

Abstract
Previously, we have shown that CCR5 transcription is regulated by CREB-1. However, the ubiquitous pattern of CREB-1 expression suggests the involvement of an additional level of transcriptional control in the cell type–specific expression of CCR5. In this study, we show that epigenetic changes (i.e. DNA methylation and histone modifications) within the context of the CCR5 P1 promoter region correlate with transcript levels of CCR5 in healthy and in malignant CD4+ T lymphocytes as well as in CD14+ monocytes. In normal naïve T cells and CD14+ monocytes the CCR5 P1 promoter resembles a bivalent chromatin state, with both repressive and permissive histone methylation and acetylation marks. The CCR5-expressing CD14+ monocytes however show much higher levels of acetylated histone H3 (AcH3) compared to the non–CCR5-expressing naïve T cells. Combined with a highly methylated promoter in CD14+ monocytes, this indicates a dominant role for AcH3 in CCR5 transcription. We also show that pharmacological interference in the epigenetic repressive mechanisms that account for the lack of CCR5 transcription in T leukaemic cell lines results in an increase in CREB-1 association with CCR5 P1 chromatin. Furthermore, RNA polymerase II was also recruited into CCR5 P1 chromatin resulting in CCR5 re-expression. Together, these data indicate that epigenetic modifications of DNA, and of histones, contribute to the control of CCR5 transcription in immune effector cells.
Keywords: chromatin remodelling, histone modifications, DNA methylation, bivalent chromatin, poised chromatin, CCR5, T cells, monocytes
Introduction
The CC chemokine receptor 5 (CCR5) regulates trafficking of lymphoid cells such as memory/effector Th1 lymphocytes, or cells of the myeloid lineage (e.g. monocytes, macrophages, immature dendritic cells) and microglia. As such, CCR5 is implicated in the pathogenesis of various inflammatory diseases such as atherosclerosis and multiple sclerosis [1-4]. Furthermore, CCR5 also functions as a co-receptor for HIV-1 [5-7]. Notably, CCR5 expression is markedly up-regulated upon T cell activation, which allows the activated T cells to migrate towards site(s) of inflammation [8-12].
Upon encountering a pathogen, antigen-presenting cells will present the antigenic peptide to resting naïve T cells, which results in the generation and activation of antigen-specific T cells [13, 14]. After activation, the T cells migrate to the site of inflammation, guided by chemokine receptors [15]. Similarly, circulating monocytes are also attracted to inflammatory sites by chemokine receptors, where they then can differentiate into, e.g. macrophages or microglia [16-18]. Atherosclerosis and multiple sclerosis are greatly characterized by inflammatory lesions, consisting of T cells and macrophages or microglia [19-21]. The chemokine receptor CCR5 has been shown to be implicated in the pathogenesis of both of these diseases [22-25].
Expression of CCR5 is under the control of a complexly organized promoter region upstream of the gene. The main transcriptional activity of the CCR5 promoter region is contained within the downstream promoter P1 [10, 12, 26]. We have previously shown that the transcription factor cAMP responsive element binding protein 1 (CREB-1) transactivates the CCR5 P1 promoter [26]. However, considering the ubiquitous expression of CREB-1 [27], we argued that additional mechanisms, including epigenetic mechanisms, could also contribute to the cell type–specific regulation of CCR5 transcription. In line with this notion is the observation that transient promoter–reporter studies in CCR5-deficient Jurkat T leukaemia cells revealed that the CCR5 promoter–reporter was activated upon transfection [10]. This observation infers that Jurkat T leukaemia cells contain all the transcription factors required for CCR5 transcription, and demonstrates that CCR5 transcription could be additionally controlled by epigenetic mechanisms.
Epigenetic mechanisms control the accessibility of DNA for transcription factors and are thought to form the basis for cell-to-cell inheritance of gene expression profiles [28]. Epigenetic mechanisms as such play an essential role in the regulation of gene transcription. Epigenetic modifications include methylation of DNA at CpG residues and post-translational modifications of histone tails such as acetylation and methylation [29]. Together these modifications form a ‘histone code,’ like the genetic code, that controls transcription levels of genes [30]. Importantly, modifications to DNA and to histone tails are functionally linked [31].
Well-studied mechanisms that underlie gene repression by histone methylation involve tri-methylation of histone H3 at lysine 9 (3MeK9H3) and at lysine 27 (3MeK27H3), and of histone H4 at lysine 20 (3MeK20H4). These modifications are catalysed, respectively, by the lysine methyltransferases (KMTases) SUVAR39H1 (hKMT1A), enhancer of Zeste homologue 2 (EZH2, hKMT6), a subunit of the polycomb repressive complex 2 (PRC-2), and SUV4-20H1/H2 (hKMT5B/C) [32-35]. The KMTase hSet1 and the MLL genes (hKMT2A/G) catalyse tri-methylation of K4-H3 (3MeK4H3) and this modification is associated with gene transcription [35, 36].
Repressive and activating chromatin marks are not mutually exclusive. Bivalent or ‘poised’ chromatin, containing both repressive and permissive histone modifications, was first described in 2006 [37]. Embryonic stem cells where shown to contain regions with both 3MeK27H3 as well as 3MeK4H3. Recently it has been reported that many more forms of bivalent, and even tri- and tetravalent chromatin exists [38]. This underscores the importance of ‘epigenetic plasticity’ and that gene regulation by epigenetic principles is dynamic rather than static.
In this study we show that induction of CCR5 transcription upon CD4+ T cell activation correlates with reduced levels of DNA methylation as well as changes in specific histone modifications within the CCR5 promoter. To establish whether the found epigenetic profiles are T cell specific, we also determined the epigenetic profile in CD14+ monocytes, being of the myeloid instead of the lymphoid lineage. It is shown that the CCR5 chromatin status in primary CD14+ monocytes correlates with the intermediate transcription levels of CCR5. Furthermore, the T leukaemia cell lines studied (Jurkat, Molt-4, HSB-2) do not express CCR5. Subsequently we established that Jurkat cells displayed a transcriptionally repressive CCR5 chromatin environment. Moreover, we show that pharmacological interference in these epigenetic silencing mechanisms in the CCR5-deficient T leukaemia cell lines results in the induction of CCR5 expression. In Jurkat T cells this is accompanied by recruitment of CREB-1 and RNA polymerase II into CCR5 P1 chromatin. Together, these data reveal that epigenetic mechanisms play a pivotal role in the control of CCR5 transcription.
