

Abstract
The current optimization of 2,4-diarylaniline analogs (DAANs) on the central phenyl ring provided a series of new active DAAN derivatives 9a–9e, indicating an accessible modification approach that could improve anti-HIV potency against wild-type and resistant strains, aqueous solubility, and metabolic stability. A new compound 9e not only exhibited extremely high potency against wild-type virus (EC50 0.53 nM) and several resistant viral strains (EC50 0.36 – 3.9 nM), but also showed desirable aqueous solubility and metabolic stability, which were comparable or better than those of the anti-HIV-1 drug TMC278 (2). Thus, new compound 9e might be a potential drug candidate for further development of novel next-generation NNRTIs.
Keywords: Diarylaniline, NNRTIs, lead optimization, anti-HIV agents
Since the first case of acquired immunodeficiency syndrome (AIDS) was reported by the U. S. in 1981, the scientific progress in HIV/AIDS research has been extraordinary, especially in the development of antiretroviral therapy (ART) that has proven to be life-saving to millions of people. Recent scientific evidence has demonstrated that ART is also effective at preventing infection1,2; thus it offers an unprecedented opportunity to control the AIDS pandemic. Therefore, the discovery and development of novel highly potent anti-HIV drugs is imperative.
Among current anti-HIV drugs, non-nucleoside reverse transcriptase inhibitors (NNRTIs) are used in combination with other drugs as key components in the highly active antiretroviral therapy (HAART)3,4. NNRTIs target an allosteric binding pocket on HIV-1 reverse transcriptase (RT) in a noncompetitive manner to cause distortion of the three-dimensional structure of the enzyme to inhibit RT catalytic function5. Currently, five NNRTIs drugs have been approved by FDA and are marketed (Figure 1) with the advantages of high potency and low toxicity. Despite the clinical high efficiency, the first-generation NNRTIs drugs (nevirapine, delavirdine, efavirenz) are limited in clinical use due to rapid emergence of drug-resistance. However, the second-generation NNRTI drugs etravirine (TMC125, 1)6 and rilpivirine (TMC278, 2)7 greatly overcome this deficiency. Both 1 and 2 possess potent antiviral activity with subnano- or low nano-molar EC50 values against wild-type and a broad spectrum of mutated viral strains. There is also a higher genetic barrier8,9 to delay the emergence of drug-resistance toward etravirine and rilpivirine10. Although both drugs are poorly water soluble over a wide pH range, 2 has the advantage of better bioavailability when compared to 1, resulting in a once-daily oral dosing. The success of etravirine and rilpivirine greatly encouraged more research to explore additional novel next-generation NNRTI agents with new scaffolds, high potencies, improved resistant profiles, and better pharmacokinetic profiles than 1 and 2 for more efficacious therapy and potential AIDS prevention.
Figure 1.

Current marketed NNRTIs
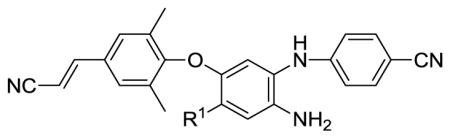
In prior studies toward the design and synthesis of novel next-generation NNRTIs, we discovered a series of new diarylanilines (DAANs, Figure 2) with nano- to subnano-molar anti-HIV potencies against wild-type and multi-RT-resistant viral strains11,12, as exemplified by compound 3 with higher potency against wild-type (EC50 0.38 nM) and RT multi-resistant viral strains (EC50 0.87 nM) than 1 in the same assays. Previous SAR results also defined the key pharmacophores of DAANs as NNRTIs: (1) a para-cyanoaniline moiety (A-ring), (2) a crucial amino group on the central phenyl ring (B-ring) ortho to the A-ring position, (3) a trisubstituted phenoxy ring (C-ring) with a para-linear hydrophobic substituent, and (4) similar molecular flexibility to 1 and 2. Their high potency and straightforward synthesis prompted us to develop DAANs as new anti-AIDS drug candidates. For orally anti-HIV drug candidates, aqueous solubility is one of the most critical physicochemical properties to be considered in the process of lead optimization, because low aqueous solubility would limit molecular absorption and bioavailability in vivo. On the other hand, most small organic molecules generally have poor water solubility. Therefore, our current optimization of DAANs must generate active DAANs with desirable molecular aqueous solubility. Our early results indicated that the nitro group (R1) on the central phenyl ring (B-ring) of DAANs was associated with antiviral potency by creating a small electrostatic interaction with the positively charged amino acid K172 on the NNRTI binding site.11 Thus, we proposed that other polar or ionizable groups, similar to the nitro group, on the B-ring might provide comparable or additional interaction point(s) with the NNRTI binding site via a “salt bridge” and/or H-bonds to enhance antiviral potency. Furthermore, the presence of R1 substituent(s) to serve as a H-bond acceptor or donor is expected to be favorable for improving aqueous solubility. Therefore, a series of new DAAN derivatives with different R1 groups on the central phenyl ring are reported herein, including chemical synthesis, antiviral activity, aqueous solubility, and metabolic stability in vitro.
