

Abstract
The C terminus (ct) of protein kinase C-α (PKCα) has a type I PDZ binding motif, whereas GluR2 has a type II PDZ binding motif. Both motifs are recognized by the PDZ domain of protein interacting with protein kinase C (PICK1), and PICK1-PKCα-controlled phosphorylation regulates the synaptic expression and function of GluR2. Here, we show that a specific mutation within the carboxylate-binding loop of the PDZ domain of PICK1 (K27E; PICK1-KE) results in a loss of interaction with GluR2 but not with PKCα. In GST pull-down studies, PICK1-WT (wild type) but not PICK1-KE was retained by GST-ct-GluR2. Furthermore, PICK1-WT co-immunoprecipitated both PKCα and GluR2, whereas PICK1-KE only co-immunoprecipitated PKCα. In heterologous cells, PICK1-WT, but not PICK1-KE, clustered GluR2 and also clustered GluR1 in a GluR2-dependent manner. However, neither PICK1-WT nor PICK1-KE altered the distribution of PKCα, even after phorbol ester-induced redistribution of PKCα to the membrane. Finally, PICK1-KE showed no mislocalization when compared with PICK1-WT in neurons. Taken together, it appears that the PDZ domain of PICK1 is less sensitive to mutations for PKCα when compared with GluR2 binding. These results suggest that the PDZ domain of PICK1 has distinct PKCα and GluR2 binding subsite(s).
α-Amino-3-hydroxy-5-methylisoxazole-4-propionate glutamate receptors (AMPARs)1 mediate a majority of excitatory synaptic neurotransmission in the central nervous system. They are crucially involved in synaptic plasticity events such as long term potentiation and long term depression, which are thought to be the molecular basis underlying learning and memory (1). AMPARs are hetero-oligomers comprised of GluR1–4 subunits (2), and in the hippocampus, AMPARs are thought to be primarily composed of GluR1/GluR2 or GluR2/GluR3 subunits (3).
The AMPA receptor subunits GluR1 and GluR2 contain type I and type II PDZ binding motifs, respectively, that interact with different sets of proteins to regulate the synaptic expression of AMPARs (4). Although the C-terminal (ct)-GluR1 interacts with PDZ domain-containing proteins such as SAP97 (5, 6), the extreme C-terminal ct-GluR2 interacts with PDZ domain proteins such as protein interacting with protein kinase C (PICK1) (7, 8), the glutamate receptor-interacting protein (GRIP) (9), and AMPAR-binding protein (ABP) (10). N-ethylmaleimide-sensitive fusion protein and the clathrin adaptor protein AP2 also bind ct-GluR2 via a non-PDZ interactions (4, 11–14).
PICK1 was first identified as a PDZ domain protein interacting with the PDZ binding motif of PKCα (15, 16). Since then, PICK1 has been shown to interact with a variety of proteins, including GluR2, the mGluR7 metabotropic glutamate receptor subtype, Eph receptor tyrosine kinases and their ephrin ligands, and many others. PICK1 can form dimers via its coiled-coil motif, leaving its PDZ domain free for protein interaction (7, 8, 17–21).
The interplay between GluR2-interacting proteins to regulate membrane expression appears complex and is still only partially understood. In simple terms, N-ethylmaleimide-sensitive fusion protein is thought to insert GluR2-containing AMPARs into the membrane, increasing AMPAR currents and playing a role in N-methyl-D-aspartate receptor-dependent long term depression (11, 13, 22). ABP and GRIP appear to anchor GluR2-containing AMPARs to the synaptic membrane and/or intracellular stores (9). PICK1 is thought to target PKCα toward its interacting proteins, for example, GluR2 (23) and mGluR7 (18). The PICK1-directed PKCα phosphorylation of the PDZ binding motif of GluR2 seems to inhibit GluR2 binding to ABP and GRIP, releasing GluR2 from the synapse and leading to AMPAR endocytosis and long term depression (23–30).
