

Abstract
Background. The mechanisms responsible for interferon α (IFN-α) production by plasmacytoid dendritic cells (pDCs) during human immunodeficiency virus type 1 (HIV-1) infection are unknown. This research examined the roles of Toll-like receptor 7 (TLR7) and autophagy in IFN-α production by pDCs during HIV-1 infection.
Methods. pDCs from human peripheral blood mononuclear cells were incubated with infectious or aldrithiol 2 (AT-2)–inactivated HIV-1 or with uridine-rich single-stranded RNA40 (ssRNA40) from the HIV-1 long terminal repeat. IFN-α was quantified by enzyme-linked immunosorbant assay. Autophagic proteins were detected by Western blot, and autophagosomes were identified using immunofluorescent and confocal microscopy. To inhibit autophagy, pDCs were treated with the phosphoinositide-3 kinase inhibitor 3-methyladenine (3-MA) or were transfected with autophagy-related protein 7 or TLR7 small interfering RNA (siRNA).
Results. Increased levels of IFN-α were present in culture supernatants following 16-hour incubation of pDCs with infectious or AT-2–inactivated HIV-1. Treatment of pDCs with ssRNA40 but not ssRNA41 resulted in high levels of IFN-α. pDCs exposed to HIV-1 gp120, rapamycin, or 3-MA alone failed to induce IFN-α. Pretreatment of pDCs with 3-MA significantly reduced the induction of IFN-α by ssRNA40. Similarly, knock down of autophagy-related protein 7 and TLR7 by use of siRNA significantly reduced the induction of IFN-α by ssRNA40 or HIV-1.
Conclusions. These findings demonstrate that IFN-α production by pDCs exposed to infectious or noninfectious HIV-1 and ssRNA40 occurs through induction of autophagy following TLR7 signaling.
Macroautophagy (autophagy) is increasingly recognized as a critical component of the innate immune response against intracellular microbial pathogens. Recently, our laboratory identified an important role for autophagy in human immunodeficiency type 1 (HIV-1) infection and pathogenesis [1, 2]. During primary infection of CD4+ T lymphocytes and macrophages, HIV-1 downregulates autophagy [1, 3–6]. Reduced autophagy enables HIV-1 to remain within nonacidified endosomes, preventing killing of the virus and prolonging survival of infected cells [7]. In contrast to permissive HIV-1 infection, inactivated HIV-1 or gp120 can induce autophagy in susceptible cells, including bystander CD4+ lymphocytes and neurons, leading to cell death [1, 7, 8].
Human plasmacytoid dendritic cells (pDCs) represent 0.2%–0.5% of circulating peripheral blood mononuclear cells (PBMCs) and are known for their ability to produce large quantities of type I interferons (IFNs), up to 1000-fold more IFN-α than other cell types in response to stimulation by DNA or RNA viruses [9–11]. In response to a wide range of enveloped viruses, including HIV-1, pDCs respond with production of a broad range of IFN-α subtypes, as well as with IFN-β, IFN-k, IFN-w, and IFN-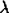 ; proinflammatory cytokines, including tumor necrosis factor α (TNF-α) and interleukin 6 (IL-6); and chemokines, including CXCL10, CCL4, and CCL5 [12]. Moreover, pDCs serve as a critical link between innate and adaptive immunity.
; proinflammatory cytokines, including tumor necrosis factor α (TNF-α) and interleukin 6 (IL-6); and chemokines, including CXCL10, CCL4, and CCL5 [12]. Moreover, pDCs serve as a critical link between innate and adaptive immunity.
HIV-1 induction of IFN-α by pDCs appears to be dependent on the interaction between gp160 and CD4 that orchestrates viral endocytosis and acidification. Once inside the endosome, HIV-1 RNA signals through Toll-like receptor 7 (TLR7), resulting in increased production of IFN-α [8, 13–19]. Whether pDCs are permissive for HIV-1 infection remains controversial, with evidence that, during infection, upregulation of the restriction factor APOBEC3G inhibits HIV-1 replication [5, 20–23]. Additionally, recently the cellular SAM domain HD domain-containing protein 1 has been identified as an important HIV-1 restriction factor present in dendritic and myeloid cells that makes them less permissive to HIV-1 infection [24, 25].
In the present study, we have examined the role of autophagy in the induction of IFN-α by pDCs. Our findings indicate that autophagy is critical for the production of IFN-α by human pDCs following challenge with HIV-1 and that signaling through TLR7 regulates this process.
MATERIALS AND METHODS
Cell Preparations From Peripheral Blood
pDCs were obtained from human PBMCs, using the Plasmacytoid Dendritic Cell Isolation Kit (MACS; MiltenyiBiotec) according to the manufacturer’s instructions. The isolated pDCs were CD303 (BDCA-2)+, CD304 (BDCA-4)+, CD123+, CD4+, CD45RA+, and CD141 (BDCA-3)dim. All lacked expression of lineage markers (CD3, CD14, CD16, CD19, CD20, and CD56) and expressed neither myeloid markers, such as CD13 and CD33, nor Fc receptors, such as CD32, CD64, and FcεRI. The purity of isolated pDCs was >90%, as determined by flow cytometric analysis (FACSCalibur; BD Biosciences) with anti–BDCA-2 (CD303) staining.
