

Abstract
UDP-galactopyranose mutase (UGM) is a flavoenzyme that catalyzes the conversion of UDP-galactopyranose to UDP-galactofuranose, the precursor of galactofuranose (Galf). Galf is found in several pathogenic organisms, including the parasite Trypanosoma cruzi, the causative agent of Chagas' disease. Galf) is important for virulence and is not present in humans, making its biosynthetic pathway an attractive target for the development of new drugs against T. cruzi. Although UGMs catalyze a non-redox reaction, the flavin must be in the reduced state for activity and the exact role of the flavin in this reaction is controversial. The kinetic and chemical mechanism of TcUGM was probed using steady state kinetics, trapping of reaction intermediates, rapid reaction kinetics, and fluorescence anisotropy. It was shown for the first time that NADPH is an effective redox partner of TcUGM. The substrate, UDP-galactopyranose, protects the enzyme from reacting with molecular oxygen allowing TcUGM to turnover ∼1000 times for every NADPH oxidized. Spectral changes consistent with a flavin iminium ion, without the formation of a flavin semiquinone, were observed under rapid reaction conditions. These data support the proposal of the flavin acting as a nucleophile. In support of this role, a flavin-galactose adduct was isolated and characterized. A detailed kinetic and chemical mechanism for the unique non-redox reaction of UGM is presented.
Introduction
The protozoan parasite Trypanosoma cruzi is the etiological agent of American trypanosomiasis or Chagas' disease. The World Health Organization estimates that 8–11 million people are infected with this disease in South and Central America [1]. Transmission occurs when a triatomine insect feeds on a human subject and defecates at the bite site [2]. Through rubbing, infected feces enter the bite wound, mouth, eyes, or open cuts. Transmission by blood transfusion, organ transplant, oral contamination, and congenital routes has also been reported [3]–[7]. T. cruzi has a dynamic life cycle, involving several morphological changes as the parasites travel from the insect vector to humans [8]. This is accompanied by several changes in cell surface sugar composition, which plays an important role in infection and resistance to the host immune system [9], [10]. Targeting the enzymes involved in biosynthesis of cell surface glycans may lead to the identification of new inhibitors that function as novel antiparasitic drugs for the treatment of Chagas' disease [11]. One unique sugar found on the cell surface of T. cruzi is galactofuranose (Galf) [11].
Galf, the five-membered ring form of galactose, is a component of the cell wall, glycolipids, and glycoproteins on the cell surface of many human pathogens including bacteria, fungi, and parasites [11]–[14]. In T. cruzi, Galf is found in glycoprotein oligosaccharides and glycoinositolphospholipids, which are involved in parasite pathogenesis [11], [15], [16]. Additionally, Galf is not present in humans. Thus, the biosynthetic pathway of Galf is an attractive drug target for T. cruzi and other eukaryotic pathogens including A. fumigatus and L. major [11], [17]. UDP-galactopyranose mutase (UGM) catalyzes the conversion of UDP-galactopyranose (UDP-Galp) to UDP-galactofuranose (UDP-Galf), the precursor of Galf found on the cell surface (Figure 1) [18]. UGM is a unique flavoprotein, as it requires the flavin to be reduced in order to catalyze a non-redox reaction (Figure 2) [19], [20]. The role of the flavin cofactor in catalysis is controversial. Experimental and structural data supports the role of the flavin acting as a nucleophile [21], [22]. Similarly, studies with flavin analogs and potentiometry experiments suggest that a single electron transfer step is necessary for catalysis [23], [24]. Here, we present a complete characterization of the recombinant form of UGM from T. cruzi (TcUGM). We utilize steady state kinetics, fluorescence anisotropy, rapid reaction kinetics, and the trapping of reaction intermediates to provide a clear view of the kinetic and chemical mechanisms employed by this unique enzyme. We also identify NAD(P)H as an effective electron donor to TcUGM, a function that is unique to eukaryotic UGMs.
Figure 1. Reaction catalyzed by TcUGM.
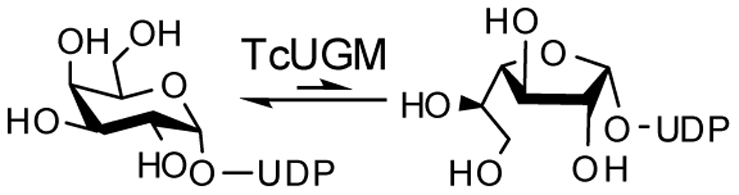
Figure 2. The two proposed chemical mechanisms for UGMs.

In one mechanism the reduced FAD (1) is depicted to act as a nucleophile forming a flavin-galactose adduct (either via SN1 or SN2) (2) and a subsequent iminium ion (3). These steps are followed by ring contraction forming the galactofuranose (4). An alternative mechanism predicts an electron transfer step, in which one electron is transferred from FADH− to an oxocarbenium ion intermediate (6) creating a flavin and a sugar radical, which react and form the galactose-FAD adduct (2), followed by ring contraction.
Materials and Methods
Materials
UDP, UDP-galactopyranose, and BL21-T1R chemical competent cells were purchased from Sigma (St. Louis, MO). Accuprime polymerase and TOP-10 chemical competent cells were obtained from Invitrogen (Carlsbad, CA). The restriction endonucleases SgfI and PmeI were obtained from Promega (Madison, WI). Plasmid miniprep and PCR purification kits were from Qiagen (Valencia, CA). All other buffers and chemicals for biochemical studies were purchased from Fisher Scientific (Pittsburgh, PA). Expression plasmids, pVP55A and pVP56K, were obtained from the Center for Eukaryotic Structural Genomics, University of Wisconsin, Madison [25]. All reagents and solvents used in the synthesis of UDP-Galf, if not specified otherwise, were obtained from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA) and were used without further purification. All the synthetic reactions were monitored to completion using thin layer chromatography (TLC) with p-anisaldehyde staining for visualizing carbohydrates along with UV detection when possible. 1H and 13C NMR were recorded on a 400MR Varian-400 MHz spectrometer. CHCl3 (7.27 ppm) or HOD (4.79 ppm) were used as an internal reference.
Cloning
T. cruzi UGM (TcUGM) was amplified by PCR from genomic DNA using 5′-GGTTGCGATCGCCATGGCAGAATTATTGACACC -3′(SgfI site is underlined) as the forward primer and 5′- CCAAGTTTAAACCATATCCTTCTGCAGTAGTC -3′ (PmeI site is underlined) as the reverse primer. The TcUGM gene was inserted into the pVP55A and pVP56K vectors as previously described [17].