In this study, we have investigated the modulation of PICK1-PKCα and PICK1-GluR2 interactions. We show that PKCα and GluR2 binding to PICK1 can be independently regulated. Using a novel PICK1 mutant, we reveal that the PDZ domain of PICK1 can be altered such that it still interacts with type I PDZ binding motifs (PKCα) but not type II PDZ binding motifs (GluR2). We propose that the PDZ domain of PICK1 contains overlapping or allosteric binding sites for PKCα and GluR2 that sterically hinder each other’s binding. Alternatively, the PKCα and GluR2 binding sites could be essentially similar but with one or more of the subsite(s) differing. The PICK1-KE mutant represents an important tool for further investigating the role(s) of PICK1.
EXPERIMENTAL PROCEDURES
Molecular Biology
The C-terminal domain of GluR2 was fused to glutathione S-transferase (GST) by subcloning into pGEX-4T-1 (Amersham Biosciences), FLAG was tagged to the N terminus of PICK1 and then subcloned into pCIneo (Promega), and hemagglutinin (HA)-PKCα was created by subcloning PKCα into pTB701-HA (see Ref. 7 for further information). Point mutations in the PDZ domain of PICK1 were constructed by PCR. The pCITE-EGFP was created by subcloning CITE (IRES) from pCITE4c(+) (Novagen) into pEGFP-N1 (Clontech). FLAG-PICK1-digested fragments from pCIneo (either wild type or KE mutant) were subcloned into pCITE-EGFP. FLAG-PICK1 was always under the more efficient cap-dependent expression, and EGFP was under the less efficient IRES-dependent expression (31). The FLAG-PICK1-IRES-EGFP (either wild type or KE mutant)-digested fragments were subcloned from pCITE-EGFP into pSinRep5 (Invitrogen). RNAs for pSin-Rep5 constructs were prepared by in vitro translation (Invitrogen) and electroporated into baby hamster kidney cells together with the helper DH(265). Sindbis virus was made according to manufacturer’s instructions (Invitrogen catalog number K750-01) and used to infect hippocampal cultures. Integrity of constructs was verified by DNA sequencing (Applied Biosystems/PerkinElmer Life Sciences), and RNA quality was visualized on agarose gels.
GST Pull-down and Co-immunoprecipitation
GST pull-down or co-immunoprecipitation experiments were performed using glutathione-Sepharose 4B (Amersham Biosciences) or anti-FLAG M2-agarose affinity gel (Sigma), respectively, as described elsewhere (7, 18). GST or GST-ct-GluR2 were expressed and extracted from Escherichia coli strain BL21. Native full-length GluR2, FLAG-PICK1 (wild type and KE mutant), and HA-PKCα were (co-)expressed and solubilized from Lipo-fectAMINE (Invitrogen) transiently transfected COS-7 cells. GST pull-down or co-immunoprecipitation samples were separated by electrophoresis on 9 or 10% SDS-polyacrylamide gels (SDS-PAGE). The amount of GluR2, FLAG-PICK1, or HA-PKCα bound to GST-coated glutathione-Sepharose 4B or anti-FLAG M2-agarose affinity gel was determined by Western blotting. Primary antibodies (Abs) were anti-FLAG M2 monoclonal mouse Ab (Sigma), anti-HA polyclonal rabbit Ab (Santa Cruz Biotechnology), and anti-ct-GluR2 polyclonal rabbit Ab (Upstate Biotechnology). Secondary antibodies were alkaline phosphatase-conjugated and were goat anti-rabbit or anti-mouse IgG (Promega). Immunoreactivity was visualized as a blue-purple color. All antibodies were used at dilutions recommended by the manufacturer. Protein concentrations were determined using the Bio-Rad assay kit with bovine serum albumin as standard (Bio-Rad).