HIV-1 Infection
HIV-1MN was propagated in H9 cells. The inactivated HIV-1MN was prepared by treating virus stocks with 1 mM aldrithiol 2 (AT-2) for 1 hour at 37°C, as previously described [1, 26]. A total of 1 × 105 pDCs per well in 96-well plates were exposed to infectious HIV-1 or inactivated AT-2 HIV-1 at a 500-ng/mL p24 antigen equivalent at 37°C and 5% CO2 for 16 hours.
pDC Treatment With Single-Stranded RNA 40 (ssRNA40)
pDCs were treated with ssRNA40 from the HIV-1 LTR (2 μg/mL) or with ssRNA41 (2 μg/mL) linked to N-[1-(2,3-dioleoyloxy) propyl]-N,N,N-trimethylammoniummethyl sulfate, using DOTAP Liposomal Transfection Reagent (Roche Diagnostics Corporation) according to the manufacturer’s instructions. Both ssRNA40 (5′-GCCCGUCUGUUGUGUGACUC-3′) and ssRNA41, in which uracil is replaced with adenosine (5′-GCCCGACAGAAGAGAGACAC-3′), were purchased from InvivoGen (San Diego, CA). Medium alone also served as a negative control [1, 26]. In all instances, transfection and cell survival were evaluated 24 hours after transfection. Transfection rates were expressed as the ratio between the number of transfected (ie, fluorescein-labeled) cells and total number of viable cells measured via DAPI nuclear staining and trypan blue exclusion test.
Gene Silencing via the DOTAP Reagent in pDCs
Freshly isolated pDCs were resuspended in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal calf serum (FCS) and subjected to transfection using a predesigned small interfering RNA (siRNA). Cells subjected to mock-siRNA and scrambled-siRNA transfection under the same experimental conditions were employed as controls. The procedure, run in a 96-well plate format or 24-well plate via triplicate tests, was performed using a 25-nM final siRNA concentration. Each well contained 0.5 × 105 cells in a volume of 100 μL, for the 96-well plates, and 2.0 × 105 cells in a volume of 500 μL, for the 24-well plates. The siRNAs were combined with DOTAP (Roche Diagnostics) and maintained for 30 minutes at room temperature to form complexes. The mixture (50 μL) was then overlaid in a dropwise motion on the cell cultures. Following 4 hours of incubation, 1.2 mL of RPMI 1640 medium containing 10% FCS was added to each well [27]. After 10 hours, cells were subjected to subsequent treatments.
Autophagy Induction and Inhibition
Autophagy was induced either by starvation of cells in EBSS alone or by treatment with rapamycin 200 nM. To inhibit autophagy, pDCs were treated with the specific phosphoinositide 3 kinase (PI3K) class III inhibitor, 3-methyladenine (3-MA) 10 nM.
Measurement of IFN-α and Western Blot Analysis
IFN-α in culture supernatants was measured by enzyme-linked immunosorbant assay (PBL Biomedical Laboratories). For Western blot analysis, cytoplasmic proteins were extracted from pDCs with M-PER reagent (ThermoFisher Scientific), separated on a 12% gradient Tris–glycine gel (Invitrogen), and blotted to polyvinylidine difluoride membrane, using a Western Breezechemiluminescent immunodetection kit (Invitrogen). Band intensity on exposed film was semiquantified using ImageJ software (National Institutes of Health). Autophagy-related protein 7 (ATG7), GAPDH, and MAP-LC3 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Intracellular Staining of LC3 Protein and Visualization of Autophagosomes
After autophagy induction, cells were fixed with 4% paraformaldehyde (PFA) and permeabilized with 0.1% Triton X-100. Rabbit anti-human LC3 monoclonal antibodies (Abgent) and Alexa Fluor 488–conjugated goat anti-rabbit monoclonal antibodies (Invitrogen) were used to detect autophagosomes in pDCs mounted on a coverslip. The number and volume of fluorescent-stained LC3-positive puncta per cell were determined using an Olympus Disk Scan confocal microscope system (Olympus America).
Imaging of IFN Regulatory Transcription Factor 7 (IRF7)
Enriched pDCs were stimulated with ssRNA40 (2 μg/mL) for 12 hours in the presence or absence of rapamycin (200 nM) at 37°C. pDCs were fixed with 4% PFA (BioLegend) for 10 minutes at room temperature and permeabilized with Perm/Wash buffer (BD Biosciences). Cells were blocked with 20% goat serum–containing blocking buffer prior to incubation with rabbit anti-IRF7 (Santa Cruz) at 4°C overnight. After washing, the cells were incubated with biotin-conjugated goat anti-rabbit immunoglobulin G (IgG; Santa Cruz), followed by the addition of strepavidin-APC (BD Biosciences). Cells were analyzed by confocal microscopy.