Protein expression and purification
Protein expression and purification of TcUGM was performed following methods previously described for A. fumigatus UGM with an additional final step of size exclusion chromatography in 25 mM HEPES, 125 mM NaCl, pH 7.5 (S-75, GE Healthcare, Piscataway, NJ) [17]. Purified TcUGM was concentrated, flash frozen in liquid N2, and stored at −80°C.
UV–visible absorbance spectrophotometry
The spectrum of recombinant TcUGM was recorded using an Agilent 8453 UV–visible spectrophotometer. The extinction coefficient was determined by dividing the absorbance value at 450 nm of the bound flavin in TcUGM by the absorbance value at 450 nm of free flavin (obtained by heat denaturation and centrifugation of the recombinant enzyme) and multiplying this value by the known extinction coefficient for FAD (εFAD = 11.3 mM−1cm−1) [26].
Solution molecular weight determination
The molecular weight of TcUGM was determined using size exclusion chromatography as previously described [17], [27].
NAD(P)H oxidation assays
Oxidation of NAD(P)H was monitored at 340 nm for 5 min. Reactions were performed at room temperature with air saturated 50 mM sodium phosphate buffer, pH 7.0, with various concentrations of NAD(P)H, in the presence or absence of 0.5 mM UDP-Galp. The reaction was initiated by addition of 1 µM enzyme.
Flavin reduction by NAD(P)H
Flavin reduction by NAD(P)H was performed anaerobically in a glove box (Coy, Grass Lake, MI) using an Applied Photophysics Stopped Flow SX20 (Leatherhead, UK). Reactions were performed at 15°C. Buffer (50 mM sodium phosphate pH 7.0) was made anaerobic by eight cycles of vacuum and argon flushing, each for 45 minutes. After this procedure, protocatechuate (400 µM) and protocatechuate dioxygenase (10 µg/mL) were added to the buffer to function as an oxygen scavenging system. All the solutions were prepared with the anaerobic buffer in the glove box. NAD(P)H solutions were prepared by dissolving the appropriate amounts in anaerobic buffer and concentrations were verified spectroscopically. The enzyme solution was made anaerobic by degassing with six 15 min cycles of vacuum and flushing with anaerobic argon. To ensure complete anaerobiosis, the solution was passed through a 2 mL desalting column previously equilibrated with anaerobic buffer. Enzyme with a final concentration of ∼7.5 µM (after mixing) was mixed with various concentrations of NAD(P)H and the reaction was monitored with a photodiode array spectrophotometer until complete reduction was achieved. Identical experiments were performed in the presence of 0.5 mM UDP-Galp. Change in absorbance at 452 nm was fitted to a single exponential equation and the resulting kobs values were plotted as a function of NAD(P)H concentration. These data was fit with equation 1 to obtain the rate constant for reduction (kred) and the Kd value.
| (1) |
Synthesis of UDP-Galf
Synthesis of α-d-Galactofuranosyl bis(triethylammonium) phosphate (Galf-1-phosphate) was performed over a series of three steps with methods adapted from previously published protocols [28]–[34]. Details of the synthesis and NMR data are outlined in the supplementary material (File S1). UDP-Galf was synthesized following published method reported by Poulin and coworkers [35].
Activity assay
The activity of recombinant TcUGM was tested with UDP-Galf as the substrate following procedures previously described [17]. Concentration of TcUGM was determined based on bound flavin. TcUGM (100 nM) was reduced with either 20 mM dithionite, 500 µM NADPH, or 2.5 mM NADH for each activity assay.
Viscosity Effects
Viscosity effects were determined using the activity assay as described above. Reactions were done in the presence of 0, 5, 10, 20, and 30% glycerol, 800 µM UDP-Galf, 20 mM dithionite, and 200 nM TcUGM. The relative viscosity (ηrel) of the reaction was determined using a reference table for different percentages of glycerol in solution [36].
Fluorescence anisotropy
Synthesis of UDP-rhodamine was performed as previously described [37]. The optimal concentration of TcUGM (30 µM) and UDP-rhodamine (30 nM) for the binding assays was determined following procedures established in our laboratory for a related UGM enzyme from Aspergillus fumigatus [37]. Binding of UDP-Galp to TcUGM was monitored in the presence and absence of 10 mM sodium dithionite by measuring the changes in anisotropy [37]. The Kd values were obtained using the equation 2, where m1 and m2 are the minimum and maximum anisotropy, respectively; m3 is the slope, and m4 is the Kd.
| (2) |
Trapping of the flavin-galactose adduct
The procedure was adapted from Gruber et al. and optimized for trapping and isolation of the adduct in TcUGM [22]. Reactions were prepared using 1 mM TcUGM, 200 mM UDP-Galp, 1 M sodium cyanoborohyride, and 100 mM sodium dithionite in a final volume of 15 µL with 100 mM sodium phosphate buffer at pH 7.0. Reactions were incubated for 45 min at room temperature then terminated with addition of 6 M HCl and neutralized with 1 M sodium phosphate buffer at pH 7.0. A final concentration of 200 mM NaCl was added. The sample was then centrifuged to precipitate denatured protein from the yellow solution containing the free flavin. The adduct was isolated using HPLC (Shimadzu) by injecting the yellow solution onto a reverse phase C18 column (Phenomenex Luna C18 5 microns, 250×4.6 mm) equilibrated with 5% B (A: Water, B: 100% acetonitrile). The column was washed for 5 min with 5% B, then a 15 min linear gradient to 15% B, followed by 20 min linear gradient to 75% B. The flavin species were eluted at ∼20–24 minutes and were characterized by high-resolution mass spectrometry (Virginia Tech Mass Spectrometry incubator).
Reaction of reduced TcUGM with UDP-Galp, UDP, and UDP-glucose
Rapid reaction kinetics were done using the methods described for the TcUGM reduction studies. However, reduced TcUGM was prepared by addition of 20 mM sodium dithionite. Excess dithionite was removed using a 2 mL desalting column. Reduced TcUGM (∼15 µM final concentration) was mixed with buffer alone for a final concentration of 0.15 mM UDP-Galf, 0.25 mM UDP-Galp, 0.25 mM UDP, and 0.25 mM UDP-glucose (UDP-Glc). Spectra were collected on a logarithmic time base from 1.3 ms to 2 s using a photodiode array. The rate of the intermediate formation was calculated by fitting the change in absorbance at 452 nm to a double exponential equation 3.
| (3) |
Results
Expression and Purification
Successful expression of soluble TcUGM was accomplished in the pVP56K vector [17]. The protein was expressed as an N-terminal fusion to maltose binding protein (MBP) with an 8×-His tag, which aids in purification. BL21-T1R cells containing the pVP56K-TcUGM plasmid were grown in auto-induction medium for 8 h at 37°C followed by overnight incubation at 25°C [38]. Approximately 9 g of cell paste per liter of media was obtained and 1 mg of purified TcUGM was isolated for each g of cell paste. Recombinant TcUGM contained noncovalently bound flavin cofactor, which had a typical flavin spectrum with peaks at 376 nm and 448 nm with a shoulder at 468 nm (Figure S1). The extinction coefficient was determined to be 10.8 mM−1 cm−1 at 450 nm.
