

Background: It is unclear how stress granule (SG) formation and cellular apoptosis are coordinately regulated.
Results: Monocyte chemotactic protein-induced protein 1 (MCPIP1) inhibited the assembly of SGs and promoted cellular apoptosis under stress.
Conclusion: MCPIP1 coordinately regulates SG formation and apoptosis during cellular stress.
Significance: MCPIP1 may play a critical role in immune homeostasis and resolution of macrophage inflammation through this mechanism.
Keywords: Apoptosis, Cell Biology, Macrophages, Signal Transduction, Stress Granule, GW Body, MCPIP1
Abstract
It is unclear how stress granule (SG) formation and cellular apoptosis are coordinately regulated. MCPIP1 (monocyte chemotactic protein-induced protein 1), also known as Zc3h12a, is a critical regulator of the inflammatory response and immune homeostasis. However, the role of MCPIP1 in stress response remains unknown. Here, we report that overexpression of MCPIP1 inhibited the assembly of SGs in response to various stresses. Conversely, MCPIP1-deficient splenocytes developed more SGs even without stress. On the other hand, overexpression of MCPIP1 sensitized RAW 264.7 cells to apoptosis under stress, whereas MCPIP1-deficient cells were resistant to stress-induced apoptosis. Mutagenesis study showed that the ability of MCPIP1 to repress SG formation is dependent on its deubiquitinating activity. Consistently, MCPIP1 negatively regulated stress-induced phosphorylation of eIF2α and thus released stress-induced inhibition of protein translation. However, MCPIP1 also inhibited 15-deoxy-Δ12,14-prostaglandin J2-induced SG formation, which was reported to be independent of eIF2α phosphorylation. Taken together, these results suggest that MCPIP1 coordinates SG formation and apoptosis during cellular stress and may play a critical role in immune homeostasis and resolution of macrophage inflammation.
Introduction
Stress granules (SGs)3 are subcellular structures that are formed in response to various environmental stresses, which is a major adaptive defense mechanism. It has been suggested that SGs function to protect housekeeping mRNAs and repress their translation by recruiting these mRNAs and certain mRNA-associated proteins. Through this mechanism, cells could allocate their available resources toward the production of dedicated stress management proteins (1–5).
SGs are dynamic cytoplasmic foci at which stalled translation complexes accumulate. Under some conditions, SG assembly and translational arrest are initiated by the phosphorylation of translation initiation factor eIF2α, which reduces the availability of the eIF2-GTP-tRNAMet ternary complex that is needed to initiate protein translation. Thereby, translationally stalled mRNAs are sequestered in dynamic cytoplasmic SGs. These granules represent a complex assembly of initiation factors, such as eIF3 or eIF4E; proteins involved in translation control, such as T-cell intracellular antigen or fragile X mental retardation protein; proteins implicated in RNA remodeling or degradation, such as HuR, tristetraprolin, or Staufen; and 40 S subunits (6–9). Recently, the endoribonuclease G3BP (Ras GTPase-activating protein SH3 domain-binding protein) and the translation regulator CPEB (cytoplasmic polyadenylation element-binding protein) have been shown to be part of SGs and induce assembly of SGs if overexpressed (10, 11). In addition, SGs also contain various polyadenylated mRNAs whose translation has been arrested. However, mRNAs encoding stress-induced proteins, such as heat shock proteins, are excluded from SGs and spared from translation inhibition. Importantly, stress defense and apoptotic dismantling tend to occur in a mutually exclusive fashion (12, 13). Recently, studies have shown that SGs can protect cells from apoptosis because actively disrupting SGs results in the release of apoptosis-inducing components and triggers cell death (14, 15).
MCPIP1 (monocyte chemotactic protein-induced protein 1), also known as Zc3h12a, is a critical regulator of the inflammatory response and immune homeostasis (16–20). We reported recently that MCPIP1 feedback controls LPS- and cytokine-induced JNK and NF-κB signaling by deubiquitinating TNF receptor-associated factors (19). Matsushita et al. (20) reported that Zc3h12a/MCPIP1 acts as an RNase to promote the mRNA degradation of some inflammatory cytokines, such as IL-6. The function of MCPIP1 in the regulation of cellular stress responses is unknown. We report here that MCPIP1 is significantly up-regulated at a later time during the stress period. MCPIP1 expression disassembles SGs and promotes cellular apoptosis. This study identifies MCPIP1 as a factor that coordinates SG formation and apoptosis during cellular stress, which may be implicated in the resolution of macrophage inflammation.
EXPERIMENTAL PROCEDURES
Cells
RAW 264.7, HeLa, and HEK293 cells were obtained from American Type Culture Collection. These cells were grown as a monolayer in DMEM (Invitrogen) containing 10% FBS, 2 mm l-glutamine, and 100 units/ml each penicillin and streptomycin at 5.0% CO2. Littermate wild-type and Mcpip1−/− day 13.5 embryos were used to generate mouse embryonic fibroblasts and maintained in DMEM containing 10% FBS at 5.0% CO2. Mouse splenocytes were isolated from 6–8-week-old wild-type and Mcpip1−/− mice according to the protocol described previously (21) and grown in lymphocyte growth medium 3 (Lonza).
Plasmids
MCPIP1-GFP, FLAG-MCPIP1, and FLAG-MCPIP1 mutants were as described previously (19). Human DCP1α (hDCP1α)-red fluorescent protein (RFP) was kindly provided by Dr. Dominique Weil (University Pierre and Marie Curie, Paris, France) and was as described previously (22). RAB5-RFP and Lamp1-RFP were obtained from Addgene and were as described previously (23, 24).