Flow Cytometry for LC3B and IRF7
Enriched pDCs were stimulated with ssRNA40 (2 μg/mL) for 12 hours in the presence or absence of rapamycin (200 nM) or 3-MA (10 mM) at 37°C. For TLR7 staining, cells were fixed and permeabilized with Cytofix/CytoPerm (BD Biosciences) and stained with anti-TLR7 mouse monoclonal antibody (Imgenex). To detect the coexpression of LC3B puncta and IRF7 phosphorylation, after 12 hours stimulation, cells were fixed with Cytofix buffer (BD Biosciences) and permeabilized with Phosflow Perm buffer III (BD Biosciences). Cells were then stained with Alexafluor 647 mouse anti-IRF7 (BD Biosciences) and anti-LC3B rabbit monoclonal IgG (Cell Signaling), biotin-conjugated goat anti-rabbit IgG (Santa Cruz), and strepavidin-FITC (BD Biosciences).
Statistical Analysis
All samples were performed in at least duplicate, with each experiment repeated at least twice. Results are expressed as means ± standard errors of the mean. Differences between groups were determined using an unpaired Student t test (with a confidence level of 95%) or 1-way analysis of variance with GraphPad Prism software, version 5.01. Two-tailed P values of <.05 were considered statistically significant.
RESULTS
IFN-α Induction in pDCs Exposed to ssRNA40 or HIV-1 Is Inhibited by TLR7 Knockdown
To determine the extent to which IFN-α production by pDCs exposed to ssRNA40 or HIV-1 is mediated through TLR7 signaling, the TLR7 gene was knocked down in pDCs. TLR7 siRNA or scrambled siRNA was transfected into pDCs, followed by stimulation with ssRNA40 or ssRNA41 for 16 hours. TLR7 siRNA transfection into pDCs resulted in marked reduction in TLR7 expression levels, as determined by Western blot analysis (Figure 1A). The amount of IFN-α released into culture supernatants was significantly decreased in TLR7 siRNA–treated pDCs followed by ssRNA40 treatment, compared with those treated with scrambled siRNA (Figure 1B). Medium alone or ssRNA41, in which uracil is replaced with adenosine, did not induce IFN-α secretion by pDCs (Figure 1B). Similarly, when pDCs transfected with TLR7 siRNA were exposed for 16 hours to infectious HIV-1 or HIV-1 inactivated by AT-2 treatment, the induction of IFN-α was significantly inhibited, compared with pDCs transfected with scrambled siRNA (Figure 1C and 1D, respectively).
Figure 1.

Knock down of Toll-like receptor 7 (TLR7) reduces interferon α (IFN-α) production by plasmacytoid dendritic cells (pDCs) following treatment with human immunodeficiency virus type 1 (HIV-1) long terminal repeat single-stranded RNA40 (ssRNA40) or with infectious or noninfectious HIV-1. TLR7 small interfering RNA (siRNA) or scrambled siRNA (5 mM) were transfected into pDCs by using DOTAP liposomal transfection reagent, followed by treatments with ssRNA40 or ssRNA41 as a control (2 μg/mL), or with infectious virus (HIV-1) or noninfectious virus (aldrithiol 2 [AT-2] HIV-1) treated with 1 mM AT-2 for 1 h at 37°C. A, The silenced expression of TLR7 protein was detected by Western blot analysis. B, IFN-α production in supernatant of TLR7-silenced pDCs treated with ssRNA40 or ssRNA41 was determined by enzyme-linked immunosorbant assay (ELISA). C and D, IFN-α production in supernatant of TLR7-silenced pDCs treated for 16 h with infectious (HIV-1) or noninfectious (AT-2 HIV-1) virus at a 500-ng/mL concentration of p24 antigens was also determined by ELISA. The error bars shown are means ± standard errors of the means (SEM) of triplicate cultures of pDCs.
TLR7 siRNA Inhibits Autophagic Protein Expression and Autophagosome Formation
During autophagy, cytosolic LC3B-I is converted to LC3B-II through lipidation by a ubiquitin-like system that involves ATG7, ATG3, and the ATG12-ATG5 complex. Therefore, to examine the association of autophagy following TLR7 activation in pDCs, levels of ATG7 and LC3B-II were detected by Western blot (Figure 2A). Our findings indicate that, compared with ssRNA41, ssRNA40 induced increased expression of ATG7 in pDCs. Additionally, there was increased autophagic flux with conversion from LC3B-I to the lipidated form, LC3B-II (Figure 2A, lanes 1 and 2) [28, 29]. To further assess the engagement of TLR7 signaling by ssRNA40 in autophagy induction in pDCs, the TLR7 gene was knocked down by siRNA, and the expression levels of ATG7 and LC3B conversion were determined by Western blot analysis. In these experiments, the increase in expression of ATG7 and in the LC3II:LC3I ratio from pDCs stimulated with ssRNA40 was inhibited by TLR7 siRNA but had no effect on scrambled siRNA (Figure 2A, lanes 3 and 4).
Figure 2.