Determination of the oligomeric state
Size exclusion chromatography was used to calculate a molecular weight of 67,800±10,000 Da for the recombinant TcUGM (Figure S2). This value is consistent with the molecular weight predicted for a single polypeptide (54,700 Da).
Oxidase activity with NADPH and NADH
The activity of all UGMs requires the flavin to be in the reduced state. Although the prokaryotic enzymes have been extensively studied, the mechanism of reduction is yet to be determined [20], [39]. Even though an NAD(P)H binding domain was not found in the primary sequence of TcUGM, we tested whether this enzyme was capable of reacting with reduced dinucleotides. The oxidation of either NADH or NADPH was monitored in the presence and absence of UDP-Galp under aerobic conditions. Under this conditions, no mutase activity is measured, instead what is measured is the steady-state reduction of the flavin cofactor in TcUGM, which is slowly oxidized by molecular oxygen (Figure 3). In the absence of UDP-Galp, a slow oxidase activity, kox, was observed for both coenzymes, while a 6-fold lower KM value was measured for NADPH. In the presence of UDP-Galp, a minor decrease in kox was measured (Table 1). In contrast, when the same activity was measured with recombinant M. tuberculosis UGM (MtUGM), there was no measurable NAD(P)H oxidation (Table 1). E. coli UGM has also been shown to be unable to react with NAD(P)H [20], [39].
Figure 3. Oxidase activity of TcUGM with NAD(P)H.

A) Scheme representing the oxidase activity of TcUGM measured in this assay. B) Activity of TcUGM measured in the presence and absence of saturating concentration of UDP-Galp (0.5 mM) under air saturating conditions at room temperature (NADPH (•), NADH (▴), NADPH+UDP-Galp (▪), and with NADH+UDP-Galp (▾)).
Table 1. Kinetic parameters of NAD(P)H oxidation reactionsa.
| NADH | NADPH | |||
| kox, s−1 | KM, µM | kox, s−1 | KM, µM | |
| TcUGM | 0.015±0.002 | 90±20 | 0.030±0.002 | 15±2 |
| TcUGM+UDP-Galp b | 0.010±0.002 | 50±20 | 0.0099±0.001 | 19±5 |
| MtUGM | 1.8×10−6 | - | 1.1×10−6 | - |
| NAD(P)H alone | 1.8×10−7 | - | 4.7×10−7 | - |
All reactions were done at room temperature with air saturated buffer (50 mM phosphate buffer pH 7.0).
In the presence of 0.5 mM UDP-Galp.
Flavin reduction monitored by rapid reaction kinetics
The decrease in absorbance at 450 nm under anaerobic conditions was measured in a stopped flow spectrophotometer to determine the rate constant of flavin reduction of TcUGM by the reduced coenzyme (Figure 4). The observed rate constant of flavin reduction, kred, with NADPH was seven-fold faster than with NADH (Table 2, Figure 4). Furthermore, the Kd value for NADPH was five-fold lower than for NADH. These results suggest that NADPH is the preferred coenzyme. Reduction of TcUGM with NADPH was also tested in the presence of saturating UDP-Galp. Under this condition, the kred value did not change, demonstrating reduction is unaffected by the presence of UDP-Galp (0.57±0.01 s−1 in the absence of UDP-Galp versus 0.57±0.04 s−1 in the presence of UDP-Galp).
Figure 4. Anaerobic reduction of TcUGM with NAD(P)H.

Reduction was monitored using the stopped flow spectrophotometer at 15°C. The data was collected from 2 ms to 100 s on a logarithmic timescale. A) Changes in the spectra of oxidized TcUGM after mixing with 0.5 mM NADPH over 23 s. B) Traces of the flavin reduction at various concentrations of NADPH. The data were fit to a single exponential decay equation. C) The kobs values were plotted as a function NADPH (•) and NADH (▴) concentrations and fitted using the equation 2.
Table 2. TcUGM Reduction by NAD(P)Ha.
| k red, s−1 | K d, µM | kred/Kd, µM−1 s−1 | |
| NADPH | 0.60±0.006 | 98±3 | 0.0061±0.0001 |
| NADH | 0.085±0.0006 | 550±10 | 0.00015±0.000002 |
Reactions were measured under anaerobic conditions at 15°C in 50 mM phosphate buffer pH 7.0.
Mutase activity
The activity of TcUGM reduced with NADPH and NADH was measured and compared to the values obtained with dithionite reduced enzyme (Table 3, Figure 5). The reverse reaction was monitored to allow significant accumulation of product, as equilibrium favors UDP-Galp over UDP-Galf (93% to 7%). Saturation kinetics was observed as a function of UDP-Galf concentration. Data were fit to the Michaelis-Menten equation and very similar kcat values were obtained independent of the source of reducing equivalents (Table 3). The KM values were also very similar when the enzyme was reduced with NADPH or dithionite. In contrast, a higher KM value was measured with NADH as the reductant, which we attributed to the slower rate of reduction by NADH (Table 3).
Table 3. Steady state kinetics of TcUGMa.
| Reductant | kcat, s−1 | KM, µM | kcat/KM, µM−1 s−1 |
| Dithionite | 13.4±0.3 | 140±10 | 0.093±0.006 |
| NADPH | 11.5±0.4 | 200±20 | 0.056±0.005 |
| NADH | 8.4±0.9 | 690±150 | 0.012±0.001 |
Reactions were incubated at 37°C for 1 min in 25 mM Hepes, 125 mM NaCl pH 7.5, and terminated by heat denaturation at 95°C. Reactions were performed using saturating amounts of reductant (20 mM dithionite, 0.5 mM NADPH, and 2.5 mM NADH).
Figure 5. TcUGM activity with UDP-Galf.

TcUGM was reduced with either 20 mM dithionite (▪), 0.5 mM NADPH(•), or 2.5 mM NADH(▴). Reactions were performed with 200 nM TcUGM incubated with varying concentrations of substrate for 1 min at 37°C. The data were fit to the Michaelis-Menten equation. Summary of data is presented in Table 3.