Immunocytochemistry
COS-7 cells were grown using Dulbecco’s modified Eagle’s medium culture media (9.5 g/liter Dulbecco’s modified Eagle’s medium, 3.5 g/liter glucose, with 0.2 g/liter sodium bicarbonate and 0.6 g/liter L-glutamine) supplemented with 10% dialyzed fetal bovine serum and 1% penicillin/streptomycin and maintained at 37 °C, 5% CO2. At 70 – 80% confluency, COS-7 cells were transfected in OPTI-MEM (serum-free media) with appropriate constructs using Lipo-fectAMINE (Invitrogen). Transiently transfected COS-7 cells were immunostained, and immunoreactivity was observed as described previously (7). Low density hippocampal cultures were prepared from E18 Sprague-Dawley rats. Briefly, hippocampi were dissected and then dissociated using trypsin for 10 min at 37 °C. After a wash procedure, cultures were plated onto poly-L-lysine-coated glass coverslips and maintained at 37 °C, 5% CO2 until required. Neurons were used after 7–21 days in vitro and were incubated with or without virus for 22 h, after which cells were collected for biochemistry or fixed using 3.7% paraformaldehyde for immunocytochemistry. FLAG-PICK1 expression was detected using anti-FLAG M2 monoclonal mouse antibody (Sigma), and for endogenous PICK1, an anti-PICK1 rabbit antibody was used (18). Secondary antibodies were Alexa Fluor 568-conjugated and were either goat anti-rabbit or goat anti-mouse IgG (Molecular Probes). Fluorescence was visualized using a Leica confocal microscope.
PKCα Trafficking Studies
The effects of TPA on PKCα membrane expression were determined as described previously with some modification (32). COS-7 cells were split into 6-well dishes and grown for 24 h (70 – 80% confluency) before treatment with or without 1 μm TPA (diluted in OPTIMEM) for 30 min at 37 °C. After treatment, cells were washed twice with phosphate-buffered saline at room temperature and scraped in 400 μl of protein lysis buffer (50 mM Tris, pH 7.4, 2 mM EDTA, 2 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, 20 μg/ml leupeptin, 80 μg/ml aprotinin, 100 μg/ml soya bean trypsin inhibitor, 0.1% 2-mercaptoethanol). Exactly 350 μl of protein lysis buffer-resuspended cells were transferred into 1.5-ml Eppendorf tubes and sonicated for 10 s. The sonicated cells (300 μl) were transferred into 1.5-ml Ultracentrifuge tubes and centrifuged at 100,000 × g for 60 min (Beckman Instruments, Ultracentrifuge TL 100). The supernatant (cytoplasmic fraction) was separated from the pellet (membrane fraction). The pellet was resuspended into 300 μl of protein lysis buffer containing 1.2% Triton X-100 and sonicated. Both cytoplasmic and membrane fractions were measured for protein content by Bio-Rad assay kit with bovine serum albumin as standard (Bio-Rad). Samples were frozen at −80 °C until Western blotting.
RESULTS AND DISCUSSION
The PICK1-KE Mutant Does Not Interact with GluR2
The binding characteristics of PICK1-WT and PICK1 carrying a double PDZ domain mutation KD27/28AA (PICK1-AA) have been reported previously; PICK1-WT binds both GluR2 and PKCα, whereas PICK1-AA binds neither of these proteins (7, 8, 15, 16, 18, 23). Here, we examined the binding properties of a PICK1 mutant in which the lysine 27 residue of the carboxylate-binding loop is mutated to glutamate (K27E; PICK1-KE). Using PICK1 from either virally transduced hippocampal cultures or transiently transfected COS-7 cells, GST-GluR2 pull-down assays were used to examine the binding properties of PICK-WT and PICK1-KE mutant (Fig. 1). Western blot analysis showed that PICK1-WT, but not PICK1-KE, was retained by GST-ct-GluR2 (Fig. 1), indicating that PICK1-KE does not interact with GluR2.
FIG.1. Mutated PICK1-KE does not bind GluR2.
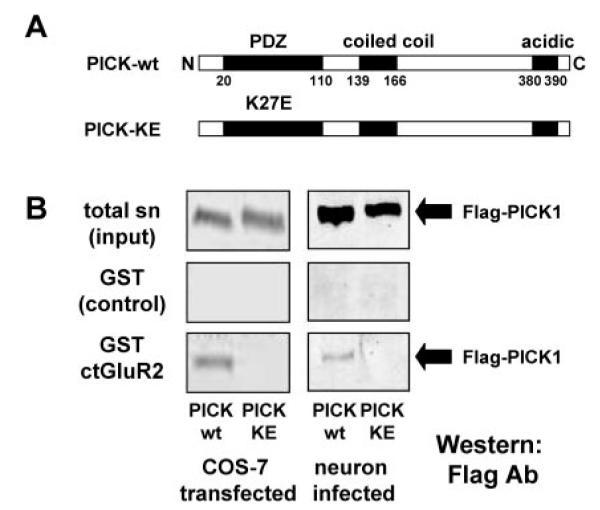
A, a schematic showing the position of the amino acid substitution in the mutant form of PICK1-KE. N, N terminus; C, C terminus. B, using the same input (top panel), Western blots show that solubilized FLAG-PICK1-WT but not FLAG-PICK1-KE obtained from transiently transfected COS-7 cells or viral-infected hippocampal cultures is retained by GST-ct-GluR2 (bottom panel) but not by GST alone (middle panel). sn, cell-solubilized supernatant.
