1, and the hypertrophy marker Col10a1. These observations were supported by experiments using MSC derived from Hmgb2−/− mice, which also showed enhanced Col10a1 expression during chondrogenesis. These findings indicate that as HMGB2 inhibits differentiation, it maintains the immature status of adult stem cells and suggest that the age-related loss of HMGB2 in articular cartilage may represent a mechanism responsible for the decline in adult cartilage stem cell populations (15).
In vivo results from mouse growth plates show that Hmgb2 is expressed in proliferating and prehypertrophic zones but not in hypertrophic cartilage where Col10a1 is strongly expressed. Because RUNX2 regulates COL10A1 and MMP-13 in growth plate as well as OA cartilage (19, 24), we examined RUNX2 expression in wild-type and Hmgb2−/− MSC during chondrocytic differentiation. We found that Runx2 was increased in Hmgb2−/− MSC compared with wild-type MSC throughout chondrogenesis culture. However, cartilage maturation in Hmgb2−/− tibia was similar to that in wild-type (15), and we also did not observe apparent differences in cartilage maturation in Hmgb1−/− mice (30). As mammalian HMGBs contain two HMG boxes arranged in tandem and share >80% identity (9), redundancy could exist between HMGB2 and HMGB1. Several lines of evidence show that β-catenin-mediated Wnt signaling induces chondrocyte maturation (26, 45, 46). In this regard, we already reported that not only HMGB2 but also HMGB1 can bind with LEF1 in vitro (25).
Ectopic expression of HMGB2 in human MSC suppressed Runx2 expression. Similarly, HMGB2 repressed the augmentation of Runx2 promoter activity by co-transfection of β-catenin and LEF1. Although HMGB2 enhanced Wnt/β-catenin signaling in chondrosarcoma cells by binding LEF1 (25), our data shows that in chicken upper sternal chondrocytes, HMGB2 functions as a co-repressor on the Runx2 promoter, suggesting an involvement of other cellular factors that can modulate the ability of HMGB2 to affect the transcriptional activity of genes containing LEF1-responsive promoters. Drissi et al. (43) reported that forced expression of RUNX2 protein down-regulates Runx2 promoter activity and that a single RUNX2 site is sufficient for transcriptional autosuppression. We found that HMGB2 can bind with RUNX2 as well as LEF1 (25), and RUNX2 also interacts with LEF1 (42). The TCF/LEF consensus sequence is located downstream of a purine-rich region of the Runx2 promoter in a highly conserved area that contains binding sequences for NKX3–2, HIF-2α, and vitamin D-responsive elements as well as RUNX2 itself (43, 47–50). As these proteins mediate Runx2 promoter activity, a possible mechanism that explains the observed modulation of LEF1-dependent transactivation by HMGB2 in vivo could be a differential interaction of HMGB2 with nuclear factors affecting transcription of genes containing LEF1-responsive elements (Fig. 8C).
RUNX2 expression initially occurs in mesenchymal cells just before differentiation into chondrocytes, and then its expression is maintained in chondrocytes until the cells complete the terminal differentiation step (51). This suggests that RUNX2 may play other roles in the regulation of chondrocyte differentiation in addition to promoting chondrocyte maturation. In fact, Runx2−/− mice have not only suppressed endochondral ossification but also retarded cartilage growth and poor organization of growth plate structures (20), and transgenic mice expressing a dominant-negative form of RUNX2 had cartilage elements of reduced size (22). Also, the expression of a native truncated form of RUNX2 inhibited chondrogenic differentiation of ATDC5 prechondrogenic cells (52), suggesting that RUNX2 may be involved in the early phase of chondrocyte differentiation. These findings indicate that RUNX2 may participate in multiple aspects of the differentiation and function of chondrocytes. Our data shows that ectopic HMGB2 expression in MSC suppresses chondrogenesis markers Col2a1 and Agc1 as well as Runx2, suggesting that suppression of the Runx2 promoter by HMGB2 in MSC might abrogate chondrocyte differentiation as well as hypertrophic differentiation.