Viscosity effects
To determine if product release is the rate-limiting step, the effect of viscosity on kcat was measured. This was done by determining the activity of TcUGM at different concentrations of glycerol. If product release was the rate-limiting step, a decrease in the kcat value would be observed as the viscosity of the medium increased. A plot of the ratio of the kcat values in water, (kcat)0, and at each concentration of viscogen, (kcat)η, should yield a straight line with a positive slope. A slope of 1 is expected if product release is the rate-liming step. With TcUGM, a slope of zero was observed, indicating that product release is not the rate determining step in the reaction (Figure 6).
Figure 6. Viscosity effect on kcat.

The effect of viscosity was determined by measuring the activity of TcUGM as a function of increasing concentrations of glycerol. The data was fit to a linear equation; the dashed line depicts the results of a diffusion controlled reaction. This line has a slope of 1.
Determination of the binding affinity of UDP-Galp to TcUGM
To determine if UDP-Galp binds to oxidized or reduced TcUGM, we synthesized UDP-rhodamine to measure binding of the substrate. Fluorescence anisotropy changes induced by release of the bound chromophore to TcUGM upon binding of the substrate can be used to calculate the dissociation constant (Figure 7A) [37].UDP-Galp was found to have very weak affinity to oxidized TcUGM. We tested concentrations of substrate as high as 100 mM and were unable to measure binding. However, when TcUGM was reduced with dithionite, a Kd of 70±40 µM was determined demonstrating UDP-Galp only binds to the reduced state of TcUGM (Figure 7B).
Figure 7. Fluorescence anisotropy assay to measure the affinity of UDP-Galp to TcUGM.

A) UDP-rhodamine chromophore used in the fluorescence anisotropy experiments. B) Fluorescence polarization binding assay. The binding of UDP-Galp to chemically reduced TcUGM was monitored by measuring the changes in anisotropy as it displaces UDP-rhodamine from the active site. The Kd values were obtained using equation 3.
Isolation of flavin-galactose adduct
In the UGM reaction, a novel role for the flavin cofactor has been proposed. It was suggested that the flavin could attack UDP-Galp at the anomeric carbon, leading to cleavage of the C-O bond (Figure 2, 2 ). In the bacterial enzymes a flavin-galactose adduct was isolated and characterized, supporting this role for the flavin cofactor [21], [22]. Involvement of a flavin sugar adduct in the reaction of eukaryotic UGMs was investigated. The adduct was trapped by performing activity assays in the presence of sodium cyanoborohydride, which reduces the iminium ion during turnover. The flavin was extracted and then isolated by HPLC (Figure 8A). Two major peaks were observed, one corresponding to native FAD, while the other had spectral peaks at 230 nm, 315 nm, and 378 nm (Figure 8B). This flavin spectrum is consistent with previous published spectra for the N5-flavin adduct however the peak at 378 nm is indicative of a C4a-oxygen-flavin adduct [21], [40]. This compound was characterized by high resolution mass spectrometry (HRMS) and shown to have a mass of 966.1946 Da (Figure 8C). This mass closely corresponds to the expected mass of the FAD-galactose adduct of 950.2340 Da; the difference corresponds to the mass of a hydroxyl group. We propose that the flavin was hydroxylated at the C4a position during the trapping and isolation process, which agrees with the observed spectrum.
Figure 8. Trapping of a covalent flavin intermediate.

A) HPLC traces of the flavin sugar adduct from free FAD. The peak eluding at 22.5 min is the adduct, while the second peak at 23.6 min is FAD. B) Spectrum of the C4a- hydroxyflavin-galactose adduct. C) High resolution mass spectrometry results of the peak containing the flavin adduct. The inset shows the structure of the adduct with a hydroxyl group at the flavin C4a-position.
Monitoring the reaction of reduced TcUGM with UDP-Galp, UDP-Galf, UDP, and UDP-Glc
The results presented in the previous section and the previously published data, establish that a flavin-galactose adduct is formed in the reaction catalyzed by UGMs. However, the mechanism leading to the formation of this intermediate can occur via the formation of a flavin and sugar radical (Figure 2, 6-2 ) or by a direct nucleophilic attack by the flavin (Figure 2, 1-2 ). Since a flavin semiquinone has distinct spectroscopic characteristics, we used rapid reaction kinetics to monitor the reaction at early time points. In this experiment, anaerobically reduced TcUGM was mixed with either buffer alone, UDP, UDP-glucose (UDP-Glc), UDP-Galp, or UDP-Galf in the stopped-flow spectrophotometer and complete spectra were collected between 350 nm and 700 nm (Figure 9). When the enzyme was mixed with buffer alone or with UDP, no major spectral changes were detected (Figure 9A, buffer alone not shown). Difference spectra of TcUGM after mixing with UDP-Glc showed a slight decrease in absorbance at ∼415 nm, which agrees with previous data with UDP-Glc binding to Klebsiella pneumoniae UGM (KpUGM) (Figure S3) [21], [41]. TcUGM mixed with either UDP-Galp or UDP-Galf showed the same spectral changes with a decrease at ∼405 nm, an isosbestic point at ∼425 nm (red shifted 10 nm with UDP-Galp), and a peak at ∼460 nm (Figure 9B, C). Equilibrium binding of UDP-Galp to KpUGM showed similar flavin spectral changes, however, it was red shifted by ∼50 nm [21]. The change in absorbance at the higher wavelengths was attributed to the formation of the iminium ion intermediate [21]. As the change at higher wavelengths does not occur upon binding of the known ligand UDP-Glc, but only upon substrate binding, we propose that these spectral changes correspond to the formation of the iminium ion in TcUGM (Figure 2, 3 ). A k1 value of 310±40 s−1 and a k2 value of 7.9±0.7 s−1 were obtained from fitting the absorbance changes at 452 nm to a double exponential equation (Figure 9D). We attributed the fast rate to the formation of the iminium ion. Since the rate of k2 closely matches the value of kcat, we concluded that it is associated with the rate limiting step, which could be either ring closing or reattachment of UDP. More importantly, upon mixing reduced TcUGM and substrate, a flavin semiquinone was not observed, suggesting either the semiquinone does not form or that the rate of decay is faster than the rate formation.
Figure 9. Rapid reaction kinetics with reduced TcUGM mixed with substrate and substrate analogs.

