

Abstract
Cerebellar long-term depression is thought to underlie motor learning and is mediated by internalization of AMPA receptors from the neuronal plasma membrane. In this issue of Neuron, Steinberg et al. provide firm evidence that PICK1 and the C terminus of GluR2 are central to this process by analyzing three different transgenic mice.
Over the past decade, the discovery and elucidation of AMPA receptor (AMPAR) interacting proteins has dramatically increased our understanding of the molecular processes that control constitutive and activity-dependent regulation of functional synaptic AMPARs. This is important because AMPAR trafficking mediates the expression of many forms of synaptic plasticity that, in turn, are believed to be the cellular processes underlying learning and memory. PICK1, an interactor that binds to the GluR2 AMPAR subunit, has attracted particular attention.
The groups of David Linden and Rick Huganir have made leading contributions toward understanding the molecular mechanisms of a particular form of synaptic plasticity, long-term depression (LTD) at the parallel fiber-Purkinje cell synapse in the cerebellum. Linden and colleagues previously demonstrated that cerebellar LTD is mediated by the removal of synaptic AMPARs by clathrin-mediated endocytosis (Wang and Linden, 2000). In collaboration with Huganir, they went on to show that GluR2-PICK1 interactions are required as part of this trafficking mechanism (Xia et al., 2000). Using cultures derived from AMPAR subunit GluR2 knockout mice transfected with mutated recombinant GluR2, they demonstrated that phosphorylation of serine880 at the carboxy-terminus of GluR2 was an absolute requirement for cerebellar LTD in vitro (Chung et al., 2003). They went on to identify PKCα as the critical kinase by using an siRNA approach to knockdown various PKC isoforms, in conjunction with transfection-based rescue (Leitges et al., 2004).
In this issue of Neuron, Steinberg et al. (2006) report the use of three transgenic mouse lines to consolidate some of their previous findings and provide further information about cerebellar LTD. First, they used a PICK1 knockout mouse to confirm that PICK1 is required for LTD. Neither cultures nor slices derived from these animals exhibit this form of plasticity unless wild-type PICK1 is transfected into the transgenic Purkinje cells to rescue the phenomenon. They next use this rescue technique to analyze the role of a specific property of PICK1 in LTD. A pair of lysine residues within the crescent-shaped BAR domain have recently been identified as crucial for phospholipid binding (Jin et al., 2006). LTD is not rescued when PICK1 mutated at these lysines is expressed in Purkinje cells, demonstrating that the interaction between PICK1 and invaginating lipid membranes is a requirement for LTD. This important finding provides a mechanism for how PICK1 stimulates receptor trafficking and begs the question of whether PICK1 senses pre-exisiting sites of membrane curvature, such as clathrin-coated pits, or actually initiates membrane invagination independently of the better-known vesicle-forming proteins.
A widely held (but formally unproven) theory of PICK1 function in AMPAR trafficking is that recruits PKCα to GluR2 to facilitate phosphorylation at Ser880. The current study suggests that this is not the case because phorbol ester-activated GluR2 phosphorylation on Ser880 in PICK1 knockout mice is similar to wild-type animals. Nonetheless, it cannot be ruled out that this intense pharmacological PKC activation could bypass the requirement for PICK1 as a scaffold. A more physiological stimulus, or a combination of stimuli, might still show enhanced Ser880 phosphorylation in the presence of a GluR2-PICK1-PKC complex. Linden and colleagues previously demonstrated a requirement for the PKCα PDZ ligand in cerebellar LTD, consistent with a requirement for PKC-PICK1 interactions (Leitges et al., 2004). However, it may be that an alternative PDZ-domain protein other than PICK1 is involved.
Steinberg et al. make use of further transgenic mice to confirm their previous findings that the GluR2 PDZ ligand and phosphorylation at Ser880 are required for LTD (Chung et al., 2003; Xia et al., 2000). Cultures and slices prepared from mice with the last seven amino acids deleted at the carboxy terminus (GluR2Δ7) or with the mutation K882A (which does not directly affect interactions with PDZ domains but does block PKC phosphorylation by disrupting the kinase consensus recognition sequence) show no cerebellar LTD. An interesting additional observation is that basal levels of GluR2 phosphorylation at Ser880 are unaffected in the K882A mice, whereas TPA-stimulated phosphorylation is completely blocked. This unexpected result suggests that PKC is specifically involved in LTD expression and that a different and as yet unidentified kinase maintains the levels of phosphorylated GluR2 under conditions of normal synaptic transmission.
The authors also analyze the subcellular distribution of GluR2 in the three transgenic mice lines with immunogold EM. Intriguingly, all three transgenics show near-identical alterations in GluR2 distribution. The fact that knocking out PICK1, truncating GluR2, and rendering GluR2 insensitive to PKC phosphorylation all result in similar effects on the distribution of this subunit infers that the same mechanism is being disrupted in all three strains of mice. Although the number of GluR2 subunits present in the PSD remains unchanged, the number of intracellular receptors in the spine and also the pool at extrasynaptic plasma membrane sites in the spine are increased in the mutant mice. This seems to be at the expense of the intracellular dendritic and somatic pool because the synaptic pool is unchanged. Given that the sites of AMPAR internalization are most likely adjacent to the synapse, rather than in the PSD itself (Blanpied et al., 2002), an interpretation of these data is that receptors that would otherwise be internalized into the dendritic shaft are held up at these sites in the absence of the PICK1-GluR2 interaction (Figure 1). These data also suggest that PICK1 is not involved in the maintenance of synaptic AMPARs, nor is it required for the lateral diffusion of receptors from PSD to extrasynaptic sites. One possibility not explored in this study is that the intracellular pool in the spine (that the authors suggest is associated with endoplasmic reticulum) may represent newly synthesized/delivered receptors that arrive at the spine but are unable to exit the ER. Indeed, PICK1 has been suggested to play a role in ER exit of GluR2-containing AMPARs (Greger et al., 2002).