Reagents
Goat anti-MCPIP1 polyclonal antibody (sc-136750), anti-GW182, anti-G3BP, anti-eIF4E, and anti-poly(A)-binding protein (PABP) antibodies were purchased from Santa Cruz Biotechnology. Anti-phospho-eIF2α, anti-phospho-JNK, anti-phospho-p38, anti-eIF2α, anti-JNK, anti-p38, anti-Ge-1, anti-DDX6, anti-eIF3, and anti-actin antibodies were purchased from Cell Signaling Technology. Anti-FLAG and anti-TGN38 antibodies, arsenite, cycloheximide, carbonyl cyanide 3-chlorophenylhydrazone, and 15d-PGJ2 were purchased from Sigma.
Transfection
Transient transfection of plasmids into cells was performed as described previously (18).
Protein Isolation and Western Blotting
Protein isolation and Western blotting were performed essentially as described previously (18).
Immunofluorescence
1 × 104 HeLa cells were seeded in a total of 400 μl of medium/well on 8-well chambered Lab-Tek slides. After stress treatment, cells were fixed by incubation with 100 μl of 4% paraformaldehyde for 20 min, permeabilized with PBS containing 0.2% Triton X-100 for 10 min, and incubated in blocking solution for 1 h at room temperature. After that, cells were incubated overnight with primary antibody at a 1:200 dilution in PBS at 4 °C. Cells were rinsed with PBS and incubated with fluorescent anti-IgG antibody (Vector Labs, Burlingame, CA) at a 1:200 dilution in PBS for 1 h at room temperature. Slides were mounted in VECTASHIELD hard set mounting medium with DAPI (Vector Labs). Photographs were taken using a Nikon C1 Plus confocal microscope.
Apoptosis Assay
The annexin V apoptosis assay (BioVision Research) was performed following the manufacturer's protocol. In brief, RAW 264.7 cells were transiently transfected with a control or MCPIP1-GFP vector. The transfected cells were seeded on 8-well chambered Lab-Tek slides at 1 × 105 cells/well. Apoptosis was induced by treating the cells with sodium arsenite for 2 h. 500 μl of 1× binding buffer (BioVision Research) and 5 μl of annexin V-Cy3 were added to each well, and cells were incubated for 10 min at room temperature and monitored using a Nikon C1 Plus confocal microscope with a ×60 oil immersion objective.
Flow Cytometry
Wild-type and Mcpip1−/− mouse embryonic fibroblasts were treated with sodium arsenite for 2 h in a 6-well plate. The cells were trypsinized and collected by centrifugation. Cells were suspended in 500 μl of 1× binding buffer. 5 μl of annexin V-Cy3 was added to the suspended cells and incubated for 10 min at room temperature. Finally, the cells were harvested by centrifugation, washed with PBS, and resuspended in 0.2% paraformaldehyde solution. Annexin V-Cy3-binding cells were analyzed by flow cytometry (excitation = 543 nm and emission = 570 nm) using a phycoerythrin emission signal detector.
Protein Synthesis Assay
The protein synthesis assay was conducted using the Click-iT AHA Alexa Fluor 488 protein assay kit (Invitrogen) following the manufacturer's instructions. Briefly, HeLa cells were seeded on 8-well chambered Lab-Tek slides. 24 h after transfection, cells were treated with or without arsenite (0.1 or 0.5 mm) in methionine-free medium (Invitrogen) supplemented with 50 μm l-azidohomoalanine (AHA; Invitrogen) for 2 h. The cells were then washed with PBS, fixed with 3.7% paraformaldehyde for 15 min, and then incubated in 3% BSA in PBS for 15 min, followed by incubation with 0.5% Triton X-100 in PBS for 20 min. Proteins that incorporated AHA were labeled with Alexa Fluor 488-conjugated alkyne (Invitrogen) and costained with anti-FLAG antibody (1:200) and DyLight 594-labeled anti-rabbit IgG (1:200; Vector Labs). Images were taken with a Nikon C1 Plus confocal microscope, and immunofluorescence density were quantified using ImageJ software.
Statistics
Data are expressed as the mean ± S.D. For comparison between two groups, Student's unpaired test was used. For multiple comparisons, analysis of variance followed by Student's test unpaired were used. A p value <0.05 was considered significant.
RESULTS
Subcellular Localization of MCPIP1
We observed previously that endogenous MCPIP1 protein is distributed in the cytoplasm and forms granule-like structures in THP-1 cells (18). Transfection of the MCPIP1-GFP expression plasmid into HeLa cells further confirmed that the MCPIP1-GFP fusion protein localized in the cytoplasm and formed many granules (Fig. 1a). To examine whether MCPIP1 protein is localized on mitochondria or Golgi bodies, HeLa cells were double-stained with anti-MCPIP1 antibody and MitoTracker or anti-TGN38 antibody (a marker of Golgi bodies). The results show that MCPIP1 overlapped with neither mitochondria nor Golgi bodies (Fig. 1b). We tested whether MCPIP1 protein is co-localized with lysosomes or endosomes. HeLa cells were transfected with MCPIP1-GFP and Lamp1-RFP (a marker of lysosomes) or RAB5-RFP (a marker of endosomes). The results show that MCPIP1 protein co-localized with neither lysosomes nor endosomes (Fig. 1b). Next, we transfected MCPIP1-GFP into HeLa cells and then treated the cells with or without cycloheximide. Again, MCPIP1-GFP protein formed granule-like structures in untreated cells. Interestingly, the granule-like structures of MCPIP1-GFP disappeared in cycloheximide-treated cells. As cycloheximide is an inhibitor of translation elongation, it can inhibit the formation of RNA granules, such as SGs and GW bodies (also called processing bodies (P bodies)), by fixing mRNA on the polysome (25). The above results suggest that MCPIP1 protein may be localized on RNA granules, such as SGs or GW/P bodies.