The initial spectrum (1.2 ms) is shown as a solid line, the final spectrum (14 ms for UDP-Galf and 1.5 s for all other analogs) as a dashed line, and intermediate time points as gray lines (spectra every 1.26 ms for UDP-Galf and every tenth spectrum collected of 400 points on logarithmic time scale over 2 s for all other analogs). Reduced TcUGM mixed with 0.25 mM UDP (A), 0.25 mM UDP-Galp (B), 0.15 mM UDP-Galf (C), and formation of the iminium ion monitored at 455 nm (D). The data was fit to equation 3.
Discussion
UGM belongs to a growing group of flavoproteins where the protein environment modulates the reactivity of the flavin to perform novel non-redox chemistries [42], [43]. There are two proposed mechanisms for UGM. The proposed mechanisms differ on the steps leading to the formation of the flavin galactose adduct. In one mechanism, the flavin acts as a nucleophile and attacks the anomeric carbon leading to the formation of the flavin-galactose adduct intermediate, which can either occur via a direct attack on UDP-Galp (SN2) or on a postulated oxocarbenium ion intermediate (SN1)(Figure 2, 1-2 ). In the other mechanism, elimination of UDP leads to the formation of an oxocarbenium ion followed by a single electron transfer from the reduced flavin (Figure 2, 6 ), forming a flavin seminquinone and sugar radical. Radical rearrangement of the N5 of the flavin and subsequent recombination leads to the formation of the flavin-galactose adduct (Figure 2, 2 ). Once the adduct is formed, the galactose ring opens leading to the formation of a sugar-flavin iminium ion (Figure 2, 3 ) [21], [22]. After recyclization, the final step in the reaction involves an attack on the anomeric carbon by UDP, leading to the formation of UDP-Galf (Figure 2, 4-5 ).
These mechanisms were developed and supported by data obtained entirely from studies on prokaryotic UGMs and several aspects of the mechanism are not completely understood. It was shown by position isotope effects (PIX) that the glycosidic bond between galactose and UDP was broken during catalysis [44]. Later studies demonstrated that the semiquinone of the flavin is stabilized in the presence of UDP and UDP-Galp and that UGM is inactive with 5-dezaflavin, supporting the single electron transfer mechanism [23], [24]. The formation of a flavin N5-C1 galactose adduct was then demonstrated by trapping the immium ion with NaCNBH3 [21], [22]. This result supports a mechanism in which the flavin acts as a nucleophile. Furthermore, the structure of bacterial enzymes in complex with UDP-Galp, show proper binding conformation for nucleophilic attack by the flavin cofactor [22], [45]. Also in support of the nucleophilic mechanism, it was also shown that the flavin was present as in the anionic hydroquinone form, a feature that is indicative of the N5 being a good nucleophile [21], [24].
Here, we expressed and purified the eukaryotic UGM from the parasite T. cruzi. Recombinant TcUGM was found to function as a monomer in solution, which differs from both the dimeric prokaryotic UGMs, and the tetrameric fungal UGM (AfUGM) [17], [20]. However, it is similar to the parasitic UGM from L. major (LmUGM), which is also a monomer [27]. This suggests that TcUGM and LmUGM are closely related, which agrees with their higher percent amino acid identity (60% with LmUGM, compared to 40% with AfUGM and 15% with EcUGM and MtUGM). TcUGM was found to be active only in the reduced form. Chemically reduced TcUGM has similar kinetic parameters with UDP-Galf as substrate as other UGMs [17], [27], [28], [45], [46].
Having stable and active TcUGM, a series of experiments were performed to address several unanswered questions about the chemical mechanism. We sought to determine the source of the reducing equivalents required for catalysis. In vivo, a common source of reducing equivalents for flavoenzymes is NAD(P)H. It was determined that recombinant TcUGM could reduced by NADPH or NADH in the absence of UDP-Galp, and was oxidized very slowly by molecular oxygen. In the presence of substrate, the reaction with oxygen was decreased. Thus, substrate binding protects the enzyme from oxidation, which is essential for activity since TcUGM is active only in the reduced form. There is no a canonical NADPH binding domain in the primary sequence of TcUGM, thus the binding site of NADPH remains to be identified. The reactivity of TcUGM with NAD(P)H is unique since prokaryotic UGMs are unable to react with reduced coenzymes. Perhaps the bacterial enzymes have specific redox partners in the cell.
The rate of flavin reduction by NAD(P)H was directly monitored in the stopped-flow spectrophotometer. It was shown that the rate constant of flavin reduction and the affinity for NADPH was higher than for NADH. These results showed a 40-fold higher kred/Kd value for NADPH over NADH, suggesting that NADPH is the preferred coenzyme.
The rate of flavin reduction was not affected by the presence of UDP-Galp, indicating that binding of UDP-Galp is not a prerequisite for reduction. Furthermore, UDP-Galp only binds to the reduced form of TcUGM, indicating that UDP-Galp binds after flavin reduction. The Kd value of UDP-Galp for the reduced UGM was determined to be 70±40 µM by fluorescence anisotropy. Thus, the kinetic mechanism starts with binding of NADPH followed by hydride transfer and UDP-Galp binding. Binding of UDP-Galp protects the enzyme from reacting with molecular oxygen. It can be estimated from the kcat value (∼12 s−1) and the rate of oxidation, (0.01 s−1), that for every NADPH oxidized the enzyme can turn over ∼1000 times.
When reduced TcUGM was mixed with UDP-Galp or UDP-Galf using stopped-flow spectroscopy only a small decrease in absorbance at 415 nm and an increase at 460 nm was observed. These changes were not observed when the enzyme was mixed with either UDP or UDP-Glc, ligands that do not turn over. Therefore, we assigned the spectral changes to the formation of the iminium ion. Similar spectral changes have been reported with bacteria UGM under equilibrium conditions, which are consistent with our conclusion [21]. Interestingly, the flavin semiquinone was not observed suggesting either it does not occur or it decays rapidly preventing detection. By addition of sodium cyanoborohydride into the reaction mixture, a flavin-sugar adduct was trapped and was isolated and characterized by mass spectroscopy. This result validates the assignment of the iminium ion intermediate observed in the stopped-flow experiments. This adduct has also been characterized in KpUGM as a flavin N5-C1 galactose intermediate by HRMS and 1H NMR [21], [22]. From our rapid reaction kinetics data, the changes in flavin absorbance best fit a double exponential equation with a fast and slow rate. We assign the fast rate to the formation of the iminium ion, which occurs at 310±42 s−1. While the slow rate occurred at 7.9±0.7 s−1, and as it closely matches the kcat value (∼12 s−1), we propose that this corresponds to the rate-limiting step. One possibility is that the rate constant that controls kcat corresponds to product release. To determine if product release is the rate-limiting step in the reaction, viscosity effects studies on kcat were performed. If product release is the slow step that determines the kcat value, a linear decrease in the kcat value should be observed as a function of increasing concentrations of viscogen. Using glycerol as a viscogen, no changes in the kcat value was observed, clearly indicating that product release is not the rate-limiting step in the reaction of TcUGM. Therefore, we conclude that the rate-limiting step must be ring closing or UDP reattachment. Further studies are needed to elucidate between the two.