Materials and methods
Cell culture and activation
Naïve human CD4+ T cells were sorted from freshly isolated peripheral blood mononuclear cell (PBMC) using a FACSAria Flow Cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Sorted cells were directly used for chromatin immunoprecipitation (ChIP) analysis, RNA extraction and DNA isolation for bisulphite analysis. Naïve CD4+ T cells were also activated in vitro as described earlier [39]. In brief, naïve CD4+ T cells were stimulated with 1 μg/ml phytohaemagglutinin (PHA; Remel Europe Ltd., Dartford, UK) and 20 U/ml interleukin-2 (IL-2) in the presence of irradiated allogeneic PBMCs (3000 rad). After 11 days of culture, cells were re-stimulated the same way and after 12 days cells were harvested for ChIP analysis and bisulphite sequencing. For RNA-extraction naïve CD4+ T cells were stimulated with anti-CD3 and anti-CD28 for 30 min. Thereafter, CD4+ T cells were cultured for 48 hrs in CFU-EC medium (Stemcell Technologies, Grenoble, France). RNA was isolated with the RNA-Bee extraction method.
The leukaemic T cell lines Jurkat (Clone E6-1; American Type Culture Collection [ATCC]) and Molt-4 (ATCC) were cultured in RPMI-1640 medium (Gibco, Invitrogen, Paisley, UK) supplemented with 10% heat-inactivated foetal calf serum (FCS; PAA), 100 IU/ml streptomycin, 100 IU/ml penicillin (both Lonza, Cologne, Germany) and 2 mM L-glutamine (Gibco). The HSB-2 cell line was cultured in Iscove's modified Dulbecco's medium (IMDM; Lonza), supplemented with 10% heat-inactivated FCS, 100 IU/ml streptomycin, 100 IU/ml penicillin and 2 mM L-glutamine.
To obtain CD14+ monocytes, PBMCs were freshly isolated from the blood of healthy volunteers by density gradient centrifugation using Ficoll-Paque™ PLUS (GE Healthcare, Buckinghamshire, UK). Monocytes were enriched from the PBMC fraction by magnetic separation with CD14 magnetic beads (MACS; Miltenyi Biotec, Bergisch Gladbach, Germany).
Fluorescence activated cell sorting analysis
CCR5 expression on Jurkat, HSB-2, Molt-4 and primary T cells was determined by fluorescence activated cell sorting (FACS) analysis, using the mouse monoclonal antibody MC-5 (kind gift of Prof. M. Mack, University of Regensburg, Regensburg, Germany) and a PE-conjugated anti-mouse IgG secondary antibody (Becton Dickinson) and the appropriate controls. FACS data acquisition was performed on a FACSCalibur flow cytometer (Becton Dickinson) using Cell Quest programming. Fluorescence activated cell sorter data were analysed using the FlowJo software package.
Bisulphite sequencing
Total genomic DNA was isolated from naïve and activated T cells, Jurkat T leukaemia cells and CD14+ monocytes. One microgram of genomic DNA was used to bisulphite convert unmethylated CpGs using the EZ DNA Methylation kit (Zymo Research, Irvine, CA, USA). CCR5 promoter DNA was then amplified using primer sets for specific CpG containing regions (Table 1, Fig. 2). PCR products were purified using the NucleoSpin Extract II kit (Macherey-Nagel, Düren, Germany), cloned into pGEM-T easy vector (Promega, Madison, WI, USA) and individual clones were sequenced at the Leiden Genome Technology Center. Results of at least 10 individual clones are represented as pie charts for each CpG analysed. The percentage of methylated clones is depicted in black.
Table 1.
Primers used for ChIP, bisulphite sequencing and qPCR
| Gene | Promoter region | Region spanning, relative to CDS | Primer sequence, 5′-3′ | Application |
|---|---|---|---|---|
| CCR5 | B1 | −3509 to −3090* | F: TGTTATTGAGTTTTGTTGTAGTATAGATA | |
| R: ACCAAACTTAAAACCTATCTTACCC | ||||
| B3 | −2625 to −2434* | F: TTTAGAAAAAGATGGGAAATTTGTT | Bisulphite | |
| R: TCCTAAACTTCACATTAACCCTATATC | ||||
| B4/5 | −2210 to −1866* | F: TTAATAGATTTTGTGTAGTGGGATGAGTA | ||
| R: CTCATCTCAAAAACTAACTAACAAAC | ||||
| −2277 to −1932* | F: TGTGGGCTTTTGACTAGATGA | ChIP | ||
| R: TAGGGGAACGGATGTCTCAG | ||||
| −47 to +188† | F: CTGAGACATCCGTTCCCCTA | |||
| R: GCTCTTCAGCCTTTTGCAGT | qPCR | |||
| RPII | +3993 to +4172#135; | F: CAGGAGTGGATCCTGGAGAC | ||
| R: GGAGCCATCAAAGGAGATGA | ||||
| CREB-1 | +276 to +609 (isoform a)# | F: AACCAGCAGAGTGGAGATGCAGCT | Semi-quantitative | |
| +276 to +659 (isoform b)§ | R: CTGTAGGAAGGCCTCCTTGAAAGA | PCR | ||
| ICER | +150 to +750¶ | F: CAGATCCGAGCTCCTACTGC | Semi-quantitative | |
| R: CAACTCGGCTCTCCAGACAT | PCR |
Based on accession number NC_000003.10. †Based on accession number NM_000579.3. ‡Based on accession number NM_000937.2. #Based on accession number NM_004379.3. §Based on accession number NM_134442.3. ¶Based on accession number NM_182717.1.
Fig 2.
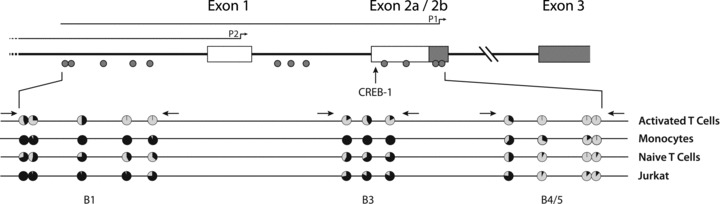
Organization of the CCR5 promoter and methylation analysis of several cell types. Each circle represents a single CpG residue. The percentage of clones methylated at a specific residue is indicated by the black colour. The distance between each circle represents the relative distance between CpG residues on the genomic sequence. Horizontal arrows indicate the relative position of primers used to amplify bisulphite modified DNA. The CREB-1 binding site most likely involved in CCR5 transactivation is indicated with a vertical arrow [26].
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as described earlier [26]. One microgram of cross-linked DNA was immunoprecipitated with antibodies (5 μg) directed to specific histone modifications (Table 2), or no antibody as background control. Quantitative PCR (qPCR) of the immune-precipitated chromatin was performed using the primer pairs shown in Table 1.
Table 2.