FIGURE 1.

MCPIP1 forms granule-like structures in the cytoplasm. a, HeLa cells were transiently transfected with GFP or MCPIP1-GFP. After 24 h, cells were fixed and observed by fluorescence microscopy. b, upper panels, HeLa cells were double-stained with anti-MCPIP1 antibody and MitoTracker (Mito) or anti-TGN38 antibody (a marker of Golgi bodies). Lower panels, HeLa cells were cotransfected with MCPIP1-GFP and RAB5-RFP or Lamp1-RFP. After 24 h, cells were fixed, stained with DAPI, and observed by confocal fluorescence microscopy. c, HeLa cells were transfected with MCPIP1-GFP and treated with or without cycloheximide. After 24 h, cells were fixed and observed by fluorescence microscopy. Scale bars = 10 μm except as indicated.
MCPIP1 Is Co-localized with GW182
SG and GW/P bodies are the major RNA granules. We first examined whether MCPIP1 is localized on GW bodies. HEK293 cells were transfected with MCPIP1-GFP and then stained with anti-GW182 antibody. As shown in Fig. 2a, the granule-like structure overlapped perfectly with GW bodies as visualized by GW182 staining. As several studies reported (2, 4), the GW body overlapped with the P body, which usually contains DCP1α, DCP2, XRN1, Ge-1, and DDX6. We tested whether MCPIP1 is also co-localized with hDCP1α. HEK293 cells were transfected with MCPIP1-GFP and hDCP1α-RFP. As shown in Fig. 2b, MCPIP1-GFP did not overlap with hDCP1α-RFP, although most of the granules were found in close juxtaposition with the DCP1 granules. Further experiments using Ge-1 and DDX6 staining also showed that, although a few MCPIP1 granules overlapped with P bodies, most of the MCPIP1 granules were juxtaposed with P bodies but not overlapped (Fig. 2, c and d). These results suggest that MCPIP1 is localized on GW bodies but not totally confined in P bodies.
FIGURE 2.

MCPIP1 is co-localized with GW182. a, HEK293 cells were transiently transfected with MCPIP1-GFP. After 24 h, cells were fixed, stained with GW182, and observed by confocal fluorescence microscopy. b, HEK293 cells were transiently transfected with MCPIP1-GFP and hDCP1α-RFP. After 24 h, cells were fixed and observed by confocal fluorescence microscopy. c and d, HeLa cells were transiently transfected with MCPIP1-GFP. After 24 h, cells were fixed, stained with anti-Ge-1 or anti-DDX6 antibodies as indicated, and observed by confocal fluorescence microscopy. Scale bars = 10 μm.
Expression of MCPIP1 Represses SG Formation under Stress
Next, we examined whether MCPIP1 is also localized on SGs under stress. HeLa cells transfected with MCPIP1-GFP were treated with arsenite (0.5 mmol/liter) for 60 min. The treated cells were visualized by immunofluorescence staining with antibodies against several markers of SGs, such as G3BP, eIF3, eIF4E, and PABP. As shown in Fig. 3 (a–d), MCPIP1-GFP was not co-localized with SGs. Surprisingly, the cells expressing MCPIP1-GFP could not assembly SGs under arsenite-induced oxidative stress. To determine whether this phenomenon is stimulus-specific, the transfected cells were incubated with 1 μm/liter carbonyl cyanide 3-chlorophenylhydrazone (an inhibitor of mitochondria) in no-glucose DMEM for 2 h. As shown in Fig. 3 (e–h), carbonyl cyanide 3-chlorophenylhydrazone treatment induced SG formation. Similar to the above results, expression of MCPIP1-GFP totally blocked the formation of SGs. Similar results were also observed in RAW 264.7 cells (data not shown). As a control experiment, expression of GFP alone did not affect the formation of SGs under both oxidative stress and energy deprivation conditions (data not shown).
FIGURE 3.

MCPIP1 expression blocks SG formation. a–d, HeLa cells were transiently transfected with MCPIP1-GFP. After 24 h, cells were stressed with 0.5 mm arsenite for 1 h; fixed; stained with anti-G3BP, anti-eIF3, anti-eIF4E, and anti-PABP antibodies, respectively; and observed by confocal fluorescence microscopy. e–h, HeLa cells were transiently transfected with MCPIP1-GFP. After 24 h, cells were cultured in DMEM without glucose; stressed with 1 μm carbonyl cyanide 3-chlorophenylhydrazone for 4 h; fixed; stained with anti-G3BP, anti-eIF3, anti-eIF4E, and anti-PABP antibodies, respectively; and observed by confocal fluorescence microscopy. Scale bar = 10 μm.
MCPIP1 Deficiency Promotes SG Formation
Next, we analyzed the ability of splenocytes from Mcpip1−/− mice to form SGs. As shown in Fig. 4a, without stress, the cells from Mcpip1+/+ mice did not form SGs. However, the cells from Mcpip1−/− mice formed SGs even without stress, as visualized by immunofluorescence staining using anti-G3BP and anti-PABP antibodies. After oxidative stress induced by arsenite, MCPIP1-deficient cells formed many more and much larger SGs compared with Mcpip1+/+ cells. Fig. 4b shows the quantitative results from three independent experiments with G3BP staining. PABP staining showed similar results (data not shown).
FIGURE 4.