The PICK1-KE Mutant Retains Interaction with PKCα
The PDZ domain of PICK1 has the capacity to accept several different PDZ binding motifs as recently described by GST pull-down studies (33) and as predicted by computer modeling (23). To test the ability of PICK1-KE to bind PKCα and to analyze whether type I and type II interactions could be independently modulated, we simultaneously tested the binding properties of PICK1-KE to GluR2 (-ESVKI; type II) and PKCα (-LQSAV; type I) in co-immunoprecipitation experiments. In these experiments, we found that although PICK1-WT interacted with both GluR2 and PKCα, the PICK1-KE co-immunoprecipitated with PKCα but showed no interaction with GluR2 (Fig. 2). These results suggest that the type I (PKCα) PDZ interactions are more tolerant of alterations in the PICK1 PDZ domain than type II (GluR2) PDZ interactions. Thus, we propose that PICK1 has the ability to differentially recognize the PDZ binding motifs of PKCα and GluR2 and that there are independent binding sites or subsite(s) within the PICK1 PDZ domain for PKCα and GluR2 ligands.
FIG.2. Mutated PICK1-KE still interacts with PKCα.
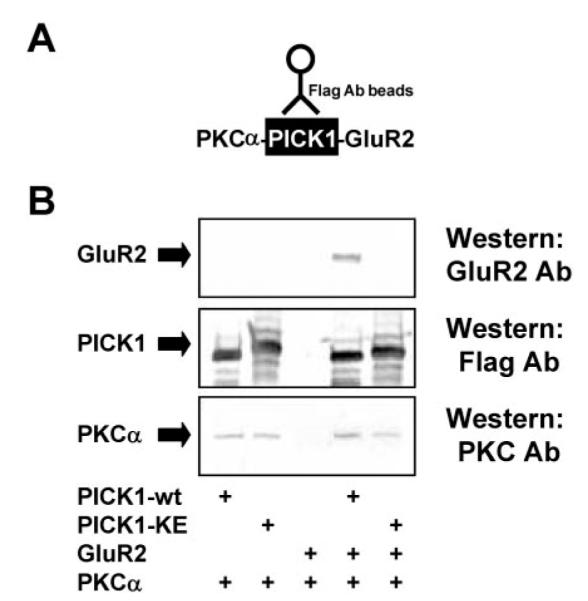
A, a schematic showing PICK1 co-immunoprecipitated complexes. B, the solubilized cell lysates of transiently transfected COS-7 cells are precipitated using FLAG-M2-agarose beads. Western blots show that both FLAG-PICK1-WT and FLAG-PICK1-KE can complex with HA-PKCa with equal ability. In contrast, GluR2 was co-immunoprecipitated with only FLAG-PICK1-WT.
A GluR2-PICK1-PKCα Complex Is Not Found in the Presence of High Amounts of GST-GluR2
Truncations of PICK1 that lack the PDZ domain (residues 1–121; PICK1-Δ121) or the negatively charged C terminus (residues 379 – 416), in addition to PICK1-AA, have been shown to dimerize with PICK1-WT (8, 23). In contrast, PICK1 fragments lacking a portion (residues 165–416) or all of the coiled-coil (residues 135–416) did not interact with PICK1-WT (23). These results indicate that the coiled-coil motif, but not the PDZ domain or the C terminus of PICK1, is responsible for PICK1 dimerization (23). Each of the two PDZ domains present in a PICK1-PICK1 dimer are available to interact with PDZ binding motifs. Such dimer-PDZ interactor complexes could be a molecular basis for targeting PKCα to PICK1-interacting proteins, assisting their phosphorylation and regulating trafficking, surface expression, and function.