Antibodies used for ChIP
| Antibody reactivity | Manufacturer | Catalogue no. |
|---|---|---|
| AcH3 | Millipore | 06-599 |
| 3MeK4H3 | Cell Signalling Technology | 97510 |
| 3MeK9H3 | Abcam | ab8898 |
| 3MeK20H4 | Abcam | ab9053 |
| 3MeK27H3 | Millipore | 07-449 |
| CREB-1 | Rockland | 100-401-195; [62] |
| RNA pol II | Santa Cruz | sc899x |
Zebularine, DZNep and MS275 treatment
For induction of expression of CCR5, Jurkat, HSB-2 and Molt-4 cells were exposed to 100 μM of Zebularine (V.E. Marquez) for 96 hrs followed by an additional treatment with 2 μM of 3-deazaneplanocin A (DZNep, V.E. Marquez) for 72 hrs and 0.5 μM MS275 (Sigma-Aldrich) for 48 hrs in IMDM (HSB-2) or RPMI-1640 (Jurkat and Molt-4) with supplements as described in previous sections.
RNA isolation and (quantitative) RT-PCR
Total RNA was isolated using the RNA-Bee extraction method (Tel-Test) from naïve and activated CD4+ T cells, from CD14+ monocytes and from Jurkat, HSB-2 and Molt-4 cells prior to and after treatment with Zebularine, DZNep and MS275. From 1 μg of RNA, cDNA was synthesized using 250 ng Random hexamers (Promega) and Superscript III reverse transcriptase (Invitrogen).
CCR5 and RNA polymerase II (RPII) transcripts were quantified on an iCycler IQ system (Bio-Rad Laboratories, Hercules, CA, USA) using the IQ SYBR Green Supermix. Relative transcript levels of CCR5 were calculated with the comparative Ct method (or ΔΔCt method) and related to RPII transcript levels. The induced levels of CCR5, after treatment of Jurkat, Molt-4 and HSB-2 cells with Zebularine, DZNep and MS275, are also depicted relative to the CCR5 expression level in in vitro activated primary T cells. The primers used in the qPCR reactions are shown in Table 1.
CREB-1 and inducible cAMP early repressor (ICER), the inducible isoform of cAMP-responsive element modulator (CREM) transcripts were analysed in triplicate by semi-quantitative PCR as previously described [26]. PCR products were separated by gel electrophoresis on a 1.5% agarose gel, run at 90 V for 45 min., and visualized by ethidium bromide staining. Densitrometric analysis was performed in ImageJ (U. S. National Institutes of Health, Bethesda, MD, USA, http://imagej.nih.gov/ij/).
Results
DNA methylation patterns of the CCR5 P1 promoter
Using flow cytometry we found that only a few naïve primary CD4+ T lymphocytes express low levels of CCR5 at the cell surface, whereas CCR5 cell surface expression is markedly up-regulated after in vitro activation of these cells (Fig. 1A). The CCR5 cell surface expression pattern of activated CD4+ T cells is accompanied by relatively high levels of CCR5 transcripts (Fig. 1B). In naïve T cells CCR5 transcripts were detected at low levels (Fig. 1B). Myeloid cells, such as monocytes express CCR5 at low to intermediate levels [40]. When compared with activated and naïve CD4+ T cells, CD14+ monocytes indeed show intermediate levels of CCR5 transcripts (Fig. 1B). In contrast, most established tumour T cell lines completely lack CCR5 surface expression, including the human CD4+ leukaemic T cell lines Jurkat, Molt-4 and HSB-2 (Figs 1A and 5A). Furthermore, these leukaemic T cell lines show only very low or undetectable CCR5 transcript levels (Fig. 1B).
Fig 1.
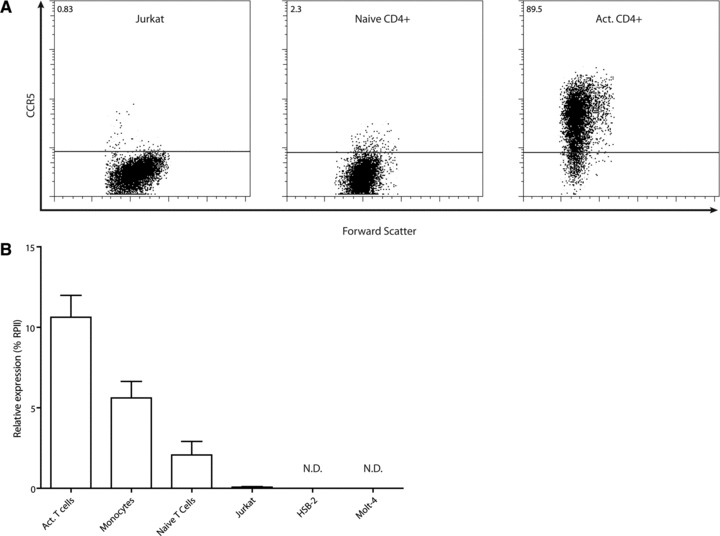
(A) Cell surface expression of CCR5 in Jurkat T leukaemia cells, naïve and activated CD4+ T cells as determined by FACS analyses. Numbers indicate percentage of CCR5 positive cells. (B) Relative transcript levels of CCR5 in various cells types. Numbers indicate expression percentage relative to RNA polymerase II (RPII). N.D. indicates transcript levels were below the detection threshold.
Fig 5.
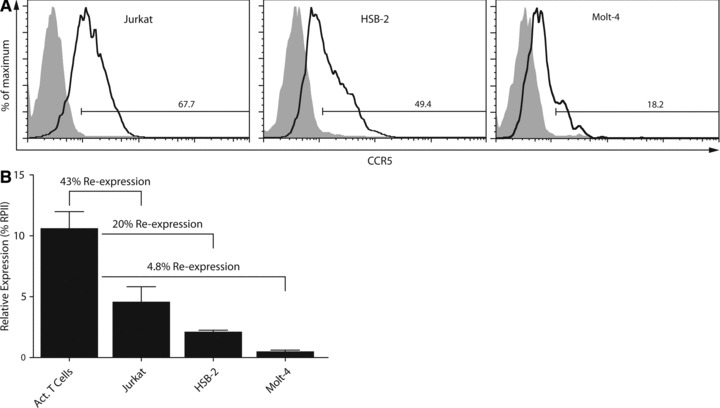
(A) Restoration of CCR5 expression in Jurkat, HSB-2 and Molt-4 T leukaemia cell lines following exposure of cells to Zebularine, DZNep and MS275 determined by FACS analysis. Numbers indicate the percentage of CCR5 positive cells; filled histograms represent the non-treated cells and open histograms the treated cells. (B) Levels of CCR5 transcripts in treated Jurkat, HSB-2 and Molt-4 T leukaemia cells relative to CCR5 transcript levels in activated CD4+ T cells. The transcript level data of activated T cells are the same as shown in Figure 1B.