Lack of MCPIP1 results in increase in SG formation. The splenocytes from Mcpip1+/+ and Mcpip1−/− mice were treated with or without arsenite for 1 h. a, SGs were visualized with anti-G3BP and anti-PABP antibodies and anti-mouse secondary antibody labeled with Alexa Fluor 594 (red). Scale bar = 10 μm. b, in G3BP-stained images, the percentage of SG-positive cells was quantified in a two-blind way and is represented as the mean ± S.D. from three independent experiments.
Ability of MCPIP1 to Repress SG Formation Is Dependent on Its Deubiquitinating Enzyme (DUB) Activity
As characterized previously (19), MCPIP1 is a multiple domain-containing protein that contains the ubiquitin-associated domain, DUB/RNase domains, the CCCH zinc-finger domain, the proline-rich domain, and the C-terminal conserved region. To analyze the domains that contribute to the repression of SG formation, FLAG-MCPIP1 and its serially truncated plasmids were transfected into HeLa cells. The transfected cells were then stimulated with 0.5 mm arsenite for 1 h. The SGs were visualized by immunofluorescence with anti-G3BP antibody. The expression of MCPIP1 and its truncated forms was visualized by immunofluorescence with anti-FLAG antibody. As shown in Fig. 5a, the MCPIP1 truncation without the C-terminal conserved region and proline-rich and ubiquitin-associated domains lost granule-like structures but still maintained the ability to inhibit SG formation. Further deletion of a partial DUB/RNase domain or a mutated CCCH zinc-finger domain resulted in maintenance granule-like structures but loss of the ability to inhibit SG formation. The truncated MCPIP1 plasmids were constructed as indicated in Fig. 5b.
FIGURE 5.

Capacity of MCPIP1 to repress SG formation is dependent on its DUB activity. a, the functional domains and conserved regions of MCPIP1 and the deletion strategy are shown. b, HeLa cells were transiently transfected with FLAG-MCPIP1 and its serial deletion as indicated. After 24 h, cells were stressed with arsenite for 1 h, fixed, stained with anti-G3BP and anti-FLAG antibodies, and observed by confocal fluorescence microscopy. UBA, ubiquitin-associated domain; ZF, CCCH zinc-finger domain; Pro, proline-rich domain; CCR, C-terminal conserved region. c and d, several point mutants of MCPIP1 previously mapped were used in this study. HeLa cells were transiently transfected with FLAG-MCPIP1 and its mutants as indicated. After 24 h, cells were stressed with arsenite for 1 h, fixed, stained with anti-G3BP and anti-FLAG antibodies, and observed by confocal fluorescence microscopy. Scale bars = 10 μm.
As reported previously (19, 20), MCPIP1 contains both DUB and RNase activities, by which it can block LPS- and cytokine-induced inflammatory signaling and promote inflammatory mRNA degradation, respectively. Both DUB and RNase active domains have been mapped on the N-terminal conserved and CCCH zinc-finger regions, which have some overlapping and distinct amino acid sites. The mutant MCPIP1-C157A lost DUB activity but maintained RNase activity. MCPIP1-D225A/D226A lost RNase activity but maintained DUB activity. MCPIP1-D141N lost both DUB and RNase activities (Fig. 5c). As shown in Fig. 5d, MCPIP1-C157A maintained the granule-like structures, but it lost the ability to inhibit SG formation. In contrast, MCPIP1-D225A/D226A lost the granule-like structures, but it maintained the ability to inhibit SG formation. MCPIP1-D141N appeared as different granule-like structures and also lost the ability to inhibit SG formation. Taken together, the results clearly suggest that the ability of MCPIP1 to repress SG formation is dependent on its DUB activity. On the other hand, its RNase activity may be responsible for its granule-like structures.
MCPIP1 Expression Sensitizes the Cells to Apoptosis under Stress
It is known that cells form SGs to prevent apoptotic death under stress (14, 15). Thus, actively disrupting SGs may result in the release of apoptosis-inducing components from SGs to trigger cell death. To test whether expression of MCPIP1 promotes cellular apoptosis, GFP or MCPIP1-GFP was transfected into RAW 264.7 cells. The transfected cells were treated with or without 0.5 mm arsenite for 2 h. Cell death was detected by annexin V-Cy3 staining. It appeared that a significantly increased cell population was annexin V-positive when the cells were transfected with MCPIP1-GFP and treated with arsenite (Fig. 6, a and b). In another experiment, mouse embryonic fibroblasts from wild-type or Mcpip1−/− mice were treated with or without 0.5 mm arsenite for 2 h. Cell death was detected by annexin V-Cy3 staining and analyzed by flow cytometry. As shown in Fig. 6 (c and d), MCPIP1-deficient cells were resistant to stress-induced apoptosis. Taken together, these results suggest that MCPIP1 expression sensitizes the cells for apoptosis under stress.
FIGURE 6.

MCPIP1 modulates stress-induced apoptosis. a, RAW 264.7 cells were transfected with GFP or MCPIP1-GFP plasmids. After 24 h, the transfected cells were quiescent and were stressed with 0.5 mm arsenite for 2 h. The cells were then fixed, stained with annexin V-Cy3, and observed by fluorescence microscopy. GFP images of the cells are also shown to serve as a transfected cell number control. b, quantification of Cy3-positive cells is shown. c, mouse embryonic fibroblasts from Mcpip1−/− mice and wild-type littermates were quiescent and were stressed with 0.5 mm arsenite for 2 h. The cells were then fixed, stained with annexin V-Cy3, and analyzed by flow cytometry. d, quantification of Cy3-positive cells is shown, and data represent triplicate determinations from three independent experiments.