In conclusion, we propose that as HMGB2 inhibits differentiation, it maintains the immature state of adult MSC. HMGB2 may play a similar role in maintaining the adult stem cell population in the normal articular cartilage SZ. Age-related loss of HMGB2 may not only contribute to SZ disruption but also to the abnormal hypertrophic differentiation that is a feature of OA cartilage.
Acknowledgments
We thank Miu Yamashita, Diana Brinson, Jean Valbracht, Lilo Creighton, and Hiroko Kawakami for technical support. We also thank Drs. K. Gokul, T. Gaur, J.B. Lian, and M. Stros for provision of the plasmid and Dr. M. E. Bianchi for sharing Hmgb2 mutant mice.
This work was supported, in whole or in part, by National Institutes of Health Grants AR056026, AG007996, RR027557 and the Consortium for Functional Glycomics GM62116. This work was also supported by the Sam and Rose Stein Endowment Fund (to M. L.) and the Arthritis National Research Foundation (to N. T.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
- SZ
- superficial zone
- OA
- osteoarthritis
- MSC
- mesenchymal stem cell(s)
- BSP
- bone sialoprotein
- LV
- lentivirus
- E14
- embryonic day 14
- LEF1
- lymphoid enhancer-binding factor 1
- TCF
- T-cell factor.
REFERENCES
- 1. Dowthwaite G. P., Bishop J. C., Redman S. N., Khan I. M., Rooney P., Evans D. J., Haughton L., Bayram Z., Boyer S., Thomson B., Wolfe M. S., Archer C. W. (2004) J. Cell Sci. 117, 889–897 [DOI] [PubMed] [Google Scholar]
- 2. Hiraoka K., Grogan S., Olee T., Lotz M. (2006) Biorheology 43, 447–454 [PubMed] [Google Scholar]
- 3. Alsalameh S., Amin R., Gemba T., Lotz M. (2004) Arthritis Rheum. 50, 1522–1532 [DOI] [PubMed] [Google Scholar]
- 4. Grogan S. P., Miyaki S., Asahara H., D'Lima D. D., Lotz M. K. (2009) Arthritis Res. Ther. 11, R85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Collas P., Noer A., Timoskainen S. (2007) J. Cell Mol. Med. 11, 602–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim C. G., Lee J. J., Jung D. Y., Jeon J., Heo H. S., Kang H. C., Shin J. H., Cho Y. S., Cha K. J., Kim C. G., Do B. R., Kim K. S., Kim H. S. (2006) Mol. Cells 21, 343–355 [PubMed] [Google Scholar]
- 7. Bhattacharya B., Miura T., Brandenberger R., Mejido J., Luo Y., Yang A. X., Joshi B. H., Ginis I., Thies R. S., Amit M., Lyons I., Condie B. G., Itskovitz-Eldor J., Rao M. S., Puri R. K. (2004) Blood 103, 2956–2964 [DOI] [PubMed] [Google Scholar]
- 8. Bustin M. (1999) Mol. Cell Biol. 19, 5237–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bianchi M. E., Agresti A. (2005) Curr. Opin. Genet. Dev. 15, 496–506 [DOI] [PubMed] [Google Scholar]
- 10. Zappavigna V., Falciola L., Helmer-Citterich M., Mavilio F., Bianchi M. E. (1996) EMBO J. 15, 4981–4991 [PMC free article] [PubMed] [Google Scholar]
- 11. Boonyaratanakornkit V., Melvin V., Prendergast P., Altmann M., Ronfani L., Bianchi M. E., Taraseviciene L., Nordeen S. K., Allegretto E. A., Edwards D. P. (1998) Mol. Cell Biol. 18, 4471–4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stros M., Ozaki T., Bacikova A., Kageyama H., Nakagawara A. (2002) J. Biol. Chem. 277, 7157–7164 [DOI] [PubMed] [Google Scholar]