Figure 2.

2,4-Diarylaniline leads
As shown in Scheme 1, the nitration of 2,4-dichlorobenzoic acid, a commercially available and inexpensive reagent, was performed with concentrated HNO3 in H2SO4 at low temperature (0–5 °C) to produce 2,4-dichloro-5-nitrobenzoic acid (4) with a 91% yield. Compound 4 was esterified with MeOH in the presence of H2SO4 to afford methyl 2,4-dichloro-5-nitrobenzoate (5). Coupling of 5 and 4-aminobenzo-nitrile was performed in DMF in the presence of excess cesium carbonate at 100 °C for about 10 h to provide methyl 2-chloro-4-(4-cyanophenyl amino)-5-nitrobenzoate (6), which was characterized by an NH signal at δ 9.76 ppm in the 1H NMR spectrum, ascribable to the chelated nitro group and consistent with previous results that the chlorine ortho to the nitro group has higher reactivity for nucleophilic substitution with an aromatic amine. Next, intermediate 6 was reacted with 4-hydroxy-3,5-dimethylbenzaldehyde under microwave irradiation in DMF in the presence of potassium carbonate with stirring at 190 °C for about 15 min to afford 7 with a three-phenyl ring skeleton in a 67% yield. Subsequently, the aldehyde group in 7 was converted into a cyanovinyl moiety by condensation with diethyl cyanomethyl phosphonate in the presence of potassium tert-butoxide to afford compound 8a. Then, the ester group in 8a was hydrolyzed under basic conditions to afford the corresponding benzoic acid compound 8b. The carboxylic acid group was then converted into an amide or N-methyl amide by treatment with SOCl2, followed with ammonia or methylamine to afford corresponding compounds 8c and 8d respectively. In addition, the ester group in 8a was reduced with LiBH4 to provide compound 8e with a hydroxymethyl substituent. Finally, the nitro group on the central ring in compounds 8a–8e was reduced by using sodium hydrosulfite dehydrate to produce corresponding target compounds 9a–9e, a series of 5-substituted 2-(4-cyanophenylamino)-4-(4-cyanovinyl-2,6-dimethylphenoxy)anilines.13
Scheme 1.

Reagent and conditions: (a) HNO3/H2SO4, 0–5 °C to r.t., 2 h; (b) H2SO4/MeOH, reflux, 2 h; (c) Cs2CO3/DMF, 100 °C, 10 h; (d) K2CO3/DMF, 190 °C, microwave, 10–15 min; (e) (EtO)2P(O)CH2CN, t-BuOK/THF, 0 °C to r.t., 3 h; (f) THF/MeOH, aq NaOH, 0.5 h; (g) (i) CH2Cl2/SOCl2, reflux, 3 h; (ii) ammonia in THF, 0 °C, 0.5 h; (h) (i) SOCl2/CH2Cl2, reflux, 3 h; (ii) methylamine in THF, 0 °C, 0.5 h; (j) LiBH4, THF/MeOH, 1 h; (k) Na2S2O4, ammonia, THF/H2O (1:1, v/v), r.t., 2 h.