MCPIP1 Expression Inhibits Arsenite-induced eIF2α Phosphorylation
To understand the molecular mechanism of MCPIP1 inhibition of SG formation, we investigated whether MCPIP1 expression suppresses stress-induced eIF2α phosphorylation because some stresses, such as arsenite, induce SG formation by phosphorylation of eIF2α (5). RAW 264.7 cells were transfected with GFP or MCPIP1-GFP expression plasmids. 24 h post-transfection, the cells were left untreated or were treated with 0.5 mm arsenite for 30, 60, and 120 min. Protein extracts were prepared and subjected to Western blotting with phospho-eIF2α and other antibodies as indicated. As shown in Fig. 7a, arsenite-induced eIF2α phosphorylation peaked at 60 min. Overexpression of MCPIP1 completely blocked arsenite-induced eIF2α phosphorylation. We reported previously that MCPIP1 inhibits LPS- and cytokine-induced JNK signaling (19), and here, MCPIP1 expression also inhibited arsenite-induced JNK phosphorylation but did not affect p38 phosphorylation. Consistently, phosphorylated eIF2α was significantly increased in Mcpip1−/− cells under both normal and stress conditions compared with that in Mcpip1+/+ cells (Fig. 7b).
FIGURE 7.
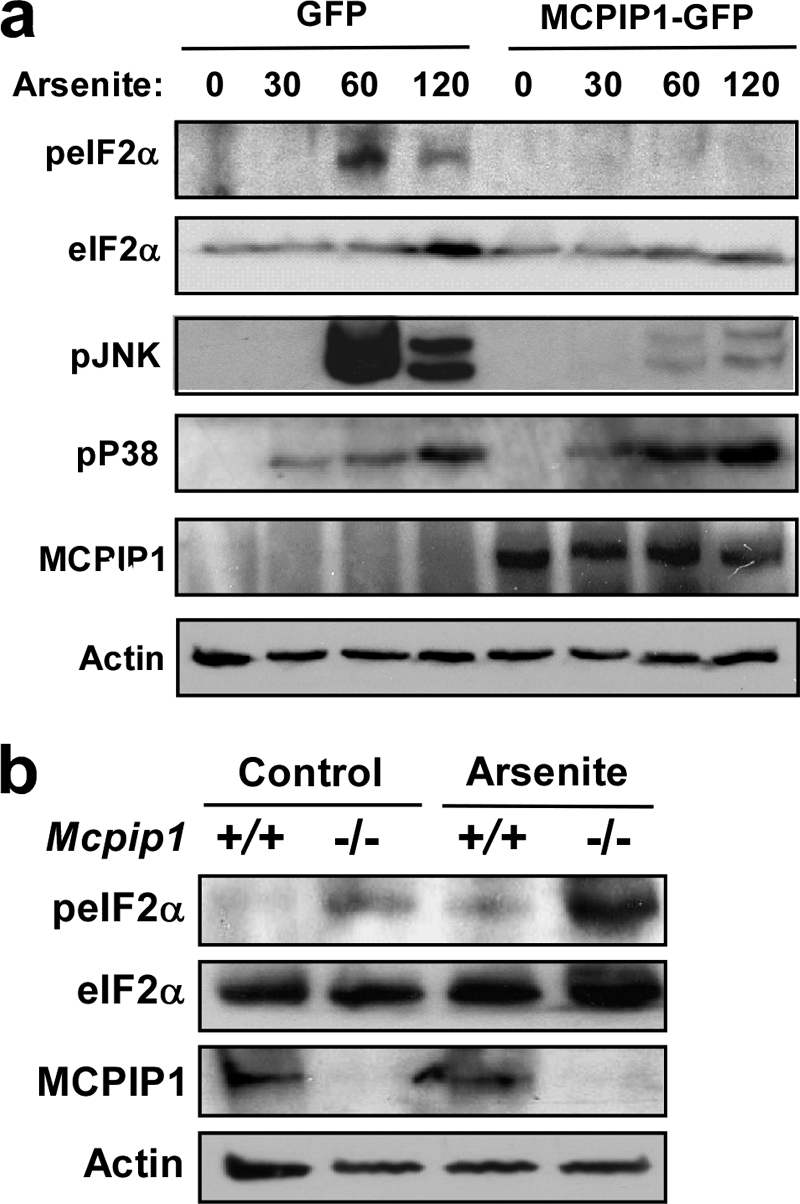
MCPIP1 inhibits stress-induced eIF2α phosphorylation. a, RAW 264.7 cells were transfected with GFP or MCPIP1-GFP plasmids. After 24 h, the transfected cells were stressed with arsenite (0.5 mm) for the indicated times (min). Whole cell extracts were subjected to Western blotting analysis with antibodies as indicated. Two independent experiments showed similar results. b, the splenocytes from Mcpip1+/+ and Mcpip1−/− mice were treated with or without arsenite for 1 h. The whole cell lysates were subjected to Western blot analysis with antibodies as indicated. Two independent experiments showed similar results.
MCPIP1 Expression Releases Stress-induced Inhibition of Protein Synthesis
As SG formation is accompanied by translational blockage, the effect of MCPIP1 expression on protein synthesis was investigated. HeLa cells were transfected with FLAG-MCPIP1 or an empty vector. The transfected cells were treated with or without arsenite (0.1 or 0.5 mm) in methionine-free medium. The newly translated proteins were labeled with the methionine analog AHA. Using click chemistry, these AHA-incorporating nascent proteins were then labeled with a fluorescent dye (Alexa Fluor 488) conjugated to an alkyne group (26, 27). The images were taken by confocal microscopy and are shown in Fig. 8a. The fluorescence intensity was quantified by ImageJ. As shown in Fig. 8b, arsenite treatment dose-dependently inhibited nascent protein synthesis. MCPIP1 expression released the stress-induced inhibition of protein synthesis.