We therefore attempted to examined the possibility of the formation of a triple complex involving PICK1, PKCα, and GluR2 using GST-ct-GluR2 pull-downs. In these GST pull-down assays, any retention of PKCα by GST-ct-GluR2 would presumably occur via a PICK1-PICK1 dimer. We reasoned that if GluR2-PICK1-PKCα complexes exist, we should pull-down a PICK1-PKCα heteromer using GST-ct-GluR2. However, GST-ct-GluR2 did not pull-down PKCα in either a PICK1-dependent or -independent manner (Fig. 3). We were initially surprised by these results since we have previously shown the complex formation of PKCα-PICK1-mGluR7 proteins by co-immunoprecipitation (18), and it has been reported that a GluR2-PICK1-PKCα macro-protein complex can exist (23). There are several possible explanations why we did not detect a GluR2-PICK1-PKCα: 1) Such a complex either does not occur or is highly transient and unstable. For example, it is possible that PKCα and GluR2 bind to PICK1 dimers sequentially rather than simultaneously. 2) The conditions of the pull-down experiments were not optimal, so it is possible that the activation state of PKCα (for example, by TPA) and/or conformation of PICK1 is a key determinant for whether a complex occurs. 3) A technical limitation of these experiments is the very high levels of immobilized GST-ct-GluR2. The large excess of GluR2 type II PDZ ligand is likely to occupy all or most of the PICK1 PDZ domains and prevent PKCα interaction with PICK1. Thus, these pull-down data are not definitive. Based on previous studies, we believe it likely that GluR2-PICK1-PKC complexes occur, but we cannot rule out the possibility that in neurons, the three proteins do not complex at all, or that if they do, the co-assembly is too transient to be detected.
FIG.3. High amounts of GST-ct-GluR2 compete with PICK1-PKC interaction.
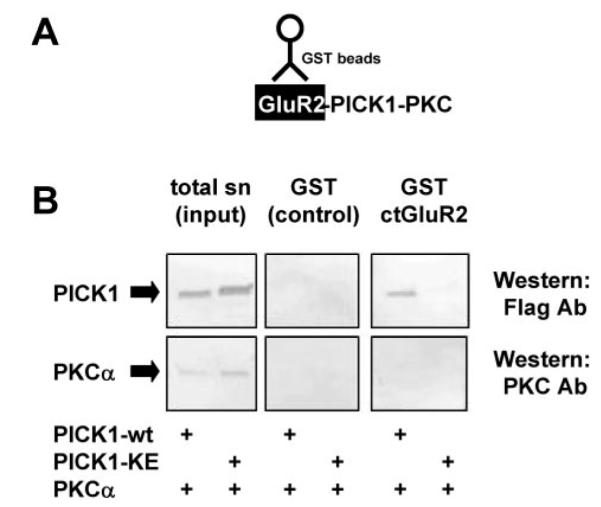
A, a schematic showing ct-GluR2-dependent GST pull-down complexes. B, GST-ct-GluR2 can retain FLAG-PICK1-WT but not FLAG-PICK-KE. GST-ct-GluR2 is likely to inhibit PICK1-PKCα interaction and was not able to retain PKCα via PICK1. sn, cell-solubilized supernatant.
PICK1-WT but Not Mutant PICK1-KE Forms Clusters with AMPARs
The interaction of PICK1 to GluR2 has previously been reported to induce perinuclear clusters of GluR2 in heterologous cells and also synaptic clusters in neurons (7, 8, 23). Here, we found that PICK1-KE had a similar distribution pattern to that observed for PICK1-WT (Fig. 4A) and PICK1-AA (23). However, in contrast to PICK1-WT, the mutant PICK1-KE showed no clustering when expressed with GluR2 (Fig. 4B). These results are consistent with our biochemical studies showing that PICK1-KE does not interact with GluR2. To further confirm our biochemical data that both PICK1 and PICK1-KE interacted with PKCα, we looked for protein clusters of both PICK1-PKCα and PICK1-KE-PKCα. No obvious protein clustering was observed when expressing PKCα and PICK1 or PICK1-KE (Fig. 4C). Since one attractive explanation of our GST pull-down data is that high levels of GluR2 inhibit PKCα binding to the PDZ domain of PICK1, we reasoned that transient overexpression of GluR2 together with PICK1 and PKCα (triple transfections) would not result in PICK1-PKCα clusters. Therefore, since TPA has been suggested to augment PICK1-PKC clusters (23), we determined whether TPA would induce PICK1-PKCα clusters in our system. However, despite observing TPA-induced movement of PKCα from cytosolic to membrane fractions in COS-7 cells (Fig. 4C), the TPA-stimulated conditions did not result in PICK1-PKCα clusters (data not shown). Although our results differ from an earlier study (23), they are in agreement with a recent report (34).