New compounds 9a–9e were first tested against wild-type HIV-1 NL4-3 infection of TZM-bl cells in parallel with TMC278 and 3. The data are summarized in Table 1. Compounds 9c (R1 = CONH2) and 9e (R1 = CH2OH) exhibited extremely high potency with EC50 values of 0.87 nM and 0.53 nM, respectively, and both showed high selective indexes (SIs) of >27,126, comparable to those of new drug 2 (TMC278) and 3 in the same assay. Compounds 9a (R1 = COOCH3) and 9d (R1 = CONHCH3) were also potent with EC50 values of 2.7 nM and 5.7 nM, respectively, and SI values of > 8464 and >1606, respectively. On the other hand, 9b with a carboxylic acid group (R1 = COOH) was significantly less potent (EC50 230 nM). Thus, the current results, even with the limited data set, demonstrated that the R1 substituent on the central phenyl ring was modifiable and could greatly affect antiviral potency. The aqueous solubility of new compounds 9a–9e at pH 7.4 and pH 2.0 were measured by a HPLC/UV method in parallel with 2 and 3, and the data are also shown in Table 1. Compared to 2 and 3, all new 9 series compounds had improved aqueous solubility at pH 7.4 and were generally more soluble at pH 2.0, likely due to the presence of a free amino group. The most active new compound 9e (R1 = CH2OH) showed better aqueous solubility (3.23 μg/mL at pH 7.4 and 21.0 μg/mL at pH 2.0) than the remaining four compounds in the series. Subsequently, the most active compounds 9c and 9e were selected for testing against NNRTI resistant HIV-1 mutants RT-multi-drug-resistant (RTMDR), K101E, and E138K. As shown in Table 2, new compounds 9c and 9e exhibited improved antiviral activity compared to 3 against the drug resistant viral strains, and a little better or comparable to those of 2. Compounds 9c and 9e were further evaluated in rat and human liver microsome (RLM and HLM, respectively) assays in vitro,14,15 respectively, to predict their metabolic stability in vivo. The two data sets in Table 2 indicated a similar metabolic stability pattern. Compounds 9c and 9e had moderate stability with half-lives of 35 min and 30 min in HLM and 122 min and 84 min in RLM assays, respectively. Compound 9c had a longer maintaining time (was more stable) than 9e in both assays, and both compounds showed comparable metabolic stability to that of 2 (t1/2 34 min) in the HLM assay.
Table 1.
Anti-HIV activities of 9a–9e in cellular assay and aqueous solubilitya
| ||||||
|---|---|---|---|---|---|---|
| R1 | EC50 (nM)b | CC50 (μM)c | SId | Aqueous Solubility (μg/mL)
|
||
| pH 7.4 | pH 2.0 | |||||
| 9a | COOCH3 | 2.74 ± 0.71 | >22.8 | >8465 | 1.30 | 1.75 |
| 9b | COOH | 230 ± 38 | 14.9 | 65 | 117 | 4.40 |
| 9c | CONH2 | 0.87 ± 0.28 | >23.6 | >27126 | 0.63 | 2.10 |
| 9d | CONHCH3 | 5.72 ± 0.94 | >9.2 | >1606 | 1.32 | 8.63 |
| 9e | CH2OH | 0.53 ± 0.13 | 16.3 | 30754 | 3.23 | 21.0 |
| 3e | NO2 | 0.38 ± 0.07 | >47.1 | >123947 | 0.29 | 0.29 |
| 2 (TMC278) | 0.52 ± 0.14 | 19.4 | 37305 | 0.24 | 156 | |
HIV-1 NL3-4 (wild-type) virus in TZM-bl cell lines.
Concentration of compound that causes 50% inhibition of viral replication and presented as mean ± standard deviation (SD) in at least triplicate.
Concentration of compound that causes cytotoxicity to 50% of cells.
Selectivity index (SI) is the ratio of CC50/EC50.
Compound and activity data were published previously12
Table 2.
Data for 9c and 9e against resistant viral strains and metabolic stability
| EC50 (nM)a | human liver microsomeb | rat liver microsomeb | |||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| HIV-1RTMDR1 | K101E | E138K | t1/2 min | CL mL/min/mg | t1/2 min | CL mL/min/mg | |
| 9c | 0.31± 0.10 | 8.8 ± 3.3 | 9.0 ± 2.6 | 35 | 0.20 | 122 | 0.057 |
| 9e | 0.36 ± 0.10 | 3.4± 1.05 | 3.9 ± 0.90 | 30 | 0.23 | 84 | 0.083 |
| 3 | 0.87 ± 0.33 | 10.8 ± 3.53 | 11.3 ± 3.76 | 47 | 0.15 | 139 | 0.050 |
| 2 | 0.49 ± 0.10 | 5.7 ± 1.4 | 5.2 ± 1.6 | 34 | 0.20 | 120 | 0.057 |
Experiments were performed at least in triplicate and data presented as mean ± SD.
Data from at least two experiments and presented as means.
In conclusion, our optimization focused on the central phenyl ring of DAANs provided a series of new active compounds 9a–9e and also verified our previous hypothesis that the R1 substitution could optimize the antiviral potency and drug-like properties. This study resulted in the discovery of a new compound 9e that not only exhibited extremely high potency against wild-type virus (EC50 0.53 nM) and several resistant viral strains (EC50 0.36 – 3.9 nM), but also showed desirable aqueous solubility and metabolic stability, which were better than those of drug 2. Thus, compound 9e could serve as a potential drug candidate for further development.