FIGURE 8.

Effect of MCPIP1 expression on protein synthesis under stress. a, HeLa cells were transfected with FLAG or FLAG-MCPIP1 plasmids. After 24 h, the transfected cells were stressed with arsenite (0.1 or 0.5 mm) and incubated with AHA for 2 h. The AHA-incorporated nascent proteins were then labeled with Alexa Fluor 488. The expression of FLAG or FLAG-MCPIP1 was determined by staining with anti-FLAG antibody, followed by DyLight 594-labeled anti-rabbit IgG. Representative images are shown. b, the fluorescence density was quantified by ImageJ. Data are from three independent experiments. *, p < 0.05 versus the FLAG group of each arsenite treatment.
Stress Induces MCPIP1 Expression
To analyze the dynamic expression of MCPIP1 protein under stress, RAW 264.7 cells were incubated with 0.5 mm arsenite or subjected to heat shock (43 °C) for different times as indicated in Fig. 9. As shown in Fig. 9a, MCPIP1 expression was significantly increased after 12 h of treatment with arsenite, and the high expression levels were maintained for up to 24 h. Interestingly, phospho-eIF2α and phospho-JNK were significantly increased after 1 h of treatment with arsenite but disappeared after 12 h, which is associated with the expression of MCPIP1 at that time. Under heat shock, MCPIP1 was induced after 4 h and declined at 6 and 12 h but increased again at 24 h. Similar to arsenite treatment, phospho-eIF2α and phospho-JNK were significant increased after 0.5 h of heat shock and disappeared after 4 h when MCPIP1 expression was increased (Fig. 9b). These results suggest that, during the early period of stress, cells quickly initiate defense mechanisms to form SGs and dictate survival; however, during the late period of stress, MCPIP1 is up-regulated, and feedback blocks stress-induced eIF2α phosphorylation and SG formation and dictates cellular apoptosis.
FIGURE 9.
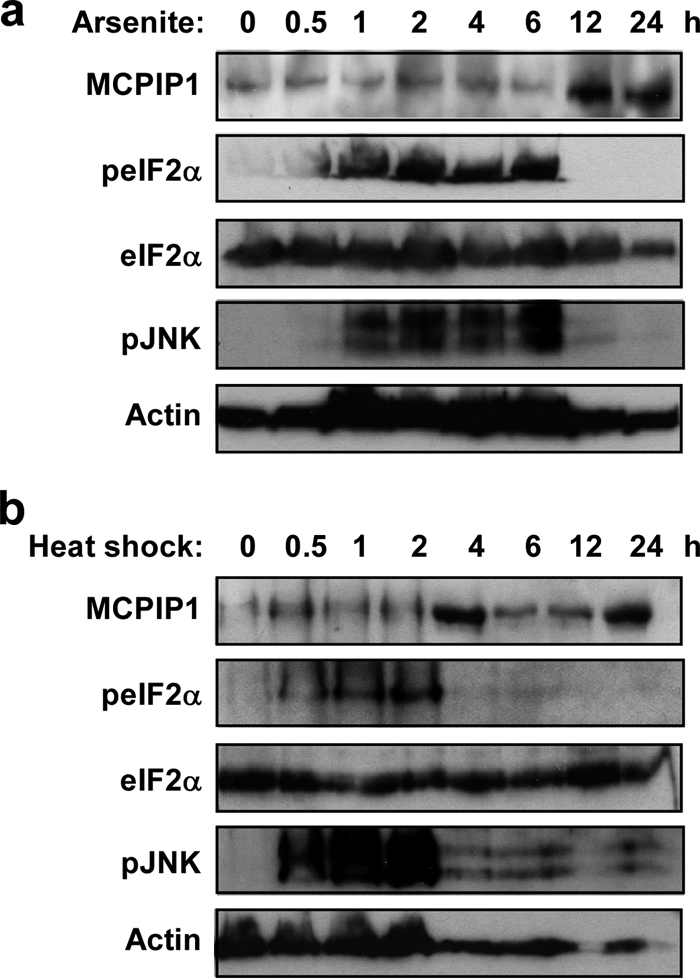
Stress-induced MCPIP1 expression and eIF2α phosphorylation. RAW 264.7 cells were stressed with arsenite (0.5 mm; a) or heat shock (43 °C; b) for the indicated times. Whole cell extracts were subjected to Western blot analysis with antibodies as indicated. Two independent experiments showed similar results.
MCPIP1 Expression Also Represses 15d-PGJ2-induced SG Formation
It was reported previously that 15d-PGJ2 blocks translation through inactivation of translation initiation factor eIF4A and results in SG formation (28). To examine whether MCPIP1 also inhibits 15d-PGJ2-induced SG formation, HeLa cells transfected with MCPIP1-GFP were treated with 15d-PGJ2 (50 μmol/liter) for 1 h. The treated cells were visualized by immunofluorescence staining with antibodies against several markers of SGs, such as G3BP, eIF3, eIF4E, and PABP. As shown in Fig. 10, a–d, MCPIP1 expression also blocked 15d-PGJ2-induced SG formation. Because 15d-PGJ2-induced SG formation is independent of eIF2α phosphorylation, these results suggest that MCPIP1 may also disassemble SGs through other mechanisms.
FIGURE 10.

MCPIP1 expression also blocks 15d-PGJ2-induced SG formation. a–d, HeLa cells were transiently transfected with MCPIP1-GFP. After 24 h, cells were treated with 50 μm 15d-PGJ2 for 1 h; fixed; stained with anti-G3BP, anti-eIF3, anti-eIF4E, and anti-PABP antibodies, respectively; and observed by confocal fluorescence microscopy. Scale bar = 10 μm.