Toll-like receptor 7 (TLR7) small interfering RNA (siRNA) inhibits autophagy protein expression and autophagosome formation in plasmacytoid dendritic cells (pDCs). pDCs were transfected with TLR7 siRNA or scrambled siRNA (5 mM), followed by treatment with single-stranded RNA40 (ssRNA40) or ssRNA41 (2 μg/mL) for 16 h. A, Autophagy-related protein 7 and the lipidated form of LC3 protein conversion were detected by Western blot with GAPDH as an internal control. B, Paraformaldehyde-fixed pDCs were permeabilized, and LC3B puncta within autophagosomes were stained with Alexa Fluor 488–conjugated goat anti-rabbit monoclonal antibodies and visualized by fluorescent microscopy. C, The number of puncta formed in autophagosomes in pDCs was quantified.
We next examined the number of LC3B puncta formed in autophagosomes to monitor autophagic flux in pDCs on TLR7 stimulation by means of intracellular staining, using Alexa Fluor 488 conjugated to rabbit anti-human LC3 antibodies. A marked increase in LC3B-II green fluorescent puncta, representing increased autophagosomes, was observed in ssRNA40-treated pDCs, compared with ssRNA41-treated cells (Figure 2B, lane 1 and 2). To further assess the involvement of TLR7 signaling in LC3B puncta formation, TLR7 gene expression in pDCs was silenced by siRNA, and puncta formation was determined. TLR7 siRNA downregulated autophagosome formation induced by ssRNA40 (Figure 2B, lane 3), whereas scrambled siRNA did not affect ssRNA40-induced autophagosome formation (Figure 2B, lane 4). Similarly, the number of LC3B puncta per pDC on exposure to ssRNA40 was significantly higher in pDCs transfected with scrambled siRNA (P < .01), compared with cells transfected with TLR7 siRNA (Figure 2C). These results suggest that the induction of autophagy is closely linked with TLR7 signaling in human pDCs.
Knock Down of ATG7 Inhibits IFN-α Expression in pDCs
To determine whether autophagy is required for the induction of IFN-α, pDCs were treated with ATG7 siRNA or scrambled siRNA (Figure 3A) as described previously, and after 16 hours of exposure to ssRNA40, IFN-α was measured in supernatants. Compared with pDCs treated with scrambled siRNA, the amount of IFN-α released into cultures of ATG7-silenced pDCs treated with ssRNA40 was significantly reduced (P < .01; Figure 3B). Similar results were obtained when ATG7-silenced pDCs were exposed to either infectious HIV-1 or inactivated AT-2 HIV-1 (Figures 3C and 3D, respectively). These findings demonstrate that when ATG7 is knocked down, IFN-α production in pDCs through TLR7 signaling is markedly reduced, suggesting that autophagy is required for the induction of IFN-α by pDCs during viral infection.
Figure 3.

Knock down of autophagy-related protein 7 (ATG7) reduces interferon α (IFN-α) production by plasmacytoid dendritic cells (pDCs) following treatment with ssRNA40, or with infectious or noninfectious human immunodeficiency virus type 1 (HIV-1). ATG7 siRNA (2 μg/mL) or scrambled siRNA (2 μg/mL) was transfected into pDCs, using DOTAP liposomal transfection reagent kit, followed by treatments with ssRNA40 or ssRNA41 as a control or with infectious virus (HIV-1) or noninfectious virus (aldrithiol 2 [AT-2] HIV-1) treated with 1 mM AT-2 for 1 h at 37°C. A, ATG7 protein detected by Western blot analysis. B, IFN-α production in supernatant of ATG7-silenced pDCs treated with ssRNA40 or ssRNA41 was determined by enzyme-linked immunosorbant assay (ELISA). C and D, IFN-α production in supernatant of ATG7-silenced pDCs treated for 16 h with infectious (HIV) or noninfectious (AT-2 HIV) virus at a 500 ng/mL of p24 antigens was also determined by ELISA. The error bars shown are means ± standard errors of the mean of triplicate cultures of pDCs.
IFN-α Induction in pDCs by Infectious or Noninfectious HIV-1 Is Dependent On Autophagy
To determine whether infectious virus is required for induction of autophagy with subsequent production of IFN-α, pDCs were treated with infectious HIV-1 or noninfectious AT-2 HIV-1 for 16 hours. Following exposure to infectious virus, a marked increase in IFN-α was observed in pDC cultures, compared with uninfected pDCs in medium alone (Figure 4A). IFN-α was detected in culture supernatants at equivalent levels in pDCs exposed to noninfectious HIV-1 alone (Figure 4B). Although these findings indicate that infectious HIV-1 is not required for induction of autophagy and IFN-α, when pDCs were treated with HIV-1 gp120 alone, there was no induction of autophagy or IFN-α (Figure 4C).
Figure 4.

