

Abstract
Multicentric carpotarsal osteolysis (MCTO) is a rare skeletal dysplasia characterized by aggressive osteolysis, particularly affecting the carpal and tarsal bones, and is frequently associated with progressive renal failure. Using exome capture and next-generation sequencing in five unrelated simplex cases of MCTO, we identified previously unreported missense mutations clustering within a 51 base pair region of the single exon of MAFB, validated by Sanger sequencing. A further six unrelated simplex cases with MCTO were also heterozygous for previously unreported mutations within this same region, as were affected members of two families with autosomal-dominant MCTO. MAFB encodes a transcription factor that negatively regulates RANKL-induced osteoclastogenesis and is essential for normal renal development. Identification of this gene paves the way for development of novel therapeutic approaches for this crippling disease and provides insight into normal bone and kidney development.
Main Text
Next-generation sequencing presents a new approach for disease-gene mapping in either small pedigrees or in small numbers of affected individuals and has been used to uncover the underlying cause for several skeletal dysplasias.1–4 Multicentric carpotarsal osteolysis (MCTO [MIM 166300]) is a rare skeletal dysplasia characterized by progressive bone resorption (osteolysis), predominantly (although not exclusively) of the carpal and tarsal bones. The early clinical appearance can mimic polyarticular juvenile idiopathic arthritis; however, subsequent clinical and radiographic appearances are characteristic (see Figure 1). Affected individuals can also have subtle craniofacial abnormalities (triangular faces, micrognathia, and exophthalmos), and many develop a progressive nephropathy leading to end-stage renal failure. Both simplex cases and families with autosomal-dominant inheritance have been reported.5–16 However, the responsible genetic defect has remained elusive. Mutations in MMP2 [MIM 120360], responsible for the autosomal-recessive Torg-Winchester syndrome [MIM 259600]17–19 that is also characterized by excessive bone resorption, have not been identified in patients with MCTO.20 Here, we report identification of the underlying genetic cause of MCTO.
Figure 1.
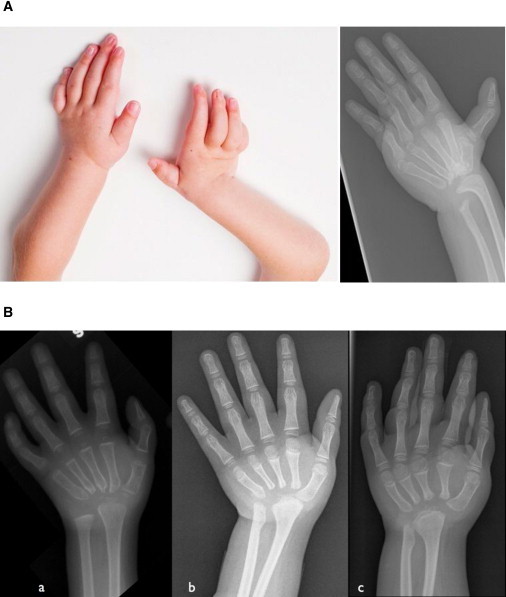
Typical Clinical and Radiographic Appearance of MCTO
(A) Complete destruction of carpal bones with relatively well-preserved metacarpals and phalanges (individual 17.3).
(B) Hand X-rays of individual 15.3 at (a) 3, (b) 8, and (c) 10 years old, respectively. Note the absence of carpal bones and progressive osteolysis of proximal end of metacarpal bones, whereas the distal end and the phalanges remain relatively well preserved.
We recruited simplex cases of MCTO and families with autosomal-dominant MCTO through the Queensland Bone Dysplasia Registry at the University of Queensland Centre for Clinical Research. Each individual had classical radiographic features of MCTO (complete destruction of carpal bones but relatively well-preserved metacarpals and phalanges) (Figure 1). Five of the 11 simplex cases had undergone renal transplantation. Of the remaining six, three had markedly impaired renal function but did not yet require dialysis. Three individuals all of whom were very young (7 years old or younger), had no evidence of renal dysfunction. The affected individuals in the two families with autosomal-dominant MCTO had no evidence of renal dysfunction. The study was approved by the University of Queensland Ethics Committee, and all subjects or their respective guardians gave informed written consent.