FIG.4. Mutated PICK1-KE does not cluster GluR2.

A, distribution patterns of single transfected proteins are shown. B, PICK1-WT but not mutated PICK-KE formed clusters with GluR2. C, neither WT- nor KE-mutated PICK1 cluster PKCα. TPA causes the movement of PKCα toward the membrane fraction but does not evoke PICK1-PKCα clustering (data not shown). D, PICK1-WT binds GluR2 and indirectly alters GluR1 distribution pattern in COS-7 cells. Small labels indicate the transient transfections performed, whereas bold labels indicate the proteins stained. In double staining, PICK1 is found in the right panel, whereas GluR1, GluR2, or PKCα are found in the left panel.
It has been suggested that GluR1 and GluR2 subunits may have independent trafficking machinery in which the trafficking of GluR1 is regulated by GluR1-interacting proteins such as SAP97 (5, 6). For GluR1/GluR2 heteromeric AMPARs, the ct-GluR1 interactions appear to dominate over GluR2 such that GluR1-containing AMPARs are inserted into the membrane during N-methyl-D-aspartate receptor activity-dependent plasticity and long term potentiation (5, 35, 36). In contrast, for GluR2/GluR3 heteromeric AMPARs, the ct-GluR2 interactions are believed to dominate and to mediate a continuous activity-independent cycling (11, 13, 22, 35). The effects of GluR2 subunit-interacting proteins, such as PICK1, on GluR1 localization have not been demonstrated. Since GluR1 and GluR2 can form heteromeric AMPARs, we further tested the effects of PICK1 on GluR1 in the presence of GluR2. In heterologous cells, we found that PICK1 formed clusters with homomeric GluR2 and heteromeric GluR1/GluR2 AMPAR complexes (Fig. 4C). Triple transfection of COS-7 cells with GluR1, GluR2, and FLAG-PICK1 resulted in a redistribution of GluR1 that was dependent upon the presence of GluR2 and followed the same pattern of PICK-GluR2 clusters (Fig. 4C). These results suggest that in the absence of GluR1 trafficking machinery (as found in COS-7 cells), GluR2-interacting proteins such as PICK1 could modulate the AMPAR complex as a whole (including GluR1). This mechanism may play an important functional role in neurons, where GluR1-interacting proteins have been down-regulated.
PICK1-KE Expression in Hippocampal Neurons
The removal of the PDZ domain from PICK1 (i.e. the removal of the first 121 residues of PICK1; PICK1-Δ121) has been shown to result in self-maintaining clusters, whereas PICK1 carrying a double mutation in its PDZ domain (KD27/28AA; PICK1-AA) showed similar distribution patterns to those of PICK1-WT (23). On the basis of these results, it was suggested that PICK1 with either an occupied PDZ domain (filled with either GluR2 or activated-PKCα) or a truncated PDZ domain would form clusters (23). Since PICK1-Δ121 showed clusters in spines, the clustering and/or targeting of PICK1 was suggested to be independent of the PDZ domain. We studied the distribution patterns of PICK1-WT and PICK1-KE in cultured hippocampal neurons. Neurons transduced with a FLAG-PICK1-IRES-GFP virus vector were identified by dendritic FLAG Ab staining (Fig. 5). FLAG-PICK1 immunoreactivity was never seen independent of nuclear GFP fluorescence (data not shown). Our results showed no observable differences in the distribution patterns and expression levels of PICK1-WT and PICK1-KE. Furthermore, the expression patterns of recombinant expressed PICK1-WT and PICK1-KE were similar to native PICK1 (Fig. 5). The FLAG-PICK1-WT protein was distributed throughout the neuron, and we also saw staining in spines, in agreement with previous studies (23), and we observed no protein clusters or protein aggregation for virally expressed PICK1-WT or PICK1-KE (Fig. 5).