Acknowledgments
This investigation was supported by grants 30930106 and 81120108022 from the Natural Science Foundation of China (NSFC) awarded to L. Xie and U.S. NIH grants awarded to C. H. Chen (AI65310) and K. H. Lee (AI33066).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Fauci AS. Science. 2011;333:13. doi: 10.1126/science.1209751. [DOI] [PubMed] [Google Scholar]
- 2.Shattock RJ, Warren M, McCormack S, Hankins C. Science. 2011;333:42. doi: 10.1126/science.1206399. [DOI] [PubMed] [Google Scholar]
- 3.Kaufmann GR, Cooper DA. Curr Opin Microbiol. 2000;3:508. doi: 10.1016/s1369-5274(00)00131-4. [DOI] [PubMed] [Google Scholar]
- 4.Vella S, Palmisano L. Antiviral Res. 2000;45:1. doi: 10.1016/s0166-3542(99)00068-6. [DOI] [PubMed] [Google Scholar]
- 5.Andries K, Azijn H, Thielemans T. Antimicrob Agents Chemother. 2004;48:4680. doi: 10.1128/AAC.48.12.4680-4686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pecora Fulco P, McNicholl IR. Pharmacotherapy. 2009;29:281. doi: 10.1592/phco.29.3.281. [DOI] [PubMed] [Google Scholar]
- 7.De Clercq E. Int J Antimicrob Agents. 2009;33:307. doi: 10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 8.Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, Arnold E. J Mol Biol. 2009;385:693. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vingerhoets J, Azijn H, Fransen E, De Baere I, Smeulders L, Jochmans D, Andries K, Pauwels R, de Bethune MP. J Virol. 2005;79:12773. doi: 10.1128/JVI.79.20.12773-12782.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pecora Fulco P, McNicholl IR. Pharmacotherapy. 2009;29:281. doi: 10.1592/phco.29.3.281. [DOI] [PubMed] [Google Scholar]
- 11.Qin BJ, Jiang XK, Lu H, Tian XT, Barbault F, Huang L, Qian K, Chen CH, Huang R, Jiang S, Lee KH, Xie L. J Med Chem. 2010;53:4906. doi: 10.1021/jm1002952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tian XT, Qin BJ, Wu ZY, Wang XF, Lu H, Morris-Natschke SL, Chen CH, Jiang S, Lee KH, Xie L. J Med Chem. 2010;53:8287. doi: 10.1021/jm100738d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Synthetic procedure for 4-substituted 1,5-diarylbenzene-1,2-diamines (9a–e). To a solution of a diaryl-nitrobenzene (8, 1 equiv) in THF and water (30 mL, v/v 1:1) was added ammonia aqueous solution (25%) and sodium hydrosulfite (10 equiv) successively with stirring at room temperature for 2 h. The reaction was monitored by TLC (CH2Cl2/MeOH 60:1) until completed. The mixture was poured into ice-water and extracted with EtOAc three times. After removal of organic solvent under reduced pressure, crude product (9) was purified by a flash silica gel column chromatograph (eluent: CH2Cl2/MeOH = 30/1) with the CombiFlash Flash chromatography system, ISCO company, Inc. to obtain pure target compounds 9a–9e respectively. HPLC analyses for purities of 9a–e were performed on an Agilent 1200 HPLC system with UV detector and a Grace Alltima HP C18 column (100 × 2.1 mm, 3 μm) eluting with a mixture of solvents A and B in two conditions: (1) acetonitrile (ACN)/water 70:30, flow rate 1.0 mL/min; (2) MeOH/water 70:30, flow rate 0.8 mL/min. The samples were detected under UV wavelength at 254 nm and an injection volume of 3 μL. Compounds: 9a, yield 64%, white solid, mp 240–242 °C; 1H NMR (CDCl3) δ ppm 2.16 (6H, s, CH3×2), 3.93 (3H, s, OCH3), 5.78 (1H, s, NH), 5.80 (1H, d, J = 16.8 Hz, =CH), 6.20 (1H, s, ArH-6), 6.67 (2H, d, J = 8.8 Hz, ArH), 7.17 (2H, s, ArH), 7.31 (1H, d, J = 16.8 Hz, CH=), 7.42 (2H, d, J = 8.8 Hz, ArH), 7.45 (1H, s, ArH-3); MS m/z (%) 439.3 (M+1, 100); HPLC-purity 96.1%. 