We performed whole-exome sequencing in five unrelated individuals with MCTO. Genomic DNA was extracted from peripheral blood samples via standard protocols. We used 5 μg DNA to construct DNA sequencing libraries with Illumina's DNA sample preparation kit for paired-end sequencing per the manufacturer's protocol. We used liquid phase hybridization for exome capture with Roche/Nimblegen SeqCap EZ Exome Library v1.0 kits (Roche/Nimblegen) according to the manufacturer's protocol. Efficiency of sequence capture was assessed via quantitative real-time PCR with standard control primers as recommended by the manufacturer. Each DNA sequencing library was quality checked and quantified with Agilent 2100 Bioanalyser and DNA 1000 chip kits. We standardized the DNA concentration for sequencing to 10 nM. Each DNA library was individually sequenced via a 56 paired-end read sequencing protocol with a single flow cell lane on an Illumina Genome Analyzer II and with the manufacturer's reagents.
Initial base calling was performed with the Illumina Data Analysis Pipeline software (CASAVA 1.7). Sequence data were aligned to the current build of the human genome (hg19, released February 2009) with the Novoalign alignment tool (V2.07.09 1); sequence alignment files were converted with SAMtools (v0.1.14) and Picard tools (v1.42).21 SNPs and indels were called with the Genome Analysis Toolkit (GATK v5506)22 and were annotated with ANNOVAR.23 An average of 1.96 Gb of sequence per individual (range 1.48–2.38 Gb) was generated, with mean on-target (exons ± 200 bp) sequence of 87.0% (range 85.5%–88.1%). Mean depth of exomic coverage was 50-fold (range 37.0–60.5) with 84.9% and 75.9% achieving 10-fold and 15-fold coverage, respectively (mapping and coverage statistics and detailed base calling information is presented in Tables S1 and S2, available online).
Custom scripts with R and Bioconductor were used to analyze and filter the sequencing data. SNPs and indels with good quality reads were retained (minimum depth of coverage for SNP calling, greater than 10-fold for homozygous SNPs and greater than 15-fold for heterozygous SNPs). Additionally, we used the GATK-provided quality parameters: we included SNPs/indels with values QUAL > 50, QD > 0.5, HRUN < 5, and SB < 0.1 (defined as: QUAL, combined recalibrated quality score at the SNP position; DP, sequencing depth at the SNP position; QD, QUAL/DP ratio at the SNP position; HRun, maximal length of the homopolymer run; SB, strand bias at the SNP position). We retained SNPs and indels with potentially damaging consequence (nonsynonymous SNV, splicing, frameshift substitution, stopgain SNV, and stoploss SNV based on either RefSeq or UCSC transcripts). We excluded SNPs observed in the National Center for Biotechnology Information (NCBI) dbSNP (release 132), 1000 Genomes, 1000 Genomes small indels (called with the DINDEL program), the SNPs of 46 genomes release by Complete Genomics and 16 other whole exomes from control samples run internally with the same capture technology that was used in this project.
We assessed the five sequenced individuals for heterozygous carriage of either a single previously unreported nonsynonymous SNP shared by all individuals or a previously unreported nonsynonymous SNP in each individual within a common gene. After quality filtering (detailed in Table 1), a single gene was identified, MAFB (v-maf musculoaponeurotic fibrosarcoma oncogene ortholog B [avian] [MIM 608968]), in which each individual was heterozygous for a single previously unreported missense mutation in a highly conserved region (Figure 2).
Table 1.
Summary of Identified Variants from Exome Sequencing
| SKDP 1.3 | SKDP 15.3 | SKDP 20.4 | SKDP 28.3 | SKDP 8.3 | Genes with Variants in All Subjects | |
|---|---|---|---|---|---|---|
| Number of variants identified | 58,734 | 58,565 | 59,195 | 50,507 | 55,802 | 8,771 |
| Number of variants after quality control and coverage filters | 24,920 | 27,818 | 28,804 | 22,545 | 24,795 | 4,974 |
| Nonsynonymous variants, indels in exons, or splice sites after quality control and filtering | 6,255 | 6,752 | 6,831 | 6,217 | 6,322 | 1,630 |
| Previously unreported nonsynonymous variants, indels in exons, or splice sites after quality control and filtering | 185 | 155 | 168 | 174 | 237 | 3a |
Variants in three genes remained. Two of these were dinucleotide repeats in splice sites and did not fit appropriate autosomal-dominant inheritance pattern. Thus variants in only one remaining gene (MAFB) fitted all the criteria.