FIG.5. Mutated PICK1-KE shows no mislocalization in neurons.
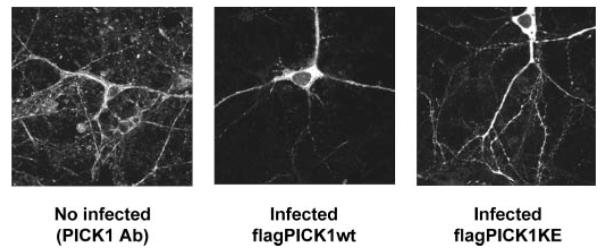
Neurons were virally transduced with FLAG-tagged versions of PICK1-WT and PICK1-KE and stained with FLAG antibody to determine distribution. Neurons not infected with virus were stained with PICK1 rabbit polyclonal antibody.
Acknowledgment
We are grateful to Dr. Naoaki Saito for providing the cDNA of HA-PKCα.
Footnotes
This work was supported in part by research grants from the Wellcome Trust, UK (to K. K. D.) and the Ministry of Education, Science and Culture of Japan (to S. N.).
The abbreviations used are: AMPA, α-amino-3-hydroxy-5-methylisoxazole-4-propionate; AMPAR, AMPA receptor; GRIP, glutamate receptor-interacting protein; ABP, AMPAR-binding protein; PKC, protein kinase C; ct, C-terminal; GluR, glutamate receptor; HA, hemagglutinin; PICK, protein interacting with protein kinase C; WT, wild type; GST, glutathione S-transferase; IRES, internal ribosome entry site; GFP, green fluorescent protein; EGFP, enhanced GFP; TPA, 12-O-tetradec-anoylphorbol-13-acetate; Ab, antibody.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Bliss T, Collingridge GL. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 2.Hollmann M, Heinemann S. Annu. Rev. Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 3.Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. J. Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henley JM. Neurosci. Res. 2003;45:243–254. doi: 10.1016/s0168-0102(02)00229-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 6.Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. J. Biol. Chem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- 7.Dev KK, Nishimune A, Henley J, Nakanishi S. Neuropharmacology. 1999;38:635–644. doi: 10.1016/s0028-3908(98)00230-5. [DOI] [PubMed] [Google Scholar]
- 8.Xia J, Zhang X, Staudinger J, Huganir RL. Neuron. 1999;22:179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- 9.Dong H, O’Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. Nature. 1997;386:279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- 10.Srivastava S, Osten P, Vilim FS, Khatri L, Inman G, States B, Daly C, DeSouza S, Abagyan R, Valtschanoff JG, Weinberg RJ, Ziff EB. Neuron. 1998;21:581–591. doi: 10.1016/s0896-6273(00)80568-1. [DOI] [PubMed] [Google Scholar]
- 11.Nishimune A, Isaac JTR, Molnar E, Noel J, Nash SR, Tagaya M, Collingridge GL, Nakanishi S, Henley JM. Neuron. 1998;21:87–97. doi: 10.1016/s0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
- 12.Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, States BA, Einheber S, Milner TA, Hanson PI, Ziff EB. Neuron. 1998;21:99–110. doi: 10.1016/s0896-6273(00)80518-8. [DOI] [PubMed] [Google Scholar]
- 13.Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Neuron. 1998;21:393–400. doi: 10.1016/s0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- 14.Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ. Neuron. 2000;28:499–510. doi: 10.1016/s0896-6273(00)00128-8. [DOI] [PubMed] [Google Scholar]
- 15.Staudinger J, Zhou J, Burgess R, Elledge SJ, Olson EN. J. Cell Biol. 1995;128:263–271. doi: 10.1083/jcb.128.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Staudinger J, Lu J, Olson EN. J. Biol. Chem. 1997;272:32019–32024. doi: 10.1074/jbc.272.51.32019. [DOI] [PubMed] [Google Scholar]