The phosphoinositide 3 kinase inhibitor 3-methyladenine (3-MA) and rapamycin inhibit interferon α (IFN-α) production in plasmacytoid dendritic cells (pDCs) treated with infectious or noninfectious human immunodeficiency virus type 1 (HIV-1) or with HIV long terminal repeat single-stranded RNA40 (ssRNA40). Isolated pDCs were exposed for 16 h to infectious HIV-1, noninfectious HIV-1 (aldrithiol 2 [AT-2] HIV-1), ssRNA40, or ssRNA41 in the presence or absence of 3-MA (10 mM). 3-MA treatment occurred 30 min prior to pDC culture with HIV-1 or ssRNAs. In addition, rapamycin (200 nM) was added to pDC culture as a control of autophagy induction. A, HIV-1 at 500 ng/mL of p24 antigen in the presence or absence of 3-MA (10 mM). B, AT-2 HIV-1 (AT-2 HIV, 500 ng/mL of p24) in the presence or absence of 3-MA (10 mM). C, ssRNA40 (RNA40, 2 μg/mL) and ssRNA41 (RNA41, 2 μg/mL) in the presence or absence of 3-MA (10 mM) and rapamycin (Rap, 200 nM). D, HIV-1 at 500 ng/mL of p24 antigen in the presence or absence of rapamycin (200 nM). pDCs cultured with medium, 3-MA, or rapamycin alone were used as negative controls. The error bars shown are means ± standard errors of the mean of triplicate cultures of pDCs.
To assess whether the inhibition of autophagy interferes with IFN-α induction by HIV-1, pDCs were pretreated with 3-MA for 30 minutes prior to addition of HIV-1, AT-2 HIV-1, or ssRNA40. Pretreatment with 3-MA significantly inhibited the quantity of IFN-α released into the supernatants in cultures exposed to infectious HIV-1 (P < .01), AT-2 HIV-1 (P < .001), and ssRNA40 (P < .001), whereas 3-MA alone had no effect on IFN-α production in untreated pDCs (Figure 4A–C).
TLR7 Expression Is Not Altered by ssRNA40 and Rapamycin
To further identify the role of autophagy in the induction of IFN-α, pDCs were treated with rapamycin prior to exposure to HIV-1 and examined for IFN-α production. Surprisingly, pDCs treated with rapamycin alone failed to produce IFN-α (Figure 4C), and in fact, rapamycin inhibited the production of IFN-α by pDCs exposed to HIV-1 (P < .01) (Figure 4D).
To explain the inhibition of IFN-α production by pDCs in the presence of rapamycin, we hypothesized that the observed inhibition reflected the multifunctional effects of rapamycin, through which interference with the PI3K/Akt/mTOR axis alters several cellular functions, including differentiation, viability, and growth. In our next experiments, we examined the mechanisms associated with the decreased IFN-α production by pDCs following rapamycin treatment. We initially questioned whether pDCs exposed to rapamycin resulted in altered TLR7 expression. For these studies, the intracellular expression of TLR7 in pDCs following exposure to ssRNA40 or ssRNA41 in the presence or absence of rapamycin was determined by flow cytometry. TLR7 constitutive expression in pDCs cultured in medium alone was consistently found to be at higher levels than those in cells cultured in medium with isotype control. No significant differences were found, however, in TLR7 expression by pDCs treated with ssRNA40 or ssRNA41, compared with resting pDCs (Figure 5A). Additionally, rapamycin had no effect on TLR7 expression in pDCs stimulated with ssRNA40 or ssRNA41 (Figure 5B). These data indicate that the altered IFN-α production by rapamycin is not the result of decreased expression of TLR7.
Figure 5.

Rapamycin and single-stranded RNA40 (ssRNA40) have no affect on Toll-like receptor 7 (TLR7) expression. A, Isolated plasmacytoid dendritic cells (pDCs) were stimulated with ssRNA40 or ssRNA41 for 12 h and then stained with TLR7-specific mouse monoclonal antibody to detect intracellular TLR7 expression. pDCs cultured in medium alone were used to determine the endogenous expression of intracellular TLR7. B, The intracellular expression of TLR7 in pDCs stimulated with ssRNA40 or ssRNA41 in the presence or absence of rapamycin (200 nM) was also determined by flow cytometry. The number above the squares represents the percentage of pDCs expressing TLR7. The mean fluorescence intensity of TLR7 expression on pDCs is shown as the means of 2 independent experiments with standard errors of the mean. Statistical significance was assessed using 1-way analysis of variance.
Inhibition of the mTOR Pathway by Rapamycin Impairs IRF7 Activation
Having determined that TLR7 expression on pDCs was not affected by ssRNA40 or rapamycin, we next examined their effect on the expression of IRF7. Activation of pDCs with ssRNA40 for 12 hours induced nuclear translocation of IRF7 between cytoplasm and nucleus, as detected by confocal microscopy. In contrast, the nuclear translocation of IRF7 was impaired when pDCs exposed to ssRNA40 were treated with rapamycin (Figure 6A). When examined by flow cytometry, the expression of phosphorylated IRF7 in pDCs with ssRNA40 in the presence or absence of rapamycin or 3-MA demonstrated that the expression of phosphorylated IRF7 was increased on stimulation with ssRNA40 but was inhibited in the presence of rapamycin. Moreover, 3-MA downregulated the expression of phosphorylated IRF7 in pDCs on ssRNA40 stimulation (Figure 6B).
Figure 6.