Our results from the rapid reaction kinetic analysis show that a flavin iminium ion is formed and suggest that a flavin semiquinone does not play a role in catalysis. Furthermore, we were able to trap and characterize a flavin-galactose adduct. These data strongly supports the role of the flavin as a nucleophile. We proposed that the formation of this adduct occurs via a direct attack of the flavin (SN2 mechanism) rather than formation of an oxocarbenium ion, followed by flavin attack (SN1 mechanism). Our conclusion is supported by data from Sun et al. where linear free energy relationship (LFER) studies with prokaryotic UGM reconstituted with various FAD analogs show changes in kcat values that correlate linearly with changes in the nucleophilicity of the flavin N5 (ρ value of −2.4), which is consistent with an SN2 mechanism [47]. Also, previous studies with E. coli UGM, it was shown that UDP-[2-deoxy-2-fluoro]Galf functions as a substrate with a kcat value 103 slower than the reported kcat with UDP-Galf [28]. In contrast, in studies with glycosyltransferases, where the formation of an oxocarbenium ion is known to occur, it has been shown that substrates with fluorine at the C2 position destabilize the formation of the oxocarbenium ion preventing the reaction from occurring [48]–[51]. Therefore, if the UGM reaction proceeded by an oxocarbenium ion, UDP-[2-deoxy-2-fluoro]Galf should act as an inhibitor instead of as a slow substrate.
Together, the data presented here provides a detailed description of the mechanism of the parasitic TcUGM, which is summarized in Figure 10. We show that TcUGM first binds to NADPH (Figure 10, b ), and after hydride transfer (Figure 10, c ), UDP-Galp binds (Figure 10,d ). We propose that an adduct forms by direct attack of the flavin to the C1 position of galactose (Figure 10, e ), without the formation of a flavin semiquinone or oxocarbenium ion. Ring opening leads to the rapid formation of an iminium ion (Figure 10, f ). Product release was determined not to be rate limiting, therefore, ring closing or formation of the glycosidic bond between Galf and UDP represent the rate determining step(s) in this reaction. Since UDP is predicted to remain bound in the active site, we believed that formation of the anomeric bond will be fast, therefore, the slow step is most probably the isomerization step (Figure 10, f to g ). The mechanism of TcUGM clearly shows a unique mode of action for the flavin cofactor, where it functions as a nucleophile and a scaffold for holding the reaction intermediates. These results will aid in the development of new drugs against human pathogens such as T. cruzi, A. fumigatus, and L. major.
Figure 10. Chemical mechanism of TcUGM.

The reaction requires the oxidized flavin cofactor (a) to be reduced for activity. First, NADPH binds to the oxidized enzyme (b), and only after the flavin is reduced (c) will UDP-Galp bind (d). The flavin then acts as a nucleophile attacking the C1 of galactose and forming a flavin sugar adduct (e), which occurs rapidly (f). This is followed by ring opening and recyclization (g). The rate limiting step in the reaction corresponds to either galactose isomerization or reattachment of the UDP (f to g). We postulate that the rate limiting step is the isomerization step. The final step is release of UDP-Galf, which occurs rapidly. The enzyme can proceed to the next reaction cycle or be slowly oxidized by molecular oxygen (h to a).
Supporting Information
Synthesis of UDP-Gal f .
(DOC)
Flavin spectrum and SDS-PAGE of purified TcUGM.
(TIF)
Size exclusion chromatography of TcUGM. The standards aprotinin (1, 6.5 kDa), ribonuclease (2, 13 kDa), ovalbumin (3, 43 kDa), conoalbumin (4, 75 kDa), aldolase (5, 158 kDa), and ferritin (6, 440 kDa) were used to calculate the Kav values using the equation, 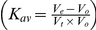 , where Vo is the void volume of the column; Vt is the total volume of the column and Ve is the elution volume of the protein). TcUGM is shown on the plot as a black square.
, where Vo is the void volume of the column; Vt is the total volume of the column and Ve is the elution volume of the protein). TcUGM is shown on the plot as a black square.
(TIF)
Rapid reaction kinetics with reduced TcUGM mixed with 0.25 mM UDP-Glc. Inset show the difference spectra.