- 17.Torres R, Firestein BL, Dong H, Staudinger J, Olson EN, Huganir RL, Bredt DS, Gale NW, Yancopoulos GD. Neuron. 1998;21:1453–1463. doi: 10.1016/s0896-6273(00)80663-7. [DOI] [PubMed] [Google Scholar]
- 18.Dev KK, Nakajima Y, Kitano J, Braithwaite SP, Henley JM, Nakanishi S. J. Neurosci. 2000;20:7252–7257. doi: 10.1523/JNEUROSCI.20-19-07252.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boudin H, Doan A, Xia J, Shigemoto R, Huganir RL, Worley P, Craig AM. Neuron. 2000;28:485–497. doi: 10.1016/s0896-6273(00)00127-6. [DOI] [PubMed] [Google Scholar]
- 20.El Far O, Airas J, Wischmeyer E, Nehring RB, Karschin A, Betz H. Eur. J. Neurosci. 2000;12:1–9. doi: 10.1046/j.1460-9568.2000.01309.x. [DOI] [PubMed] [Google Scholar]
- 21.Takeya R, Takeshige K, Sumimoto H. Biochem. Biophys. Res. Comm. 2000;267:149–155. doi: 10.1006/bbrc.1999.1932. [DOI] [PubMed] [Google Scholar]
- 22.Luthi A, Chittajallu R, Duprat F, Palmer MJ, Benke TA, Kidd FL, Henley JM, Isaac JT, Collingridge GL. Neuron. 1999;24:389–399. doi: 10.1016/s0896-6273(00)80852-1. [DOI] [PubMed] [Google Scholar]
- 23.Perez PL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. J. Neurosci. 2001;21:5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. J. Neurosci. 2000;20:7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, Collingridge GL, Isaac JTR. Neuron. 2000;28:873–886. doi: 10.1016/s0896-6273(00)00160-4. [DOI] [PubMed] [Google Scholar]
- 26.Kim CH, Chung HJ, Lee HK, Huganir RL. Proc. Natl. Acad. Sci. U. S. A. 2001;98:11725–11730. doi: 10.1073/pnas.211132798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li P, Kerchner GA, Sala C, Wei F, Huettner JE, Sheng M, Zhuo M. Nat. Neurosci. 1999;2:972–977. doi: 10.1038/14771. [DOI] [PubMed] [Google Scholar]
- 28.Matsuda S, Mikawa S, Hirai H. J. Neurochem. 1999;73:1765–1768. doi: 10.1046/j.1471-4159.1999.731765.x. [DOI] [PubMed] [Google Scholar]
- 29.Matsuda S, Launey T, Mikawa S, Hirai H. EMBO J. 2000;9:2765–2774. doi: 10.1093/emboj/19.12.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osten P, Khatri L, Kohr G, Giese G, Daly C, Schulz TW, Wensky A, Lee LM, Ziff EB. Neuron. 2000;27:313–325. doi: 10.1016/s0896-6273(00)00039-8. [DOI] [PubMed] [Google Scholar]
- 31.Mizuguchi H, Xu Z, Ishii-Watabe A, Uchida E, Hayakawa T. Mol. Ther. 2000;1:376–382. doi: 10.1006/mthe.2000.0050. [DOI] [PubMed] [Google Scholar]
- 32.Goodnight J-A, Mischak H, Kolch W, Mushinski JF. J. Biol. Chem. 1995;270:9991–10001. doi: 10.1074/jbc.270.17.9991. [DOI] [PubMed] [Google Scholar]
- 33.Hirbec H, Francis JC, Lauri SE, Braithwaite SP, Dev KK, Couthino V, Meyer G, Isaac JTR, Collingridge GL, Henley JM. Neuron. 2003;37:625–638. doi: 10.1016/s0896-6273(02)01191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang W-L, Yeh S-F, Chang Y-I, Hsiao S-F, Lian W-N, Lin C-H, Huang C-YF, Lin W-J. J. Biol. Chem. 2003;278:37705–37712. doi: 10.1074/jbc.M304619200. [DOI] [PubMed] [Google Scholar]
- 35.Shi S-H, Hayashi Y, Esteban JA, Malinow R. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- 36.Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Nat. Neurosci. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]