Figure 2.

Sanger Sequencing and Comparative Genomics of MAFB
(Upper panel) Sanger sequencing of MAFB, demonstrating mutations in all 11 simplex cases with MCTO. Labels in bold refer to the original five probands sequenced with NGS.
(Lower panel) Protein coding and comparative genomics of MAFB (derived from UCSC genome browser).
We used Sanger sequencing to validate the observed mutations. Because of the presence of the adjacent large deletion polymorphism (rs71854600), PCR primers were designed to amplify 170 bp of the coding region surrounding the mutation hotspot. The primer sequences for initial PCR amplification were as follows: MAFB_ShF 5′-(M13-21) GCTCAAGTTCGACGTGAAGA−3′, and MAFB_ShR 5′-CGAGGTGTGTCTTCTGTTCG−3′. Sequencing was performed with standard M13 (−21) sequencing primer and BigDye v3.1 cycle sequencing reagents, and capillary electrophoresis was performed on Genetic Analyzer 3130 (Applied Biosystems).
In addition to the five probands who had undergone exomic sequencing, we also sequenced the same region in their (clinically unaffected) parents; a further six unrelated simplex cases with MCTO and their parents (with a single exception, individual 81.3, for whom no parental DNA sample was available); and all available family members of two families with autosomal-dominant inheritance of MCTO (Figure 3). We also sequenced 164 unrelated white European controls. Sanger sequencing confirmed the mutations identified through exomic sequencing in the five probands. Further, the six additional simplex cases with MCTO also demonstrated previously unreported heterozygous missense mutations in the same region of MAFB (Figure 2). None of the parents of the affected probands carried the mutations present in their child, demonstrating that these are previously unreported autosomal-dominant mutations in the affected individuals. In the two families with autosomal-dominant inheritance of MCTO, heterozygous missense mutations were also demonstrated in the same region and segregated with disease status (Figure 3). None of the 164 control samples carried any mutations in this region (328 alleles). The location of the mutated amino acids within the protein domain is shown in Figure 4. We used three SNP function prediction algorithms (Polyphen [PolyPhen-2 v2.1.0r367]; SIFT, PMut) to predict the effect of the observed previously unreported SNPs upon protein function. All observed mutations were predicted to be probably or possibly damaging with Polyphen (full results presented in Table 2). We assessed conservation of this region of MafB against UCSC genome browser data and demonstrated conservation of this region back to the zebra fish (Figure 2).
Figure 3.
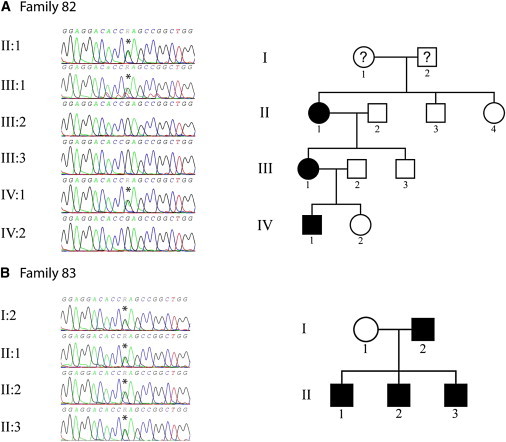
Sanger Sequencing and Family Trees for Families 82 and 83
(A) Family 82.
(B) Family 83.
Black indicates known affected and white indicates known unaffected; a question mark indicates unknown status.
Figure 4.
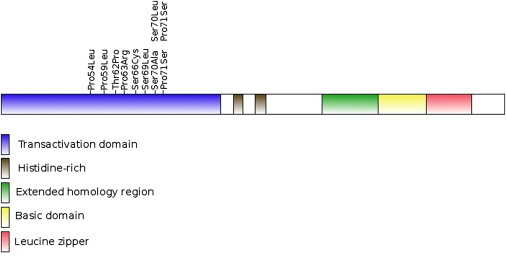
Schematic picture of MafB, showing protein domains and positions of the observed mutations in individuals and families with MCTO
Table 2.