Evaluating the role of epigenetic mechanisms in the regulation of CCR5 expression we first assessed the CpG methylation status of three subregions of the CCR5 downstream promoter P1 (Fig. 2). The most downstream subregion (B4/5), which is known to be transactivated by CREB-1 [26], appears to be mostly unmethylated and displays only marginal differences in DNA methylation between the various cell types (Fig. 2). The upstream subregions B1 and B3 display remarkable differences in DNA methylation status. In activated T cells, the CpG residues in these subregions of the P1 promoter display low levels of DNA methylation. In monocytes, which express intermediate levels of CCR5, the promoter subregions B1 and B3 are highly methylated, while the B4/5 region displays low levels of DNA methylation (Fig. 2). By contrast, in naïve CD4+ T cells these subregions are mainly methylated and almost completely methylated in Jurkat T cells. Together, these data reveal that the intermediate, low and lack of CCR5 transcription levels, in monocytes, unstimulated CD4+ T cells and in Jurkat T leukaemia cells respectively, are associated with high levels of DNA methylation in the subregions B1 and B3 of the P1 promoter but not in the B4/5 subregion.
Histone modifications of the CCR5 P1 promoter
Next we determined the association of specific histone acetylation and methylation modifications within chromatin of the CCR5 P1 promoter by chromatin immunoprecipitation (ChIP) (Fig. 3A–C). CCR5 expressing, activated, CD4+ T cells display relative high levels of AcH3 (Fig. 3A). Interestingly, monocytes display AcH3 levels in chromatin of the CCR5 P1 promoter, which are similar to activated T cells (Fig. 3A). This is in contrast to the non–CCR5-expressing naïve T cells and Jurkat T cells, which display markedly lower levels of AcH3 in CCR5 P1 chromatin.
Fig 3.
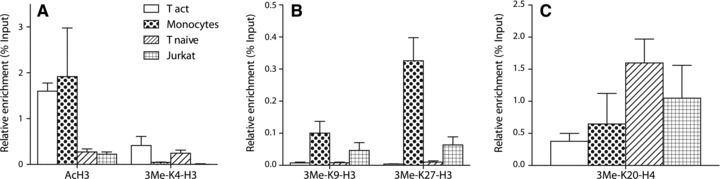
Chromatin environment at the CCR5 promoter as determined by ChIP analysis. CCR5 expressing activated CD4+ T cells clearly show higher levels of transcriptionally permissive chromatin marks AcH3 and 3MeK4H3 (A) whereas there is an opposite association with transcriptionally repressive chromatin marks 3MeK9H3, 3MeK27H3 (B) and 3MeK20H4 (C). These non-permissive marks are clearly present in higher amounts in CCR5 non-expressing cells (Jurkat T leukaemia cells, naïve CD4+ T cells) versus expressing activated CD4+ T cells. Naïve T cells and CD14+ monocytes show a poised chromatin state, encompassed by both transcriptionally permissive and non-permissive marks. Whereas naïve T cells show relatively low levels of AcH3, monocytes have high levels of AcH3.
CCR5-expressing activated T cells display relatively high levels of the permissive 3MeK4H3 mark in CCR5 P1 chromatin. Interestingly, naïve T cells expressing low levels of CCR5 show similar levels of the permissive 3MeK4H3 mark (Fig. 3A). In contrast, CCR5-deficient Jurkat T cells display low levels of the permissive 3MeK4H3 modification (Fig. 3A).
The repressive marks 3MeK9H3 and 3MeK27H3 are only present at very low levels in chromatin of low CCR5-expressing naïve T cells (Fig. 3B). In contrast, the repressive mark 3MeK20H4 is highly enriched at the CCR5 P1 promoter region of naïve T cells (Fig. 3C). The presence of both an activating mark (3MeK4H3) and a repressive mark (3MeK20H4) indicates a bivalent, so-called ‘poised’ state of the CCR5 promoter chromatin of naïve CD4+ T cells.
Activated CD4+ T cells show a two-fold higher CCR5 transcription level as compared to monocytes. Assessing the chromatin status of CD14+ monocytes, we observe the presence of relative high levels of the repressive marks 3MeK9H3 and 3MeK27H3 in the monocytic CCR5 P1 promoter (Fig. 3B). Conversely, the repressive mark 3MeK20H4 is only slightly enriched in monocytes as compared to activated T cells (Fig. 3C). Furthermore, hardly any of the permissive 3MeK4H3 mark could be detected, yet monocytes show high levels of AcH3 in the CCR5 promoter (Fig. 3A). This indicates that also monocytes display a chromatin state in which repressive and permissive histone modification marks co-exist. Compared to naïve CD4+ T cells however the chromatin state of CD14+ monocytes is markedly different, permitting transcription of CCR5.
CCR5-deficient Jurkat T cells show relative high levels of the repressive 3MeK9H3 and 3MeK27H3 histone marks, when compared with naïve and activated T cells (Fig. 3B). Similar to naïve T cells, Jurkat T leukaemia cells also show higher levels of the repressive 3MeK20H4 modification when compared to activated T cells (Fig. 3C). The presence of these repressive marks in the absence of activating histone modifications clearly shows a repressive chromatin conformation encompassing the CCR5 P1 promoter in Jurkat T cells.
Taken together, these data show that there is a differential pattern of chromatin conformation of the CCR5 P1 promoter region in the different cell populations investigated in this study. Our observations also indicate that the CCR5 transcription profiles could not be explained by a single epigenetic modification, but rather the sum of modifications appears to determine the level of CCR5 transcripts in the various cell types investigated.
Re-expression of CCR5 through pharmacologic interference in epigenetic mechanisms in Jurkat, Molt-4 and HSB-2 T cell lines
To show that DNA methylation, and histone acetylation/methylation mechanisms control CCR5 transcription, we aimed to induce CCR5 transcription in non–CCR5-expressing cells through pharmacologic interference in the catalytic activities of the various enzymes involved in these epigenetic regulatory processes. Figure 4 presents a schematic overview of the working mechanisms of the agents used for this purpose. Zebularine is a potent inhibitor of DNA-methylation showing much lower toxicity than the widely used inhibitor 5-Aza-dC [41, 42]. First recognized as an inhibitor with specificity for the KMTase EZH2, DZNep is now regarded as a more general lysine methyltransferase inhibitor, with a high affinity for the enzymes that triple-methylate K20H4 and K27H3 (Refs. [43, 44] and own observations). Finally, MS275 is a potent inhibitor of histone deacetylase activities (HDACs), with high affinity for the class I HDACs 1 and 3 [45].
Fig 4.
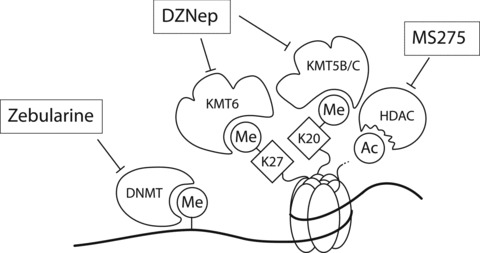
Schematic representation of the working mechanism for the pharmacological intervention in CCR5 transcription. Zebularine inhibits DNMTs, whereas MS275 inhibits HDACs. DZNep is a more general KMTase inhibitor [43].