(TIF)
Acknowledgments
We thank Dr. Janet Webster for critical reading of the manuscript
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by National Institutes of Health grant R01 GM094469 (P. S.). M.O. was supported by a fellowship from the American Heart Association. A.L.V was supported by a fellowship from the Ministry of Science and Technology of Costa Rica. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.WHO. World Health Report. 2002. (World Health Organization, Geneva, 2002)
- 2.Bern C, Montgomery SP, Herwaldt BL, Rassi A, Jr, Marin-Neto JA, et al. Evaluation and treatment of chagas disease in the United States: a systematic review. J Am Med Assoc. 2007;298:2171–2181. doi: 10.1001/jama.298.18.2171. [DOI] [PubMed] [Google Scholar]
- 3.Dorn PL, Perniciaro L, Yabsley MJ, Roellig DM, Balsamo G, et al. Autochthonous transmission of Trypanosoma cruzi, Louisiana. Emerg Infect Dis. 2007;13:605–607. doi: 10.3201/eid1304.061002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buekens P, Almendares O, Carlier Y, Dumonteil E, Eberhard M, et al. Mother-to-Child Transmission of Chagas' Disease in North America: Why Don't We Do More? Matern Child Health J. 2008;12:283–286. doi: 10.1007/s10995-007-0246-8. [DOI] [PubMed] [Google Scholar]
- 5.Hanford EJ, Zhan FB, Lu Y, Giordano A. Chagas disease in Texas: recognizing the significance and implications of evidence in the literature. Soc Sci Med. 2007;65:60–79. doi: 10.1016/j.socscimed.2007.02.041. [DOI] [PubMed] [Google Scholar]
- 6.De Souza W. Basic cell biology of Trypanosoma cruzi. Curr Pharm Des. 2002;8:269–285. doi: 10.2174/1381612023396276. [DOI] [PubMed] [Google Scholar]
- 7.Coura JR, Borges-Pereira J. Chagas disease: 100 years after its discovery. A systemic review. Acta Trop. 2010;115:5–13. doi: 10.1016/j.actatropica.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Prata A. Evolution of the clinical and epidemiological knowledge about Chagas disease 90 years after its discovery. Mem Inst Oswaldo Cruz. 1999;94(Suppl 1):81–88. doi: 10.1590/s0074-02761999000700008. [DOI] [PubMed] [Google Scholar]
- 9.Buscaglia CA, Campo VA, Frasch AC, Di Noia JM. Trypanosoma cruzi surface mucins: host-dependent coat diversity. Nat Rev Microbiol. 2006;4:229–236. doi: 10.1038/nrmicro1351. [DOI] [PubMed] [Google Scholar]
- 10.Roper JR, Guther ML, Milne KG, Ferguson MA. Galactose metabolism is essential for the African sleeping sickness parasite Trypanosoma brucei. Proc Natl Acad Sci U S A. 2002;99:5884–5889. doi: 10.1073/pnas.092669999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oppenheimer M, Valenciano AL, Sobrado P. Biosynthesis of galactofuranose in kinetoplastids: novel therapeutic targets for treating leishmaniasis and chagas' disease. Enzyme Res. 2011;2011:415976. doi: 10.4061/2011/415976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crick DC, Mahapatra S, Brennan PJ. Biosynthesis of the arabinogalactan-peptidoglycan complex of Mycobacterium tuberculosis. Glycobiology. 2001;11:107R–118R. doi: 10.1093/glycob/11.9.107r. [DOI] [PubMed] [Google Scholar]
- 13.Stevenson G, Neal B, Liu D, Hobbs M, Packer NH, et al. Structure of the O antigen of Escherichia coli K-12 and the sequence of its rfb gene cluster. J Bacteriol. 1994;176:4144–4156. doi: 10.1128/jb.176.13.4144-4156.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Latge JP. Galactofuranose containing molecules in Aspergillus fumigatus. Med Mycol. 2009;47(Suppl 1):S104–109. doi: 10.1080/13693780802258832. [DOI] [PubMed] [Google Scholar]
- 15.Pereira-Chioccola VL, Acosta-Serrano A, Correia de Almeida I, Ferguson MA, Souto-Padron T, et al. Mucin-like molecules form a negatively charged coat that protects Trypanosoma cruzi trypomastigotes from killing by human anti-alpha-galactosyl antibodies. J Cell Sci. 2000;113:1299–1307. doi: 10.1242/jcs.113.7.1299. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki E, Mortara RA, Takahashi HK, Straus AH. Reactivity of MEST-1 (antigalactofuranose) with Trypanosoma cruzi glycosylinositol phosphorylceramides (GIPCs): immunolocalization of GIPCs in acidic vesicles of epimastigotes. Clin Diagn Lab Immunol. 2001;8:1031–1035. doi: 10.1128/CDLI.8.5.1031-1035.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oppenheimer M, Poulin MB, Lowary TL, Helm RF, Sobrado P. Characterization of recombinant UDP-galactopyranose mutase from Aspergillus fumigatus. Arch Biochem Biophys. 2010;502:31–38. doi: 10.1016/j.abb.2010.06.035. [DOI] [PubMed] [Google Scholar]
- 18.Nassau PM, Martin SL, Brown RE, Weston A, Monsey D, et al. Galactofuranose biosynthesis in Escherichia coli K-12: identification and cloning of UDP-galactopyranose mutase. J Bacteriol. 1996;178:1047–1052. doi: 10.1128/jb.178.4.1047-1052.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Q, Liu HW. Studies of UDP-Galactopyranose Mutase from Escherichia coli: An Unusual Role of Reduced FAD in Catalysis. J Am Chem Soc. 2000;122:6. [Google Scholar]
- 20.Sanders DAR, Staines AG, McMahon SA, McNeil MR, Whitfield C, et al. UDP-galactopyranose mutase has a novel structure and mechanism. Nat Struct Mol Biol. 2001;8:858–863. doi: 10.1038/nsb1001-858. [DOI] [PubMed] [Google Scholar]
- 21.Soltero-Higgin M, Carlson EE, Gruber TD, Kiessling LL. A unique catalytic mechanism for UDP-galactopyranose mutase. Nat Struct Mol Biol. 2004;11:539–543. doi: 10.1038/nsmb772. [DOI] [PubMed] [Google Scholar]
- 22.Gruber TD, Westler WM, Kiessling LL, Forest KT. X-ray crystallography reveals a reduced substrate complex of UDP-galactopyranose mutase poised for covalent catalysis by flavin. Biochemistry. 2009;48:9171–9173. doi: 10.1021/bi901437v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang ZH, Zhang QB, Liu HW. Reconstitution of UDP-galactopyranose mutase with 1-deaza-FAD and 5-deaza-FAD: analysis and mechanistic implications. Bioorg Chem. 2003;31:494–502. doi: 10.1016/j.bioorg.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Fullerton SWB, Daff S, Sanders DAR, Ingledew WJ, Whitfield C, et al. Potentiometric analysis of UDP-galactopyranose mutase: Stabilization of the flavosemiquinone by substrate. Biochemistry. 2003;42:2104–2109. doi: 10.1021/bi027077f. [DOI] [PubMed] [Google Scholar]