DISCUSSION
In response to stress, cells form SGs to prevent damage that environmental forces inflict on DNA or proteins. However, if the stress is intense and sustained, SGs will be disassembled, and apoptosis will be initiated. The molecular mechanisms that coordinate the two opposite responses remain largely unknown. Our previous work demonstrated that MCPIP1 acts as a DUB to negatively regulate JNK and NF-κB signaling and macrophage inflammation (19). However, its role in the cellular stress response remains unknown. In this study, we found that expression of MCPIP1 completely blocked SG formation and promoted macrophage apoptosis under stress conditions, including arsenite-induced oxidative stress, heat shock, and energy deprivation, which is dependent on its DUB activity. Consistently, MCPIP1-deficient cells spontaneously formed aggregation of SGs even in the absence of stress and were resistant to apoptosis under stress. Furthermore, MCPIP1 was induced by stress in macrophages during the late period, which is associated with the decrease in phosphorylation of eIF2α. These results suggest that MCPIP1 is critically involved in the coordination of SG formation and apoptosis under stress.
In most cases, the stress-induced phosphorylation of eIF2α leads to SG assembly by preventing or delaying translational initiation. Cells that express a non-phosphorylatable form of eIF2α (S51A) cannot assemble SGs in response to arsenite-induced oxidative stress and are hypersensitive to the toxic effect of low doses of arsenite (7). The mechanism by which MCPIP1 blocks arsenite-induced SG formation is probably by repressing stress-induced phosphorylation of eIF2α. As evident in this study, overexpression of MCPIP1 significantly inhibited arsenite-induced eIF2α phosphorylation. Conversely, stress-induced eIF2α phosphorylation was significantly increased in MCPIP1-deficient cells. It was noted that MCPIP1 expression also inhibited 15d-PGJ2-induced SG formation. 15d-PGJ2 has been reported to inhibit translation by blocking the interaction between eIF4A and eIF4G and results in SG formation (28). These results suggest that MCPIP1 may modulate SG assembly by multiple mechanisms.
One interesting finding is that overexpressed MCPIP1 forms many granule-like structures in the cytoplasm and overlaps perfectly with GW182, suggesting co-localization with GW/P bodies. Previous reports indicate that SGs and GW/P bodies are spatially, compositionally, and functionally linked. Overexpression of another CCCH zinc-finger domain-containing protein, tristetraprolin, promotes the interaction and composition interchanges between SGs and GW/P bodies (29). It is possible that overexpression of MCPIP1 promotes the formation of GW/P bodies, by which it interferes with the formation of SGs.
MCPIP1 granules overlap perfectly with GW-182 but only partially with hDCP1α, Ge-1, and DDX6. GW182 has been identified as a major component of the RNA-induced silencing complex and is dynamically recruited into P bodies (30, 31), including hDCP1α, Ge-1, and DDX6. However, there are also many observations that the RNA-induced silencing complex does not completely overlap with P bodies (32, 33), suggesting the dynamic nature of fusion of the RNA-induced silencing complex and P bodies. Importantly, the granule-like structure of MCPIP1 is associated with its RNase activity. In addition, a previous report suggests that MCPIP1 promotes mRNA degradation of IL-6 and other inflammatory cytokines (20). These findings suggest that MCPIP1 may be involved in the RNAi/microRNA functional pathway.
How does MCPIP1 promote cellular apoptosis? As reported previously (14, 15), actively disrupting SGs would result in the release of apoptosis-inducing components from SGs to trigger cell death. Thus, MCPIP1 may promote cellular apoptosis by disassembling SGs and releasing apoptosis-inducing components. Two reports showed that ROCK1 and RACK1 can be released from disassembled SGs and activate JNK to trigger cellular apoptosis (14, 15). In this study, MCPIP1 not only suppressed SG formation but also inhibited JNK phosphorylation, suggesting that cellular apoptosis may be triggered by other signaling pathways.
As we and others have reported recently, MCPIP1 is a critical regulator for controlling the inflammatory response and immune homeostasis; this is demonstrated by the finding that mice with an Mcpip1 disruption spontaneously develop inflammatory syndrome and the autoimmune response (19, 20). The mechanisms by which MCPIP1 controls the inflammatory response may be involved in repressing inflammatory signaling through its DUB activity and promoting inflammatory mRNA degradation through its RNase activity. As apoptosis is also a major mechanism used to resolve inflammation, our study suggests another mechanism, that MCPIP1 may control inflammation by suppressing SG formation, which determines macrophage apoptosis under oxidative stress. In summary, we have reported here that MCPIP1 functions as a factor to coordinate formation and apoptosis during cellular stress, which may be implicated in the resolution of macrophage inflammation and immune homeostasis.
Acknowledgments
We thank Drs. D. Weil, A. Helenius and W. Mothes for providing plasmids used in this work.
This work was supported, in whole or in part, by National Institutes of Health Grants HL-098794 (to M. F.), CA-137126 and AR-055353 (to J. L.), HL-106325 and RR-016434 (to D. F.), and HL-092421, HL-068878, and HL-89544 (to Y. E. C.). This work was also supported by an American Heart Association beginning grant-in-aid award (to M. F.).
- SG
- stress granule
- RFP
- red fluorescent protein
- hDCP1α
- human DCP1α
- PABP
- poly(A)-binding protein
- 15d-PGJ2
- 15-deoxy-Δ12,14-prostaglandin J2
- AHA
- l-azidohomoalanine
- P body
- processing body
- DUB
- deubiquitinating enzyme
- GW
- glycine-tryptophan.