Originally we found that inhibition of DNA-methylation by 5-Aza-dC treatment resulted in only a modest and time-dependent induction of CCR5 mRNA expression levels in Jurkat cells (results not shown). However, combining inhibition of DNA and histone methylation by inclusion of DZNep resulted in a clear synergistic induction of CCR5 mRNA expression, whereas inhibition of histone methylation alone was found only marginally effective (results not shown). Additional treatment with the HDAC inhibitor MS275 mainly potentiated the effect obtained by the other inhibitors (results not shown).
We therefore combined all of the above-mentioned inhibitors to induce CCR5 expression in Jurkat, Molt-4 and HSB-2 T leukaemia cells and included Zebularine rather than 5-Aza-dC for the aforementioned reasons. After treatment with Zebularine, in combination with DZNep and MS275, 67.7% of Jurkat cells are CCR5 positive as determined by FACS staining (Fig. 5A). In untreated Jurkat cells, only 0.83% of the cells stain positive for CCR5 (Fig. 1A). Correspondingly, after treatment the levels of CCR5 transcripts found in Jurkat T cells increased to 43% of the CCR5 transcript levels found in activated CD4+ T cells (Fig. 5B). HSB-2 and Molt-4 were more refractory to this combined epigenetic treatment; however, still 49.4% and 18.2% of the cells, respectively, were expressing CCR5 at the cell surface after treatment (Fig. 5A), whereas transcript levels were 20% and 4.8% relative to activated T cell transcript levels in HSB-2 and Molt-4, respectively (Fig. 5B).
Next we evaluated the effect of the epigenetic drug treatment on the expression characteristics of CREB-1 and ICER in Jurkat cells by semi-quantitative RT-PCR as we have previously explored [26]. Inducible cAMP early repressor, the inducible cAMP early repressor, which is induced by forskolin, competes with CREB-1 for DNA binding. We and more recently others also have shown that induction of ICER by forskolin treatment indeed reduces CCR5 expression [26, 46]. In Figure 6 it is shown that pharmacological induction of CCR5 expression did neither result in the induction of CREB-1, nor in a reduction of ICER transcript levels in Jurkat T cells (Fig. 6). Notably, when compared with naïve or activated CD4+ T cells, Jurkat cells do express CREB-1, but hardly any ICER could be detected. In contrast, naïve T cells show low levels of CREB-1, with relatively high levels of ICER. Upon activation, the levels of ICER are reduced while on the other hand CREB-1 levels are induced (Fig. 6). These observations indicate that in Jurkat T cells induction of CCR5 expression most likely is not resulting from alterations in the interplay of CREB-1 and ICER.
Fig 6.
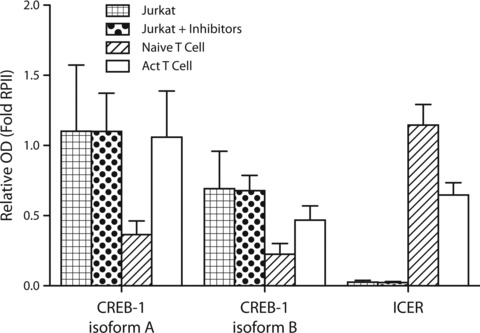
Semi-quantitative RT-PCR for CREB-1 isoforms and ICER was performed in triplicate. Activated T cells show higher levels of both CREB-1 isoforms, when compared to naïve T cells, whereas naïve T cells show higher levels of ICER then activated T cells. Jurkat T leukaemia cells show virtually undetectable levels of ICER. Treatment of Jurkat cells with Zebularine, DZNep and MS275 does not influence CREB-1 or ICER transcript levels.
We also investigated whether the pharmacological induction of CCR5 expression was associated with alterations in the histone acetylation/methylation profile and recruitment of CREB-1 and RNA polymerase II in CCR5 promoter chromatin. As shown in Figure 7(A) there is a clear increase in the AcH3 mark (associated with gene expression) after treatment, whereas histone marks associated with gene repression appear to be more resistant to the treatment. Shown in Figure 7(B) is that the permissive CCR5 chromatin structure in activated T cells (Fig. 3) results in increased recruitment of CREB-1 and RNA polymerase II into CCR5 promoter chromatin when compared with naïve T cells. Similarly, the induction of CCR5 expression after epigenetic treatment of Jurkat cells is also accompanied by an increase in the recruitment of CREB-1 and RNA polymerase II into CCR5 promoter chromatin (Fig. 7B). Together, the pharmacological inhibition of the activities of the various epigenetic enzymes that account for the repressive chromatin state of CCR5 in Jurkat T cells has resulted in a shift into a more open chromatin structure. This is accompanied by an increase in promoter association of the transcription factor CREB-1 and recruitment of RNA polymerase II.
Fig 7.
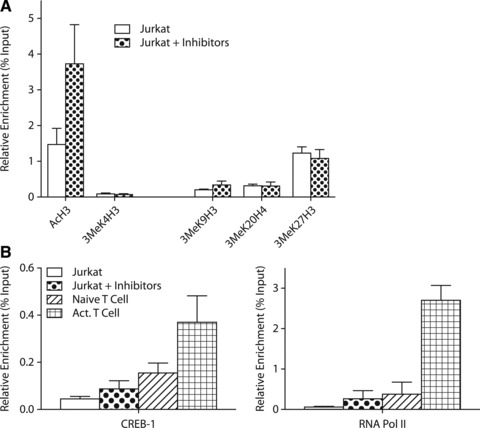
(A) ChIP analysis of histone modification at the CCR5 promoter in Jurkat cells, after treatment with Zebularine, DZNep and MS275. The treatment of Jurkat cells results in an increase of AcH3 at the CCR5 promoter. Repressive marks at the CCR5 chromatin are not influenced much by the treatment, although a minor decrease in 3MeK27H3 can be noted. (B) ChIP analysis of the CCR5 promoter for CREB-1 and RNA polymerase II after treatment with small molecule inhibitors in Jurkat, compared to both naïve and activated T cells. Treatment of Jurkat cells with Zebularine, DZNep and MS275 slightly increases CREB-1 in chromatin of the CCR5 promoter. In both naïve and activated T cells higher levels of chromatin-associated CREB-1 can be found. Compared to naïve T cells, there is an increase of CREB-1 in activated T cells. Treatment of Jurkat cells with small molecule inhibitors increases RNA polymerase II recruitment to the CCR5 promoter to levels similar of naïve T cells. In comparison to activated T cells, the levels of RNA polymerase II in the CCR5 promoter region of treated Jurkat cells are modest.