mTOR-inhibition by rapamycin impairs interferon (IFN) regulatory transcription factor 7 (IRF7). A, Enriched plasmacytoid dendritic cells (pDCs) were stimulated for 12 h with single-stranded RNA40 (ssRNA40) in the presence or absence of rapamycin (200 nM), followed by fixation with 4% paraformaldehyde and permeabilization with 0.5% saponin. pDCs were stained with anti-IRF7 antibody followed by Alexa Fluor 647–conjugated secondary antibody, mounted with ProLong Gold anti-fade reagent with DAPI, and detected by confocal microscopy. B, Enriched pDCs were stimulated for 12 h with ssRNA40 (2 μg/mL) in the presence or absence of rapamycin (200 nM) or 3-methyladenine (3-MA; 10 mM); cells were fixed, permeabilized, and stained to detect IRF7 phosphorylated at Ser477 and Ser479 (phospho-IRF7) by flow cytometry. MFI, mean fluorescence intensity. C, In enriched pDCs upon stimulation with ssRNA40 in the presence or absence of rapamycin (200 nM) or 3-MA (10 mM), co-expression of LC3B autophagic puncta formation and phosphorylated IRF7 was detected by flow cytometry. The gated population shows bright expression of both LC3B and phosphorylated IRF7, and the numbers next to the square boxes represents the percentage of double-positive gated population. Fluorescence Minus One (FMO), a background fluorescence control, is used to determine positive gates showing expression of LC3B and phosphorylated IRF7. D, The bar chart shows the means of 2 independent experiments with standard errors of the mean, showing the co-expression of LC3B and phospho-IRF7 by flow cytometry. Statistical significance was assessed using 1-way analysis of variance (*P < .05, **P < .01, and ***P < .001, by the Tukey test for multiple comparisons).
To further understand the relationship of autophagic LC3B puncta formation and IRF7 expression in pDCs following TLR7 signaling, the level of co-expression of LC3B puncta and phosphorylated IRF7 in pDCs was examined following ssRNA40 stimulation by flow cytometry. ssRNA40 elicited increased expression of both LC3B and phosphorylated IRF7 in pDCs (31.4%), compared with medium-alone control (7.6%; P < .01). Of note, this expression was partially inhibited by addition of rapamycin (20.2%; P < .05) and, to an even greater extent, by addition of 3-MA (2.2%; P < .001) (Figures 6C and 6D). Our data suggest that IRF7 activation is mTOR dependent; thus, inhibition of mTOR impairs the expression of phosphorylated IRF7, resulting in diminished IFN-α production by pDCs. Moreover, these findings indicate that 3-MA downregulates the expression of phosphorylated IRF7, suggesting that inhibition of autophagy impairs the TLR7 signaling pathway.
DISCUSSION
IFN-α has been identified as a key cytokine in HIV-1 pathogenesis. During acute viral infection, TLR stimulation leads to a rapid increase in IFN-α production, followed by the induction of a cascade of other cytokines, including TNF-α, interleukin 10, and IFN-γ, that are associated with the generation of an adaptive immunologic response [30]. IFN-α has also been shown to inhibit HIV-1 [21]. However, an increase in IFN-α may be a double-edged sword: whereas during acute infection it contributes to the control of viral spread, during chronic infection it may contribute to persistent T-cell activation and CD4+ T-cell depletion [31]. Moreover, the ability of peripheral blood cells to produce IFN-α has been found to be markedly diminished in persons with AIDS, reflecting the loss of pDCs [12].
These studies were designed to examine the mechanisms through which HIV-1 induces the production of IFN-α by pDCs. Our findings, using primary human pDCs, indicate that signaling through TLR7 by HIV-1 results in triggering of autophagy and production of IFN-α. These data provide several insights into HIV-1 pathogenesis. Activation of TLR7 signaling and IFN-α production when pDCs were exposed to noninfectious virus occurred as efficiently as when pDCs were exposed to infectious HIV-1, a result suggesting that viral replication is not necessary to elicit a robust IFN-α response by pDCs. This finding further supports a previous study demonstrating that endocytosis of HIV-1 at entry leads to pDC activation and IFN-α production through TLR7 signaling [15]. It still remains controversial whether gp120 itself, in the absence of viral nucleic acid, can elicit the activation and IFN-α secretion of pDCs [32]. However, our results showing that gp120 alone failed to trigger IFN-α production by pDCs further supports the findings of Martinelli et al [33] that multisubtype IFN-α is not detected in freshly isolated pDCs exposed to gp120 recombinant protein derived from both R5 and X4 HIV-1.
Additionally, our data provide evidence that autophagy is required for the induction of IFN-α in human pDCs exposed to HIV-1. Support for this conclusion includes the following: (1) knock down of autophagic protein ATG7 markedly decreased the ability of pDCs to produce IFN-α on exposure to HIV-1; (2) TLR7 silencing inhibited ATG7 expression and conversion of lipidated LC3B-II and suppressed LC3B puncta formation in autophagosomes; and (3) treatment of pDCs with 3-MA, a PI3K inhibitor and a potent inhibitor of autophagy, failed to induce IFN-α production in pDCs following treatment with ssRNA40 or HIV-1.