REFERENCES
- 1. Mosser D. D., Morimoto R. I. (2004) Oncogene 23, 2907–2918 [DOI] [PubMed] [Google Scholar]
- 2. Kedersha N., Anderson P. (2002) Biochem. Soc. Trans. 30, 963–969 [DOI] [PubMed] [Google Scholar]
- 3. Thomas M. G., Loschi M., Desbats M. A., Boccaccio G. L. (2011) Cell. Signal. 23, 324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buchan J. R., Parker R. (2009) Mol. Cell 36, 932–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anderson P., Kedersha N. (2009) Nat. Rev. Mol. Cell Biol. 10, 430–436 [DOI] [PubMed] [Google Scholar]
- 6. Gilks N., Kedersha N., Ayodele M., Shen L., Stoecklin G., Dember L. M., Anderson P. (2004) Mol. Biol. Cell 15, 5383–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kedersha N., Chen S., Gilks N., Li W., Miller I. J., Stahl J., Anderson P. (2002) Mol. Biol. Cell 13, 195–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kedersha N. L., Gupta M., Li W., Miller I., Anderson P. (1999) J. Cell Biol. 147, 1431–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mazroui R., Huot M. E., Tremblay S., Filion C., Labelle Y., Khandjian E. W. (2002) Hum. Mol. Genet. 11, 3007–3017 [DOI] [PubMed] [Google Scholar]
- 10. Tourrière H., Chebli K., Zekri L., Courselaud B., Blanchard J. M., Bertrand E., Tazi J. (2003) J. Cell Biol. 160, 823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11. Wilczynska A., Aigueperse C., Kress M., Dautry F., Weil D. (2005) J. Cell Sci. 118, 981–992 [DOI] [PubMed] [Google Scholar]
- 12. Garrido C., Gurbuxani S., Ravagnan L., Kroemer G. (2001) Biochem. Biophys. Res. Commun. 286, 433–442 [DOI] [PubMed] [Google Scholar]
- 13. Candé C., Vahsen N., Métivier D., Tourrière H., Chebli K., Garrido C., Tazi J., Kroemer G. (2004) J. Cell Sci. 117, 4461–4468 [DOI] [PubMed] [Google Scholar]
- 14. Tsai N. P., Wei L. N. (2010) Cell. Signal. 22, 668–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arimoto K., Fukuda H., Imajoh-Ohmi S., Saito H., Takekawa M. (2008) Nat. Cell Biol. 10, 1324–1332 [DOI] [PubMed] [Google Scholar]
- 16. Zhou L., Azfer A., Niu J., Graham S., Choudhury M., Adamski F. M., Younce C., Binkley P. F., Kolattukudy P. E. (2006) Circ. Res. 98, 1177–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liang J., Song W., Tromp G., Kolattukudy P. E., Fu M. (2008) PLoS ONE 3, e2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liang J., Wang J., Azfer A., Song W., Tromp G., Kolattukudy P. E., Fu M. (2008) J. Biol. Chem. 283, 6337–6346 [DOI] [PubMed] [Google Scholar]
- 19. Liang J., Saad Y., Lei T., Wang J., Qi D., Yang Q., Kolattukudy P. E., Fu M. (2010) J. Exp. Med. 207, 2959–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsushita K., Takeuchi O., Standley D. M., Kumagai Y., Kawagoe T., Miyake T., Satoh T., Kato H., Tsujimura T., Nakamura H., Akira S. (2009) Nature 458, 1185–1190 [DOI] [PubMed] [Google Scholar]
- 21. Caton M. L., Smith-Raska M. R., Reizis B. (2007) J. Exp. Med. 204, 1653–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Serman A., Le Roy F., Aigueperse C., Kress M., Dautry F., Weil D. (2007) Nucleic Acids Res. 35, 4715–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vonderheit A., Helenius A. (2005) PLoS Biol. 3, e233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sherer N. M., Lehmann M. J., Jimenez-Soto L. F., Ingmundson A., Horner S. M., Cicchetti G., Allen P. G., Pypaert M., Cunningham J. M., Mothes W. (2003) Traffic 4, 785–801 [DOI] [PubMed] [Google Scholar]
- 25. Parker R., Sheth U. (2007) Mol. Cell 25, 635–646 [DOI] [PubMed] [Google Scholar]
- 26. Dieterich D. C., Link A. J., Graumann J., Tirrell D. A., Schuman E. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 9482–9487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dieterich D. C., Lee J. J., Link A. J., Graumann J., Tirrell D. A., Schuman E. M. (2007) Nat. Protoc. 2, 532–540 [DOI] [PubMed] [Google Scholar]
- 28. Kim W. J., Kim J. H., Jang S. K. (2007) EMBO J. 26, 5020–5032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kedersha N., Stoecklin G., Ayodele M., Yacono P., Lykke-Andersen J., Fritzler M. J., Scheuner D., Kaufman R. J., Golan D. E., Anderson P. (2005) J. Cell Biol. 169, 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jakymiw A., Lian S., Eystathioy T., Li S., Satoh M., Hamel J. C., Fritzler M. J., Chan E. K. (2005) Nat. Cell Biol. 7, 1267–1274 [DOI] [PubMed] [Google Scholar]
- 31. Sen G. L., Blau H. M. (2005) Nat. Cell Biol. 7, 633–636 [DOI] [PubMed] [Google Scholar]
- 32. Rossi J. J. (2005) Nat. Cell Biol. 7, 643–644 [DOI] [PubMed] [Google Scholar]
- 33. Chang W. L., Tarn W. Y. (2009) Nucleic Acids Res. 37, 6600–6612 [DOI] [PMC free article] [PubMed] [Google Scholar]