9b: yield 35%, brown solid, mp 226–228 °C. 1H NMR (CDCl3) δ ppm 2.19 (6H, s, CH3×2), 5.84 (1H, d, J = 16.8 Hz, =CH), 6.04 (1H, s, NH), 6.27 (1H, s, ArH-6), 6.75 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.22 (2H, s, ArH-3″,5″), 7.32 (1H, d, J = 16.8 Hz, CH=), 7.44 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.72 (1H, s, ArH-3); MS m/z (%) 423.2 (M-1, 100); HPLC purity 100.0%. 9c: yield 63%, white solid, mp 290–292 °C; 1H NMR (DMSO-d6) δ ppm 2.10 (6H, s, CH3×2), 4.75 (2H, s, NH2), 6.01 (1H, s, ArH-6), 6.39 (1H, d, J = 16.8 Hz, =CH), 6.63 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.45 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.47 (2H, s, ArH-3′,5′), 7.57 (1H, d, J = 16.8 Hz, CH=), 7.61 (1H, s, ArH-3), 8.20 (1H, s, NH); MS m/z (%) 424.2 (M+1, 100); purity (HPLC) 98.2%. 9d: yield 31%, white solid, mp 112–114 °C; 1H NMR (CDCl3) δ ppm 2.16 (6H, s, CH3×2), 3.07 (3H, d, NCH3), 5.80 (1H, s, NH), 5.83 (1H, d, J = 16.8 Hz, =CH), 6.18 (1H, s, ArH-6), 6.65 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.21 (2H, s, ArH-3″,5″), 7.32 (1H, d, J = 16.8 Hz, CH=), 7.41 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.81 (1H, s, ArH-3); MS m/z (%) 438.4 (M+1, 100); HPLC-purity 100.0%. 9e: yield 81%, white solid, mp 186–188 °C; 1H NMR (CDCl3) δ ppm 2.13 (6H, s, CH3×2), 4.87 (2H, s, CH2), 5.50 (1H, s, NH), 5.79 (1H, d, J = 16.8 Hz, CH=), 6.03 (1H, s, ArH-6), 6.55 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.94 (1H, s, ArH-3), 7.17 (2H, s, ArH-3″,5″), 7.30 (1H, d, J = 16.8 Hz, CH=), 7.40 (2H, d, J = 8.8Hz, ArH-3′,5′); MS m/z (%) 411.3 (M+1, 100); HPLC-purity 99.9%.
- 14.Microsomal stability assay. Stock solutions of test compounds (1 mg/mL) were prepared by dissolving the pure compound in DMSO and stored at 4 °C. Before assay, the stock solution was diluted with ACN to 0.1 mM concentration. For measurement of metabolic stability, all test compounds were brought to a final concentration of 1 μM with 0.1 M potassium phosphate buffer at pH 7.4, which contained 0.1 mg/mL human liver microsomes and 5 mM MgCl2. The incubation volumes were 300 μL, and reaction temperature was 37 °C. Reactions were started by adding 60 μL of NADPH (final concentration of 1.0 mM) and quenched by adding 600 μL of ice-cold ACN to stop the reaction at 5, 15, 30, 60 min time points. Samples at 0 min time point were prepared by adding 600 μL ice-cold ACN first, followed by 60 μL NADPH. Incubations of all samples were conducted in duplicate. After quenching, all samples were centrifuged at 12,000 rpm for 5 min at 0 °C. The supernatant was collected, and 20 μL of the supernatant was directly injected onto a Shimadzu LC-MS-2010 system with an electrospray ionization source (ESI) for further analysis. The following controls were also conducted: 1) positive control incubation containing liver microsomes, NADPH, and reference compound; 2) negative control incubation omitting NADPH; and 3) baseline control containing only liver microsomes and NADPH. The peak heights of test compounds at different time points were converted to percentage of remaining, and the peak height values at initial time (0 min) served as 100%. The slope of the linear regression from log percentage remaining versus incubation time relationships (−k) was used to calculate in vitro half-life (t1/2) value by the formula of in vitro t1/2 = 0.693/k, regarded as first-order kinetics. Conversion to in vitro CLint (in units of ml/min/mg protein) was calculated by the formula15: CLint = (0.693/in vitro t1/2) × (ml incubation/mg microsomes).
- 15.Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, MacIntyre F, Rance DJ, Wastall P. J Pharmacol Exp Ther. 1997;283:46. [PubMed] [Google Scholar]