The TLR7 and TLR9 signaling–mediated production of type I IFN in pDCs require MyD88 activation, which is subsequently followed by phosphorylation and nuclear translocation of IRF7 [34–36]. TLR7/8 triggering has been shown to inhibit HIV-1 replication while also inducing the release of HIV virions from latently infected cells [37]. Our finding that the knock down of autophagic protein ATG7 markedly reduces IFN-α production induced by HIV-1 in pDCs provides additional support for an important role of autophagy in IFN-α production in pDCs. These findings are consistent with those of Lee et al [38], who observed with vesicular stomatitis virus and influenza virus in murine pDCs that the ability of pDCs to produce IFN-α on viral recognition via TLR7 or TLR9 signaling is defective in the absence of autophagic protein ATG5. These results, when combined with our findings, suggest that TLR7-mediated IFN-α production in human pDCs occurs through an autophagy-dependent pathway.
The serine-threonine protein kinase mTOR is a downstream mediator of the PI3K-Akt signaling pathway and a key regulator of a broad range of cellular processes, such as cell growth, proliferation, aging, synaptic plasticity, memory metabolism, and disease development [39]. mTOR responds to signals from extracellular stimuli, such as growth factors and nutrients, or to intracellular cues, such as energy status and oxygen [40]. An important molecular mechanism of mTOR activity is the regulation of translation, which is central to gene expression [39]. Cao et al [41] demonstrated that the TLR9-mediated production of IFN-α in pDCs is mTOR dependent, and use of mouse pDCs demonstrated that phosphorylation of mTOR and its downstream mediators, elF4E binding protein and S6 kinases 1 and 2, proteins that are critical for the control of translation rates, are blocked by rapamycin, resulting in a decrease in IFN-α, TNF-α, and interleukin 6. In addition, a DNA microarray using pDCs stimulated by a TLR9 ligand showed that a subset of genes encoding interferons and cytokines was repressed by rapamycin [41]. Our results demonstrating that rapamycin failed to induce IFN-α production in pDCs and inhibited the ability of pDCs to produce IFN-α on HIV-1 infection are consistent with the findings of Cao et al [41]. Although rapamycin is a well-known potent autophagy inducer, it is also a strong immunosuppressant, a property often exploited in allogeneic transplantation or anticancer treatment [42]. It appears that the downregulation of IRF by rapamycin in pDCs overrides its role as a potent inducer of autophagy, resulting in the inhibition of IFN-α production.
In summary, our findings demonstrate that infectious or noninfectious HIV-1, through TLR7 signaling, induces pDCs to produce IFN-α through an autophagy-dependent pathway. The modulation of autophagy in pDCs, leading to the production of IFN-α and, subsequently, other cytokines, likely plays a central role in T-cell activation and HIV-1 pathogenesis and provides insights into possible new targets to control HIV-1 infection.
Notes
Acknowledgments.
We thank Jennifer Meerloo of the University of California–San Diego Neuroscience Microscopy Center for technical support.
Financial support.
This work was supported in part by a grant from the National Institute of Allergy and Infectious Diseases to the IMPAACT Network (AI068632 and R21 AI084573).
Potential conflicts of interest.
All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Zhou D, Spector SA. Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS. 2008;22:695–9. doi: 10.1097/QAD.0b013e3282f4a836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou D, Masliah E, Spector SA. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J Infect Dis. 2011;203:1647–57. doi: 10.1093/infdis/jir163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanchet FP, Moris A, Nikolic DS, et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity. 2010;32:654–69. doi: 10.1016/j.immuni.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell GR, Spector SA. Hormonally active vitamin D3 (1alpha, 25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J Biol Chem. 2011;286:18890–902. doi: 10.1074/jbc.M110.206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheney KM, McKnight A. Interferon-alpha mediates restriction of human immunodeficiency virus type-1 replication in primary human macrophages at an early stage of replication. PLoS One. 2010;5:e13521. doi: 10.1371/journal.pone.0013521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Grol J, Subauste C, Andrade RM, Fujinaga K, Nelson J, Subauste CS. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS One. 2010;5:e11733. doi: 10.1371/journal.pone.0011733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Espert L, Varbanov M, Robert-Hebmann V, et al. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS One. 2009;4:e5787. doi: 10.1371/journal.pone.0005787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Espert L, Denizot M, Grimaldi M, et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest. 2006;116:2161–72. doi: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fanning SL, George TC, Feng D, et al. Receptor cross-linking on human plasmacytoid dendritic cells leads to the regulation of IFN-alpha production. J Immunol. 2006;177:5829–39. doi: 10.4049/jimmunol.177.9.5829. [DOI] [PubMed] [Google Scholar]