- 13. Ronfani L., Ferraguti M., Croci L., Ovitt C. E., Schöler H. R., Consalez G. G., Bianchi M. E. (2001) Development 128, 1265–1273 [DOI] [PubMed] [Google Scholar]
- 14. Calogero S., Grassi F., Aguzzi A., Voigtländer T., Ferrier P., Ferrari S., Bianchi M. E. (1999) Nat. Genet. 22, 276–280 [DOI] [PubMed] [Google Scholar]
- 15. Taniguchi N., Caramés B., Ronfani L., Ulmer U., Komiya S., Bianchi M. E., Lotz M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 1181–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Husa M., Liu-Bryan R., Terkeltaub R. (2010) Nat. Med. 16, 641–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saito T., Fukai A., Mabuchi A., Ikeda T., Yano F., Ohba S., Nishida N., Akune T., Yoshimura N., Nakagawa T., Nakamura K., Tokunaga K., Chung U. I., Kawaguchi H. (2010) Nat. Med. 16, 678–686 [DOI] [PubMed] [Google Scholar]
- 18. Kamekura S., Kawasaki Y., Hoshi K., Shimoaka T., Chikuda H., Maruyama Z., Komori T., Sato S., Takeda S., Karsenty G., Nakamura K., Chung U. I., Kawaguchi H. (2006) Arthritis Rheum. 54, 2462–2470 [DOI] [PubMed] [Google Scholar]
- 19. Komori T. (2010) Cell Tissue Res. 339, 189–195 [DOI] [PubMed] [Google Scholar]
- 20. Inada M., Yasui T., Nomura S., Miyake S., Deguchi K., Himeno M., Sato M., Yamagiwa H., Kimura T., Yasui N., Ochi T., Endo N., Kitamura Y., Kishimoto T., Komori T. (1999) Dev Dyn. 214, 279–290 [DOI] [PubMed] [Google Scholar]
- 21. Kim I. S., Otto F., Zabel B., Mundlos S. (1999) Mech. Dev. 80, 159–170 [DOI] [PubMed] [Google Scholar]
- 22. Ueta C., Iwamoto M., Kanatani N., Yoshida C., Liu Y., Enomoto-Iwamoto M., Ohmori T., Enomoto H., Nakata K., Takada K., Kurisu K., Komori T. (2001) J. Cell Biol. 153, 87–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stricker S., Fundele R., Vortkamp A., Mundlos S. (2002) Dev. Biol. 245, 95–108 [DOI] [PubMed] [Google Scholar]
- 24. Higashikawa A., Saito T., Ikeda T., Kamekura S., Kawamura N., Kan A., Oshima Y., Ohba S., Ogata N., Takeshita K., Nakamura K., Chung U. I., Kawaguchi H. (2009) Arthritis Rheum. 60, 166–178 [DOI] [PubMed] [Google Scholar]
- 25. Taniguchi N., Caramés B., Kawakami Y., Amendt B. A., Komiya S., Lotz M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 16817–16822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dong Y. F., Soung do Y., Schwarz E. M., O'Keefe R. J., Drissi H. (2006) J. Cell Physiol. 208, 77–86 [DOI] [PubMed] [Google Scholar]
- 27. Pittenger M. F. (2008) Methods Mol. Biol. 449, 27–44 [DOI] [PubMed] [Google Scholar]
- 28. Grogan S. P., Olee T., Hiraoka K., Lotz M. K. (2008) Arthritis Rheum 58, 2754–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taniguchi N., Kawahara K., Yone K., Hashiguchi T., Yamakuchi M., Goto M., Inoue K., Yamada S., Ijiri K., Matsunaga S., Nakajima T., Komiya S., Maruyama I. (2003) Arthritis Rheum. 48, 971–981 [DOI] [PubMed] [Google Scholar]
- 30. Taniguchi N., Yoshida K., Ito T., Tsuda M., Mishima Y., Furumatsu T., Ronfani L., Abeyama K., Kawahara K., Komiya S., Maruyama I., Lotz M., Bianchi M. E., Asahara H. (2007) Mol. Cell Biol. 27, 5650–5663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hashimoto S., Ochs R. L., Rosen F., Quach J., McCabe G., Solan J., Seegmiller J. E., Terkeltaub R., Lotz M. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 3094–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Syres K., Harrison F., Tadlock M., Jester J. V., Simpson J., Roy S., Salomon D. R., Cherqui S. (2009) Blood 114, 2542–2552 [DOI] [PubMed] [Google Scholar]