Discussion
This study reveals that epigenetic mechanisms involving DNA methylation, histone acetylation and methylation modifications all contribute to the transcriptional regulation of CCR5 expression. In CCR5-deficient T leukaemia cells we show that the promoter region is mainly characterized by repressive histone marks in the presence of methylated DNA. In CCR5-expressing activated T cells this region is mainly associated with activating histone marks and low levels of DNA methylation. Interestingly, the B4/5 region in the CCR5 promoter, which was previously attributed to CREB-1–mediated transactivation, is mostly unmethylated both in Jurkat and activated T cells.
Intermediate or low CCR5-expressing CD14+ monocytes and naïve CD4+ T cells, respectively, are characterized by both repressive histone methylation marks and permissive histone acetylation marks. In naïve T cells, an intermediate level of DNA methylation accompanies these histone modifications. However, in monocytes the level of DNA methylation is markedly higher as compared to naïve T cells, with the B4/5 region in a mostly unmethylated state in both cell types. Together, the cell types investigated here show that the B4/5 region is mostly unmethylated, irrespective of CCR5 transcription. This suggests that the B1 and B3 regions could also contribute to the transcriptional regulation of CCR5 as has been argued previously [26, 47, 48].
Notably, CD14+ monocytes and naïve CD4+ T cells represent a bivalent chromatin state recognized by the presence of relative high levels of both repressive (3MeK27H3 and/or 3MeK20H4) and permissive (AcH3) histone marks. This state differs from the originally defined bivalent chromatin structure recognized by high levels of 3MeK4H3 and 3MeK27H3 [37]. However, the bivalent chromatin structure observed in this study might reflect the existence of additional multivalent epigenetic marks, including co-existence of AcH3 and 3MeK27H3, as has been appreciated more recently [38]. Bivalent chromatin structures were first described to be important for genes that play an essential role in development. This bivalent state would be lost upon differentiation [37]. Here we show, as has been argued before [38], that multivalent chromatin states also occur in differentiated cells. These multivalent states may be of importance for the control of gene expression in the activation of T cells and the differentiation of monocytes [49]. The observed multivalent chromatin state of CCR5 therefore might reflect the central role of CCR5 in the regulation of lymphoid cell trafficking.
Considering the various histone triple-methylation modifications investigated, we conclude that acetylation of histone H3 is the critical factor for CCR5 expression as is illustrated in naïve CD4+ T cells and in CD14+ monocytes as well as in re-expressing Jurkat cells. The dominant role of histone modifications is further underscored by the fact that monocytes show high levels of DNA methylation. Although DNA methylation is usually interpreted as a repressive chromatin mark, this study as well as a number of recent other studies show that DNA methylation in the absence of repressive histone marks permits active gene transcription [38, 50-55]. This is also in line with previous studies showing that the presence of the 3MeK27H3 histone modification correlated with lack of transcription despite the absence of DNA methylation [51, 56, 57]. Interestingly, the monocyte population presented in this study shows transcription in presence of DNA methylation, 3MeK27H3, 3MeK9H3 and AcH3, but notably low levels of 3MeK20H4. This underscores, as has been previously noted [50] that not all epigenetic histone marks contribute equally to a specific chromatin status. Rather, the sum of epigenetic modifications, or ‘epigenetic profile,’ is more important than individual modifications to allow gene transcription.
The role of epigenetic regulatory mechanisms in the control of CCR5 transcription is also underscored by the pharmacological interference in the identified components of epigenetic machinery. As the epigenetic modifications were observed in both DNA and in histones encompassing the CCR5 promoter, as exemplified in Jurkat T cells, we combined the various inhibitors to induce re-expression. This intervention resulted in the re-expression of CCR5 in Jurkat (and also in HSB-2 and Molt-4) T leukaemia cells, albeit that the levels of re-expression differ between the cell lines investigated and were never on par with activated T cells. This re-expression of CCR5 correlated with an increased recruitment of CREB-1 and RNA polymerase II into CCR5 promoter chromatin as shown in Jurkat T cells.
In initial experiments it was possible to restore CCR5 expression in modest amounts using a single small molecule inhibitor. However, in order to induce CCR5 expression on Jurkat cells to more substantial levels, it was necessary to combine multiple small molecule inhibitors interfering in the activities of both DNA and histone modifying enzymes. The existence of multivalent chromatin marks may necessitate the use of multiple inhibitors. By using a single inhibitor, usually only a single chromatin mark is being assessed. Yet expression or repression is determined by the epigenetic profile, which is composed of multiple marks. Furthermore, in the case of DZNep, being a global histone methylation inhibitor, both repressing and activating marks are being influenced at the same time.
Changing the DNA methylation status through pharmacological disruption with Zebularine requires incorporation of Zebularine into the DNA [41, 42]. Demethylation through usage of Zebularine thus requires replication of DNA and therefore proliferation of cells. Jurkat, HSB-2 and Molt-4 cell lines show different doubling times. The difference in re-expression levels of CCR5 after combined epigenetic therapy can therefore be explained by this difference in cell doubling times. Furthermore, the relative toxicity of MS275 and DZNep may lower the proliferative capacity of the cells, thereby influencing the efficacy of Zebularine treatment. This may result in a situation where 100% re-expression of the gene of interest might prove to be a challenge, especially because DNA methylation and histone modifications are intimately linked [31, 58]. Yet despite these drawbacks, interference in the epigenetic machinery still results in a dramatic rise of CCR5 transcripts in T leukaemia cells as shown in this study.
In addition to epigenetic regulatory mechanisms we now also show in normal T cells that upon T cell activation the ratio in expression of CREB-1 and ICER is altered, which has a bearing on the interplay of CREB-1 and ICER in the regulation of CCR5 transcription. Therefore, in normal T cells alterations in expression of CREB-1 and ICER in conjunction with chromatin modifications correlate with induction of CCR5 expression. In Jurkat T cells, which lack expression of ICER, chromatin modification by the epigenetic treatment seems to be sufficient to induce CREB-1 mediated CCR5 expression.
Together, these data strongly indicate that histone acetylation and methylation modification mechanisms contribute to the transcriptional control of CCR5. In addition, we show that chromatin in a bivalent state allows for the fine-tuning of transcription levels, as has been shown before for other genes [31, 59]. Moreover, our data suggest that epigenetic deregulation could be one of the mechanisms leading to enhanced CCR5 expression as observed in a variety of inflammatory conditions [60, 61]. Although we demonstrate in this study the re-expression of CCR5, it could be envisioned that interference in the epigenetic processes that mediate increased CCR5 transcription as found in inflammatory conditions may have a beneficiary effect. As it is increased CCR5 expression that aggravates diseases such as atherosclerosis and multiple sclerosis [1-4]. As such, CCR5-mediated trafficking of lymphoid and myeloid cells is a possible target for pharmacological intervention. Interference in these deregulated epigenetic processes may therefore be a promising therapy for the treatment of inflammatory diseases.