List of Variants Observed in MAFB (NM_005461.3), and Predicted Protein Changes
| Individual | Variant | Amino Acid Substitution |
Functional Prediction |
||
|---|---|---|---|---|---|
| Polyphen | PMut | SIFT | |||
| SKDP 81.3 | c.176C>T | p.Pro59Leu | probably damaging | pathologic | tolerated |
| SKDP 20.4 | c.184A>C | p.Thr62Pro | possibly damaging | pathologic | tolerated |
| SKDP 25.3 | c.188C>G | p. Pro63Arg | probably damaging | pathologic | damaging |
| SKDP 27.3 | c.197C>G | p.Ser66Cys | probably damaging | neutral | damaging |
| SKDP 15.3 | c.206C>T | p.Ser69Leu | possibly damaging | neutral | tolerated |
| SKDP 8.3 | c.208T>G | p.Ser70Ala | probably damaging | neutral | damaging |
| SKDP 17.3 and 28.3 | c.209C>T | p.Ser70Leu | probably damaging | pathologic | damaging |
| SKDP 22.3 and 43.3 | c.211C>T | p.Pro71Ser | probably damaging | neutral | tolerated |
| SKDP 1.3 | c.212C>T | p.Pro71Leu | probably damaging | Pathologic | damaging |
| SKDP 82 and 83 (affected members) | c.161C>T | p.Pro54Leu | probably damaging | pathologic | damaging |
Thus we have identified previously unreported missense mutations, all predicted to be damaging to protein function and clustering within a narrow region of the single exon of MAFB in all cases of MCTO; we have also demonstrated segregation of mutations with disease in two multigenerational families with autosomal-dominant MCTO. MAFB codes for a member of the V-MAF musculoaponeurotic fibrosarcoma oncogene family of transcription factors. It is a single exon gene with two known transcripts, a 3.0 kb transcript expressed ubiquitously and a shorter 1.8 kb transcript expressed predominantly in bone marrow and skeletal muscle,24 coding for a 323 amino acid protein (UniProt: Q9Y5Q3). In common with other Maf proteins, the carboxy-terminal of MafB contains a well-conserved basic region for DNA binding (amino acid position 238–264) and a leucine zipper domain involved in dimerization (amino acid position 266-287). The amino-terminal region (amino acid positions 1-110) is a Pro-Ser-Thr- rich acidic transcriptional activation domain.24 Overall homology between human and mouse MAFB is 93% and that between human and mouse MafB is 97%, and the region containing the observed mutations is highly conserved (Figure 2).
MafB is known to play critical roles in both osteoclast differentiation and activation25 and renal development,26 consistent with the major phenotypic features of MCTO. It is expressed in myeloid cells in the bone marrow and is essential for normal hematopoieisis. Specifically, MafB suppresses erthyroid differentiation while upregulating myeloid differentiation to monocytes and macrophages.27 Osteoclasts are derived from hematopoietic stem cells of the macrophage/monocyte lineage under the combined influence of receptor activator of nuclear factor κB ligand (RANKL) and macrophage colony stimulating factor (M-CSF). MafB negatively regulates RANKL-induced osteoclastogenesis.25 In bone marrow-derived monocyte/macrophage cell cultures, MafB overexpression inhibited osteoclastogenesis by interfering with the key transcription factors normally activated by RANKL, whereas reduced MafB expression enhanced osteoclastogenesis. Conversely, RANKL downregulated expression of MafB.25 Thus, MafB might regulate the development of pluripotent myeloid precursors by enhancing macrophage differentiation while repressing osteoclastogenesis. MafB is itself regulated by the transcription factor CCAAT/enhancer-binding protein β (C/EBPβ); and osteoclastic bone resorption is increased in mice with C/EBPβ deficiency and in mice expressing only the short isoform of C/EBPβ in both of which MafB expression levels are attenuated.28,29
The observed mutations in MAFB all lie within a short region of the amino-terminal transcriptional activation domain (amino acids 54-71; see Table 2 and Figure 4). Of note, amino-terminal truncation of MAFB (residual amino-acids 195-311 fused to the N terminus) exerted a dominant negative effect upon myeloid colony formation and macrophage differentiation.27 The observed heterozygous missense mutations in MAFB in individuals with MCTO have a dominant effect; however, whether the mechanism for this is haploinsufficiency or a dominant negative effect requires further study.