- 33. Gaur T., Lengner C. J., Hovhannisyan H., Bhat R. A., Bodine P. V., Komm B. S., Javed A., van Wijnen A. J., Stein J. L., Stein G. S., Lian J. B. (2005) The Journal of biological chemistry 280, 33132–33140 [DOI] [PubMed] [Google Scholar]
- 34. Tetsunaga T., Nishida K., Furumatsu T., Naruse K., Hirohata S., Yoshida A., Saito T., Ozaki T. (2011) Osteoarthritis Cartilage 19, 222–232 [DOI] [PubMed] [Google Scholar]
- 35. Nakade K., Pan J., Yamasaki T., Murata T., Wasylyk B., Yokoyama K. K. (2009) The Journal of biological chemistry 284, 10808–10817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee H. J., Choi B. H., Min B. H., Park S. R. (2009) Arthritis Rheum 60, 2325–2332 [DOI] [PubMed] [Google Scholar]
- 37. Nadri S., Soleimani M., Hosseni R. H., Massumi M., Atashi A., Izadpanah R. (2007) Int. J. Dev. Biol. 51, 723–729 [DOI] [PubMed] [Google Scholar]
- 38. Stiehler M., Bünger C., Baatrup A., Lind M., Kassem M., Mygind T. (2009) J. Biomed Mater Res. A 89, 96–107 [DOI] [PubMed] [Google Scholar]
- 39. Komori T. (2010) Adv. Exp. Med. Biol. 658, 43–49 [DOI] [PubMed] [Google Scholar]
- 40. Zheng Q., Zhou G., Morello R., Chen Y., Garcia-Rojas X., Lee B. (2003) J. Cell Biol. 162, 833–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Itou J., Taniguchi N., Oishi I., Kawakami H., Lotz M., Kawakami Y. (2011) Dev. Dyn. 240, 1151–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kahler R. A., Westendorf J. J. (2003) J. Biol. Chem. 278, 11937–11944 [DOI] [PubMed] [Google Scholar]
- 43. Drissi H., Luc Q., Shakoori R., Chuva De Sousa Lopes S., Choi J. Y., Terry A., Hu M., Jones S., Neil J. C., Lian J. B., Stein J. L., Van Wijnen A. J., Stein G. S. (2000) J. Cell Physiol. 184, 341–350 [DOI] [PubMed] [Google Scholar]
- 44. Xiao Z. S., Liu S. G., Hinson T. K., Quarles L. D. (2001) J. Cell Biochem. 82, 647–659 [DOI] [PubMed] [Google Scholar]
- 45. Hartmann C., Tabin C. J. (2000) Development 127, 3141–3159 [DOI] [PubMed] [Google Scholar]
- 46. Daumer K. M., Tufan A. C., Tuan R. S. (2004) J. Cell Biochem. 93, 526–541 [DOI] [PubMed] [Google Scholar]
- 47. Drissi H., Pouliot A., Stein J. L., van Wijnen A. J., Stein G. S., Lian J. B. (2002) J. Cell Biochem. 86, 403–412 [DOI] [PubMed] [Google Scholar]
- 48. Lengner C. J., Hassan M. Q., Serra R. W., Lepper C., van Wijnen A. J., Stein J. L., Lian J. B., Stein G. S. (2005) The Journal of biological chemistry 280, 15872–15879 [DOI] [PubMed] [Google Scholar]
- 49. Tamiya H., Ikeda T., Jeong J. H., Saito T., Yano F., Jung Y. K., Ohba S., Kawaguchi H., Chung U. I., Choi J. Y. (2008) Gene 416, 53–60 [DOI] [PubMed] [Google Scholar]
- 50. Drissi H., Pouliot A., Koolloos C., Stein J. L., Lian J. B., Stein G. S., van Wijnen A. J. (2002) Exp. Cell Res. 274, 323–333 [DOI] [PubMed] [Google Scholar]
- 51. Goldring M. B., Tsuchimochi K., Ijiri K. (2006) J. Cell Biochem. 97, 33–44 [DOI] [PubMed] [Google Scholar]
- 52. Akiyama H., Kanno T., Ito H., Terry A., Neil J., Ito Y., Nakamura T. (1999) J. Cell Physiol. 181, 169–178 [DOI] [PubMed] [Google Scholar]