Acknowledgments
The authors gratefully acknowledge the financial support of the Translation of Excellence in Regenerative Medicine (TeRM) Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science. This research was further supported by the Dutch MS Research Foundation (MS 00-407 and MS 04-543), the Macropa Foundation, the Department of Immunohematology and Blood Transfusion, The European Union Erasmus Program (to S.C.) and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (to V.E.M.). We thank Prof. Dr. Jeremy Boss for the gift of the CREB-1 antibody, Prof. M. Mack for the gift of the MC-5 antibody and Prof. Dr. W.E. Fibbe for his support.
Author contributions
R.J.W., H.F.K., M.C.J.A.v.E., A.B., J.C.v.L., S.C. and S.B.G. performed experiments. R.J.W. and H.F.K. wrote the paper. V.E.M. provided essential reagents. J.W.J., P.H.A.Q., and P.J.v.d.E. critically discussed and reviewed the paper. P.J.v.d.E. supervised the project.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Bursill CA, Channon KM, Greaves DR. The role of chemokines in atherosclerosis: recent evidence from experimental models and population genetics. Curr Opin Lipidol. 2004;15:145–9. doi: 10.1097/00041433-200404000-00007. [DOI] [PubMed] [Google Scholar]
- 2.Ribeiro S, Horuk R. The clinical potential of chemokine receptor antagonists. Pharmacol Ther. 2005;107:44–58. doi: 10.1016/j.pharmthera.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Biber K, Zuurman MW, Dijkstra IM, et al. Chemokines in the brain: neuroimmunology and beyond. Curr Opin Pharm. 2002;2:63–8. doi: 10.1016/s1471-4892(01)00122-9. [DOI] [PubMed] [Google Scholar]
- 4.Schober A. Chemokines in vascular dysfunction and remodeling. Arterioscler Thromb Vasc Biol. 2008;28:1950–9. doi: 10.1161/ATVBAHA.107.161224. [DOI] [PubMed] [Google Scholar]
- 5.Wu L, Paxton WA, Kassam N, et al. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J Exp Med. 1997;185:1681–91. doi: 10.1084/jem.185.9.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oswald-Richter K, Grill SM, Leelawong M, et al. Identification of a CCR5-expressing T cell subset that is resistant to R5-tropic HIV infection. PLoS Path. 2007;3:0553–65. doi: 10.1371/journal.ppat.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrington M, Dean M, Martin MP, et al. Genetics of HIV-1 infection: chemokine receptor CCR5 polymorphism and its consequences. Hum Mol Genet. 1999;8:1939–45. doi: 10.1093/hmg/8.10.1939. [DOI] [PubMed] [Google Scholar]
- 8.Ebert LM, McColl SR. Up-regulation of CCR5 and CCR6 on distinct subpopulations of antigen-activated CD4+ T lymphocytes. J Immunol. 2002;168:65–72. doi: 10.4049/jimmunol.168.1.65. [DOI] [PubMed] [Google Scholar]
- 9.Mummidi S, Adams LM, VanCompernolle SE, et al. Production of specific mRNA transcripts, usage of an alternate promoter, and octamer-binding transcription factors influence the surface expression levels of the HIV coreceptor CCR5 on primary T cells. J Immunol. 2007;178:5668–81. doi: 10.4049/jimmunol.178.9.5668. [DOI] [PubMed] [Google Scholar]
- 10.Mummidi S, Ahuja SS, McDaniel BL, et al. The human CC chemokine receptor 5 (CCR5) gene. Multiple transcripts with 5’-end heterogeneity, dual promoter usage, and evidence for polymorphisms within the regulatory regions and noncoding exons. J Biol Chem. 1997;272:30662–71. doi: 10.1074/jbc.272.49.30662. [DOI] [PubMed] [Google Scholar]
- 11.Bleul CC, Wu L, Hoxie JA, et al. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci USA. 1997;94:1925–30. doi: 10.1073/pnas.94.5.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guignard F, Combadiere C, Tiffany HL, et al. Gene organization and promoter function for CC chemokine receptor 5 (CCR5) J Immunol. 1998;160:985–92. [PubMed] [Google Scholar]
- 13.van der Merwe PA, Davis SJ. The immunological synapse-a multitasking system. Science. 2002;295:1479–80. doi: 10.1126/science.1069896. [DOI] [PubMed] [Google Scholar]
- 14.Grakoui A, Bromley SK, Sumen C, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–7. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 15.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–42. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 16.Serbina NV, Jia T, Hohl TM, et al. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–52. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmitz G, Leuthauser-Jaschinski K, Orso E. Are circulating monocytes as microglia orthologues appropriate biomarker targets for neuronal diseases? Cent Nerv Syst Agents Med Chem. 2009;9:307–30. doi: 10.2174/187152409789630424. [DOI] [PubMed] [Google Scholar]
- 18.Chan WY, Kohsaka S, Rezaie P. The origin and cell lineage of microglia: new concepts. Brain Res Rev. 2007;53:344–54. doi: 10.1016/j.brainresrev.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Hansson GK, Robertson AKL, Soderberg-Naucler C. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- 20.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 21.Noseworthy JH, Lucchinetti C, Rodriguez M, et al. Multiple sclerosis. N Engl J Med. 2000;343:938–52. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 22.Zernecke A, Shagdarsuren E, Weber C. Chemokines in atherosclerosis: an update. Arterioscler Thromb Vasc Biol. 2008;28:1897–908. doi: 10.1161/ATVBAHA.107.161174. [DOI] [PubMed] [Google Scholar]
- 23.Zernecke A, Liehn EA, Gao J-L, et al. Deficiency in CCR5 but not CCR1 protects against neointima formation in atherosclerosis-prone mice: involvement of IL-10. Blood. 2006;107:4240–3. doi: 10.1182/blood-2005-09-3922. [DOI] [PubMed] [Google Scholar]
- 24.Fox RJ, Kivisakk P, Fisher E, et al. Multiple sclerosis: chemokine receptor expression on circulating lymphocytes in correlation with radiographic measures of tissue injury. Mult Scler. 2008;14:1036–43. doi: 10.1177/1352458508092261. [DOI] [PubMed] [Google Scholar]
- 25.Trebst C, Konig F, Ransohoff R, et al. CCR5 expression on macrophages/microglia is associated with early remyelination in multiple sclerosis lesions. Mult Scler. 2008;14:728–33. doi: 10.1177/1352458508089359. [DOI] [PubMed] [Google Scholar]
- 26.Kuipers HF, Biesta PJ, Montagne LJ, et al. CC chemokine receptor 5 gene promoter activation by the cyclic AMP response element binding transcription factor. Blood. 2008;112:1610–9. doi: 10.1182/blood-2008-01-135111. [DOI] [PubMed] [Google Scholar]
- 27.Meyer TE, Habener JF. Cyclic adenosine 3′,5′-monophosphate response element binding protein (CREB) and related transcription-activating deoxyribonucleic acid-binding proteins. Endocr Rev. 1993;14:269–90. doi: 10.1210/edrv-14-3-269. [DOI] [PubMed] [Google Scholar]