- 25.Blommel PG, Martin PA, Wrobel RL, Steffen E, Fox BG. High efficiency single step production of expression plasmids from cDNA clones using the Flexi Vector cloning system. Protein Expr Purif. 2006;47:562–570. doi: 10.1016/j.pep.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 26.Macheroux P. UV-visible spectroscopy as a tool to study flavoproteins. Methods Mol Biol. 1999;131:1–7. doi: 10.1385/1-59259-266-X:1. [DOI] [PubMed] [Google Scholar]
- 27.Oppenheimer M, Valenciano AL, Sobrado P. Isolation and characterization of functional Leishmania major virulence factor UDP-galactopyranose mutase. Biochem Biophys Res Commun. 2011;407:552–556. doi: 10.1016/j.bbrc.2011.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Q, Liu H. Mechanistic investigation of UDP-galactopyranose mutase from Escherichia coli using 2- and 3-fluorinated UDP-galactofuranose as probes. J Am Chem Soc. 2001;123:6756–6766. doi: 10.1021/ja010473l. [DOI] [PubMed] [Google Scholar]
- 29.Cattiaux L, Sendid B, Collot M, Machez E, Poulain D, et al. Synthetic biotinylated tetra beta(1→5) galactofuranoside for in vitro aspergillosis diagnosis. Bioorg Med Chem. 2011;19:547–555. doi: 10.1016/j.bmc.2010.10.062. [DOI] [PubMed] [Google Scholar]
- 30.D'Accorso N, Thiel IME, Schüller M. Proton and C-13 nuclear magnetic resonance spectra of some benzoylated aldohexoses. Carbohydr Res. 1983;124:177–184. [Google Scholar]
- 31.Davis CB. Development of Novel Carbohydrate-Based Anti-Bacterial Agents. QLD, Australia: Griffith University; 2006. [Google Scholar]
- 32.Ness RK, Fletcher HG, Hudson CS. 1,4-Anhydro-D-galactitol. J Am Chem Soc. 1951;73:3742–3744. doi: 10.1021/ja01181a095. [DOI] [PubMed] [Google Scholar]
- 33.Tsvetkov YE, Nikolaev AV. The chemical synthesis of UDP-a-D-galactofuranose. J Chem Soc, Perkin Transactions. 2000;1:889–891. [Google Scholar]
- 34.de Lederkremer RM, Nahmad VB, Varela O. Synthesis of a-D-Galactofuranosyl Phosphate. J Org Chem. 1994;59:690–692. [Google Scholar]
- 35.Poulin MB, Lowary TL. Chapter Nineteen - Methods to Study the Biosynthesis of Bacterial Furanosides. In: Minoru F, editor. Methods in Enzymology. Academic Press; 2010. pp. 389–411. [DOI] [PubMed] [Google Scholar]
- 36.Lide DR. Handbook of Chemistry and Physics 2000–2001. Boca Raton, FL: CRC Press; 2000. [Google Scholar]
- 37.Qi J, Oppenheimer M, Sobrado P. Fluorescence Polarization Binding Assay for Aspergillus fumigatus Virulence Factor UDP-Galactopyranose Mutase. Enzyme Res. 2011;2011:513905. doi: 10.4061/2011/513905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blommel PG, Becker KJ, Duvnjak P, Fox BG. Enhanced bacterial protein expression during auto-induction obtained by alteration of lac repressor dosage and medium composition. Biotechnol Prog. 2007;23:585–598. doi: 10.1021/bp070011x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barlow JN, Marcinkevicience J, Blanchard JS. The Enzymatic Conversion of UDP-galactopyranose to UDP-galactopyranose. In: Frey PA, Northrop DB, editors. Enzymatic Mechanisms. Amsterdam: IOS Press; 1999. [Google Scholar]
- 40.Chocklett SW, Sobrado P. Aspergillus fumigatus SidA is a highly specific ornithine hydroxylase with bound flavin cofactor. Biochemistry. 2010;49:6777–6783. doi: 10.1021/bi100291n. [DOI] [PubMed] [Google Scholar]
- 41.Gruber TD, Borrok MJ, Westler WM, Forest KT, Kiessling LL. Ligand binding and substrate discrimination by UDP-galactopyranose mutase. J Mol Biol. 2009;391:327–340. doi: 10.1016/j.jmb.2009.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller SM. A new role for an old cofactor. Nat Struct Mol Biol. 2004;11:497–498. doi: 10.1038/nsmb0604-497. [DOI] [PubMed] [Google Scholar]
- 43.Bornemann S. Flavoenzymes that catalyse reactions with no net redox change. Nat Prod Rep. 2002;19:761–772. doi: 10.1039/b108916c. [DOI] [PubMed] [Google Scholar]
- 44.Barlow JN, Girvin ME, Blanchard JS. Positional Isotope Exchage Catalyzed by UDP-Galactopyranose Mutase. J Am Chem Soc. 1999;121:12. [Google Scholar]
- 45.Partha SK, van Straaten KE, Sanders DA. Structural basis of substrate binding to UDP-galactopyranose mutase: crystal structures in the reduced and oxidized state complexed with UDP-galactopyranose and UDP. J Mol Biol. 2009;394:864–877. doi: 10.1016/j.jmb.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 46.Chad JM, Sarathy KP, Gruber TD, Addala E, Kiessling LL, et al. Site-directed mutagenesis of UDP-galactopyranose mutase reveals a critical role for the active-site, conserved arginine residues. Biochemistry. 2007;46:6723–6732. doi: 10.1021/bi7002795. [DOI] [PubMed] [Google Scholar]
- 47.Sun HG, Ruszczycky MW, Chang WC, Thibodeaux CJ, Liu HW. Nucleophilic participation of the reduced flavin coenzyme in the mechanism of UDP-Galactopyranose mutase. J Biol Chem. 2012;287:4602–4608. doi: 10.1074/jbc.M111.312538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eppe G, Peltier P, Daniellou R, Nugier-Chauvin C, Ferrieres V, et al. Probing UDP-galactopyranose mutase binding pocket: a dramatic effect on substitution of the 6-position of UDP-galactofuranose. Bioorg Med Chem Lett. 2009;19:814–816. doi: 10.1016/j.bmcl.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 49.Lai EC, Morris SA, Street IP, Withers SG. Substituted glycals as probes of glycosidase mechanisms. Bioorg Med Chem. 1996;4:1929–1937. doi: 10.1016/s0968-0896(96)00175-7. [DOI] [PubMed] [Google Scholar]
- 50.Gordon RD, Sivarajah P, Satkunarajah M, Ma D, Tarling CA, et al. X-ray crystal structures of rabbit N-acetylglucosaminyltransferase I (GnT I) in complex with donor substrate analogues. J Mol Biol. 2006;360:67–79. doi: 10.1016/j.jmb.2006.04.058. [DOI] [PubMed] [Google Scholar]
- 51.Tarling CA, Withers SG. The synthesis of a series of modified mannotrisaccharides as probes of the enzymes involved in the early stages of mammalian complex N-glycan formation. Carbohydr Res. 2004;339:2487–2497. doi: 10.1016/j.carres.2004.08.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synthesis of UDP-Gal f .
(DOC)
Flavin spectrum and SDS-PAGE of purified TcUGM.
(TIF)
Size exclusion chromatography of TcUGM. The standards aprotinin (1, 6.5 kDa), ribonuclease (2, 13 kDa), ovalbumin (3, 43 kDa), conoalbumin (4, 75 kDa), aldolase (5, 158 kDa), and ferritin (6, 440 kDa) were used to calculate the Kav values using the equation, , where Vo is the void volume of the column; Vt is the total volume of the column and Ve is the elution volume of the protein). TcUGM is shown on the plot as a black square.
(TIF)
Rapid reaction kinetics with reduced TcUGM mixed with 0.25 mM UDP-Glc. Inset show the difference spectra.
(TIF)