Figure 1.
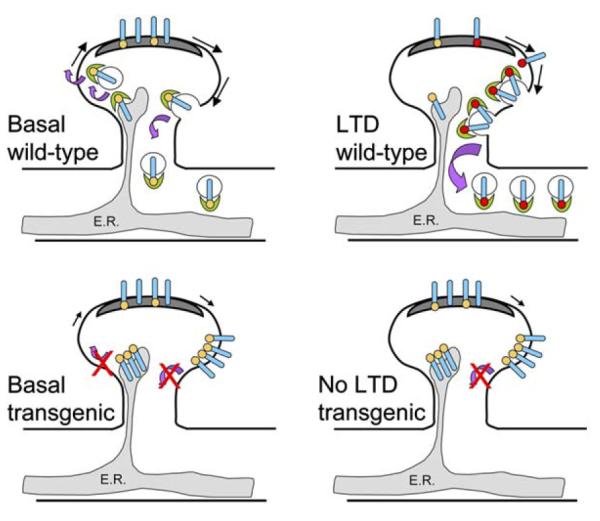
Schematic of Effects of In Vivo Blockade of PICK1-GluR2 Interaction
Wild-type: induction of LTD results in PICK1 (green crescent) mediated internalization of PKCα phosphorylated Ser880 (red dot) GluR2-containing AMPARs (blue cylinder) from the extrasynaptic spine plasma membrane into the dendrite. There is basal phosphorylation (orange dot) of GluR2 at Ser880 mediated by a kinase other than PKCα. PICK1 does not recruit PKCα to GluR2; rather, the BAR domain of PICK1 is involved in sensing or creating the membrane curvature required for vesicle formation. PICK1 may also be necessary for the export of new AMPARs from the ER (gray shaded region). Representation of forward traffic has been omitted in the LTD diagram for clarity. Transgenic animals: in animals where the PICK1-GluR2 interaction is prevented, there is no change in the number of AMPARs in the PSD (dark gray). However, there is an increase in surface AMPARs on the spine outside the PSD and a decrease in AMPARs in the dendritic shaft presumably because AMPARs fail to internalize from the extrasynaptic spine plasma membrane in the absence of GluR2-PICK1 interactions. The build up of AMPARs inside the spine reflects a role for PICK1 in exit of GluR2-containing AMPARs from the ER. There is no LTD because AMPAR internalization is PICK1 dependent.
This paper puts PICK1 firmly at the core of the protein machinery involved in expression of cerebellar LTD. PICK1 has also been implicated in hippocampal LTD (Kim et al., 2001), so it would be informative to investigate whether this form of synaptic plasticity is absent in the knockout mice. Certain functional aspects of PICK1 have now been studied in hippocampal neurons, and others in cerebellum. It remains to be seen whether PICK1 plays exactly the same role in these two important brain regions. The precise molecular mechanisms of PICK1’s action will surely be a topic of intense future research, particularly given reports of key properties besides its ability to bind GluR2. PICK1 binds lipid membranes and likely senses membrane curvature via the BAR domain (Peter et al., 2004; Jin et al., 2006; Steinberg et al., 2006). In this way, it may direct AMPARs destined for internalization to membrane invaginations such as clathrin-coated pits. In a similar manner, the BAR domain could be involved in the budding of GluR2-containing vesicles from the ER. PICK1 is also a calcium sensor; stimulated calcium influx enhances PICK1-GluR2 binding to initiate AMPAR endocytosis (Hanley and Henley, 2005). It will be of great interest to investigate whether PICK1 plays an active role in vesicle movement after membrane invagination.
In order to fully define the roles of PICK1, a great deal of further work is needed, from the systems level to the molecular level. For example, behavioral analyses of the mutant mice reported here is essential and should be extremely informative. Complete deletion of PICK1 may be too drastic a genotype to readily assess, especially given PICK1 has numerous binding partners. However, the more subtle K882A GluR2 mutant may well provide a valuable resource. Such behavioral studies are likely to provide long-awaited links between the molecules of synaptic plasticity and behavioral learning paradigms.
Selected Reading
- Blanpied TA, Scott DB, Ehlers MD. Neuron. 2002;36:435–449. doi: 10.1016/s0896-6273(02)00979-0. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Steinberg JP, Huganir RL, Linden DJ. Science. 2003;300:1751–1755. doi: 10.1126/science.1082915. [DOI] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Ziff EB. Neuron. 2002;34:759–772. doi: 10.1016/s0896-6273(02)00693-1. [DOI] [PubMed] [Google Scholar]
- Hanley JG, Henley JM. EMBO J. 2005;24:3266–3278. doi: 10.1038/sj.emboj.7600801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Ge W-P, Xu J, Cao M, Peng L, Yung W, Liao D, Duan S, Zhang M, Xia J. J. Neurosci. 2006;26:2380–2390. doi: 10.1523/JNEUROSCI.3503-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, Lee HK, Huganir RL. Proc. Natl. Acad. Sci. USA. 2001;98:11725–11730. doi: 10.1073/pnas.211132798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitges M, Kovac J, Plomann M, Linden DJ. Neuron. 2004;44:585–594. doi: 10.1016/j.neuron.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL. Neuron. 2006;49(this issue):845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Wang YT, Linden DJ. Neuron. 2000;25:635–647. doi: 10.1016/s0896-6273(00)81066-1. [DOI] [PubMed] [Google Scholar]
- Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ. Neuron. 2000;28:499–510. doi: 10.1016/s0896-6273(00)00128-8. [DOI] [PubMed] [Google Scholar]