- 28.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 29.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 30.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 31.Vaissiere T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res. 2008;659:40–8. doi: 10.1016/j.mrrev.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Rice JC, Briggs SD, Ueberheide B, et al. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell. 2003;12:1591–8. doi: 10.1016/s1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- 33.Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev. 2004;14:155–64. doi: 10.1016/j.gde.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 34.Schotta G, Lachner M, Sarma K, et al. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251–62. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–49. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 36.Wang P, Lin C, Smith ER, et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol. 2009;29:6074–85. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 38.Bapat SA, Jin V, Berry N, et al. Multivalent epigenetic marks confer microenvironment-responsive epigenetic plasticity to ovarian cancer cells. Epigenetics. 2010;5:716–29. doi: 10.4161/epi.5.8.13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holling TM, van der Stoep N, Quinten E, et al. Activated human T cells accomplish MHC class II expression through T cell-specific occupation of class II transactivator promoter III. J Immunol. 2002;168:763–70. doi: 10.4049/jimmunol.168.2.763. [DOI] [PubMed] [Google Scholar]
- 40.Ubogu EE, Callahan MK, Tucky BH, et al. CCR5 expression on monocytes and T cells: modulation by transmigration across the blood-brain barrier in vitro. Cell Immunol. 2006;243:19–29. doi: 10.1016/j.cellimm.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stresemann C, Brueckner B, Musch T, et al. Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res. 2006;66:2794–800. doi: 10.1158/0008-5472.CAN-05-2821. [DOI] [PubMed] [Google Scholar]
- 42.Marquez VE, Barchi JJ, Jr, Kelley JA, et al. Zebularine: a unique molecule for an epigenetically based strategy in cancer chemotherapy. The magic of its chemistry and biology. Nucleosides Nucleotides Nucl Acids. 2005;24:305–18. doi: 10.1081/ncn-200059765. [DOI] [PubMed] [Google Scholar]
- 43.Miranda TB, Cortez CC, Yoo CB, et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8:1579–88. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan J, Yang X, Zhuang L, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–63. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khan N, Jeffers M, Kumar S, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–9. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 46.Banerjee A, Pirrone V, Wigdahl B, et al. Transcriptional regulation of the chemokine co-receptor CCR5 by the cAMP/PKA/CREB pathway. Biomed Pharmacother. 2011;65:293–7. doi: 10.1016/j.biopha.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moriuchi H, Moriuchi M, Fauci AS. Cloning and analysis of the promoter region of CCR5, a coreceptor for HIV-1 entry. J Immunol. 1997;159:5441–9. [PubMed] [Google Scholar]
- 48.Liu R, Zhao X, Gurney TA, et al. Functional analysis of the proximal CCR5 promoter. AIDS Res Hum Retroviruses. 1998;14:1509–19. doi: 10.1089/aid.1998.14.1509. [DOI] [PubMed] [Google Scholar]
- 49.De Gobbi M, Garrick D, Lynch M, et al. Generation of bivalent chromatin domains during cell fate decisions. Epigenetics Chromatin. 2011;4:9. doi: 10.1186/1756-8935-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ke X-S, Qu Y, Cheng Y, et al. Global profiling of histone and DNA methylation reveals epigenetic-based regulation of gene expression during epithelial to mesenchymal transition in prostate cells. BMC Genomics. 2010;11:669. doi: 10.1186/1471-2164-11-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holling TM, Bergevoet MWT, Wilson L, et al. A role for EZH2 in silencing of IFN-gamma inducible MHC2TA transcription in uveal melanoma. J Immunol. 2007;179:5317–25. doi: 10.4049/jimmunol.179.8.5317. [DOI] [PubMed] [Google Scholar]
- 52.Holling TM, Bergevoet MWT, Wierda RJ, et al. Genetic and epigenetic control of the major histocompatibility complex class Ib gene HLA-G in trophoblast cell lines. Ann N Y Acad Sci. 2009;1173:538–44. doi: 10.1111/j.1749-6632.2009.04660.x. [DOI] [PubMed] [Google Scholar]
- 53.Gonzalgo ML, Hayashida T, Bender CM, et al. The role of DNA methylation in expression of the p19/p16 locus in human bladder cancer cell lines. Cancer Res. 1998;58:1245–52. [PubMed] [Google Scholar]
- 54.Ten Haaf A, Franken L, Heymann C, et al. Paradox of sonic hedgehog (SHH) transcriptional regulation: Alternative transcription initiation overrides the effect of downstream promoter DNA methylation. Epigenetics. 2011;6:465–77. doi: 10.4161/epi.6.4.14952. [DOI] [PubMed] [Google Scholar]
- 55.Noguchi T, Takeno S, Kimura Y, et al. FHIT expression and hypermethylation in esophageal squamous cell carcinoma. Int J Mol Med. 2003;11:441–7. [PubMed] [Google Scholar]
- 56.Kondo Y, Shen L, Cheng AS, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet. 2008;40:741–50. doi: 10.1038/ng.159. [DOI] [PubMed] [Google Scholar]
- 57.Yu Q. Cancer gene silencing without DNA hypermethylation. Epigenetics. 2008;3:315–7. doi: 10.4161/epi.3.6.7202. [DOI] [PubMed] [Google Scholar]
- 58.Cheng X, Blumenthal RM. Coordinated chromatin control: structural and functional linkage of DNA and histone methylation. Biochemistry. 2010;49:2999–3008. doi: 10.1021/bi100213t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fuks F. DNA methylation and histone modifications: teaming up to silence genes. Curr Opin Genet Dev. 2005;15:490–5. doi: 10.1016/j.gde.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 60.Gong X, Feng H, Zhang S, et al. Increased expression of CCR5 in experimental autoimmune myocarditis and reduced severity induced by anti-CCR5 monoclonal antibody. J Mol Cell Cardiol. 2007;42:781–91. doi: 10.1016/j.yjmcc.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 61.Trebst C, Konig F, Ransohoff R, et al. CCR5 expression on macrophages/microglia is associated with early remyelination in multiple sclerosis lesions. Mult Scler. 2008;14:728–33. doi: 10.1177/1352458508089359. [DOI] [PubMed] [Google Scholar]
- 62.Moreno CS, Beresford GW, Louis-Plence P, et al. CREB regulates MHC class II expression in a CIITA-dependent manner. Immunity. 1999;10:143–51. doi: 10.1016/s1074-7613(00)80015-1. [DOI] [PubMed] [Google Scholar]