- 10.Fitzgerald-Bocarsly P, Dai J, Singh S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 2008;19:3–19. doi: 10.1016/j.cytogfr.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siegal FP, Kadowaki N, Shodell M, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–7. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 12.Fitzgerald-Bocarsly P, Jacobs ES. Plasmacytoid dendritic cells in HIV infection: striking a delicate balance. J Leukoc Biol. 2010;87:609–20. doi: 10.1189/jlb.0909635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hornung V, Rothenfusser S, Britsch S, et al. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–7. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 14.Asselin-Paturel C, Brizard G, Chemin K, et al. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J Exp Med. 2005;201:1157–67. doi: 10.1084/jem.20041930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beignon AS, McKenna K, Skoberne M, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. 2005;115:3265–75. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferbas JJ, Toso JF, Logar AJ, Navratil JS, Rinaldo CR., Jr CD4+ blood dendritic cells are potent producers of IFN-alpha in response to in vitro HIV-1 infection. J Immunol. 1994;152:4649–62. [PubMed] [Google Scholar]
- 17.Schmidt B, Ashlock BM, Foster H, Fujimura SH, Levy JA. HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology. 2005;343:256–66. doi: 10.1016/j.virol.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt B, Scott I, Whitmore RG, et al. Low-level HIV infection of plasmacytoid dendritic cells: onset of cytopathic effects and cell death after PDC maturation. Virology. 2004;329:280–8. doi: 10.1016/j.virol.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 19.Langenkamp A, Nagata K, Murphy K, Wu L, Lanzavecchia A, Sallusto F. Kinetics and expression patterns of chemokine receptors in human CD4+ T lymphocytes primed by myeloid or plasmacytoid dendritic cells. Eur J Immunol. 2003;33:474–82. doi: 10.1002/immu.200310023. [DOI] [PubMed] [Google Scholar]
- 20.Trapp S, Derby NR, Singer R, et al. Double-stranded RNA analog poly(I: C) inhibits human immunodeficiency virus amplification in dendritic cells via type I interferon-mediated activation of APOBEC3G. J Virol. 2009;83:884–95. doi: 10.1128/JVI.00023-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang FX, Huang J, Zhang H, Ma X. APOBEC3G upregulation by alpha interferon restricts human immunodeficiency virus type 1 infection in human peripheral plasmacytoid dendritic cells. J Gen Virol. 2008;89:722–30. doi: 10.1099/vir.0.83530-0. [DOI] [PubMed] [Google Scholar]
- 22.Levy JA. HIV pathogenesis: knowledge gained after two decades of research. Adv Dent Res. 2006;19:10–6. doi: 10.1177/154407370601900104. [DOI] [PubMed] [Google Scholar]
- 23.Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol. 2009;83:9474–85. doi: 10.1128/JVI.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hrecka K, Hao C, Gierszewska M, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–61. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laguette N, Sobhian B, Casartelli N, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–7. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossio JL, Esser MT, Suryanarayana K, et al. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J Virol. 1998;72:7992–8001. doi: 10.1128/jvi.72.10.7992-8001.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martino S, di Girolamo I, Tiribuzi R, D'Angelo F, Datti A, Orlacchio A. Efficient siRNA delivery by the cationic liposome DOTAP in human hematopoietic stem cells differentiating into dendritic cells. J Biomed Biotechnol. 2009;2009:410260. doi: 10.1155/2009/410260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. doi: 10.1007/978-3-642-00302-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang JJ, Altfeld M. Innate immune activation in primary HIV-1 infection. J Infect Dis. 2010;202(Suppl 2):S297–301. doi: 10.1086/655657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herbeuval JP, Hardy AW, Boasso A, et al. Regulation of TNF-related apoptosis-inducing ligand on primary CD4+ T cells by HIV-1: role of type I IFN-producing plasmacytoid dendritic cells. Proc Natl Acad Sci U S A. 2005;102:13974–9. doi: 10.1073/pnas.0505251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Del Corno M, Gauzzi MC, Penna G, Belardelli F, Adorini L, Gessani S. Human immunodeficiency virus type 1 gp120 and other activation stimuli are highly effective in triggering alpha interferon and CC chemokine production in circulating plasmacytoid but not myeloid dendritic cells. J Virol. 2005;79:12597–601. doi: 10.1128/JVI.79.19.12597-12601.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinelli E, Cicala C, Van Ryk D, et al. HIV-1 gp120 inhibits TLR9-mediated activation and IFN-{alpha} secretion in plasmacytoid dendritic cells. Proc Natl Acad Sci U S A. 2007;104:3396–401. doi: 10.1073/pnas.0611353104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–21. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carty M, Bowie AG. Recent insights into the role of Toll-like receptors in viral infection. Clin Exp Immunol. 2010;161:397–406. doi: 10.1111/j.1365-2249.2010.04196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 37.Schlaepfer E, Audige A, Joller H, Speck RF. TLR7/8 triggering exerts opposing effects in acute versus latent HIV infection. J Immunol. 2006;176:2888–95. doi: 10.4049/jimmunol.176.5.2888. [DOI] [PubMed] [Google Scholar]
- 38.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 39.Costa-Mattioli M, Sonenberg N. RAPping production of type I interferon in pDCs through mTOR. Nat Immunol. 2008;9:1097–9. doi: 10.1038/ni1008-1097. [DOI] [PubMed] [Google Scholar]
- 40.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 41.Cao W, Manicassamy S, Tang H, et al. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat Immunol. 2008;9:1157–64. doi: 10.1038/ni.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saemann MD, Haidinger M, Hecking M, Horl WH, Weichhart T. The multifunctional role of mTOR in innate immunity: implications for transplant immunity. Am J Transplant. 2009;9:2655–61. doi: 10.1111/j.1600-6143.2009.02832.x. [DOI] [PubMed] [Google Scholar]