In common with other large Maf proteins, MafB is phosphorylated by glycogen synthase kinase-3. The conserved phosphorylation sites shared by Maf proteins lie in the same region as the observed mutations; there are four mutations predicted to affect putative phosphorylation sites (amino acids 54, 62, 66, and 70). Mutations of the corresponding amino acids in MafA prevented phosphorylation and decreased its transcriptional activity.30 It is possible that the mutations we observed might have a similar effect upon MafB phosphorylation and subsequent function. However, functional studies are required to confirm such speculation.
It is not clear why the bony changes in MCTO are seen predominantly in the carpal and tarsal bones. Systemic bone mineral density (BMD) has not been studied comprehensively in patients with MCTO; however, one case report noted osteopaenia in a child before significant renal dysfunction had developed.16 In adult patients with advanced renal failure, it is not possible to partition the effects of renal osteodystrophy from direct systemic bone effects from MAFB mutations. Whether polymorphisms in MAFB are associated with variation in BMD in the normal population is unclear. In our GWAS of 2,055 postmenopausal women with either extremely high or low BMD, three SNPs in MAFB were associated with lumbar spine BMD with p value < 10−3 (not significant at a genome-wide level),31 a finding sufficiently suggestive to warrant further investigation in larger data sets.
Patients with MCTO commonly develop renal impairment and frequently progress to end-stage kidney disease. Renal biopsy is often only obtained at a late stage of disease, by which time nonspecific findings of glomerulosclerosis and severe tubulointerstitial fibrosis dominate the biopsy appearance. However, two studies report renal biopsies obtained from children with MCTO and early renal dysfunction. One child, with isolated proteinuria, had focal and segmental glomerulosclerosis with distortion, fusion, and effacement of the podocyte foot processes.16 Another child, only 2 years old but with hypertension, proteinuria, and mildly elevated serum creatinine, had a normal biopsy with light microscopy, but electron microscopy also showed abnormal fusion of the podocyte foot processes and discrete abnormalities of the glomerular basement membrane.32 MafB is essential for normal development of podocyte foot processes26,33 and is expressed in both the podocyte late in renal development and in the mature glomerulus. MafB−/− homozygous knockout mice have renal dysgenesis, including loss of normal foot processes of podocytes, abnormal glomerular differentiation with decreased mature glomeruli, tubular dysgenesis due to abnormal tubular survival, and renal cysts. The mice have decreased renal expression of nephrin, podocin, and CD2AP, critical proteins for normal podocyte development, and glomerular function.26,33 Although abnormalities in these basement membrane proteins have not been studied in patients with MCTO, the histopathological features that have been demonstrated in homozygous mice are consistent with the renal biopsy findings observed in patients with MCTO and renal impairment.
Renal disease was present in most but not all patients in our cohort. Individuals 43.3, 81.3, and 22.3 have as yet no evidence of renal disease. However, these patients are only 3, 6 and 7 years old, respectively, and it is as yet unclear whether their renal function will remain normal. Of note, individual 81.3 has a unique mutation (c.176C>T [p.Pro59Leu]); and individuals 22.3 and 43.3 share the same mutation (c.211C>T [p.Pro71Ser]) although at the same location as another individual who has manifested renal disease. The affected individuals in family 82 and family 839 (who carry the same mutation, c.161C>T [p.Pro54Leu]), many of whom are adults, do not have renal disease, with the exception of individual 1.2 in Family 83 who at a very old age developed renal dysfunction of unknown aetiology. Thus it seems likely that MAFB mutations are also responsible for MCTO in the absence of renal disease.
Patients with MCTO also have a subtle craniofacial abnormalities that include micrognathia, a slim nose, maxillary hypoplasia, and consequent exophthalmos.8,12 A recent GWAS of cleft lip with and without cleft palate found significant association with multiple variants near MAFB and expression studies demonstrated MafB expression in palatal tissue.34 Cleft lip and/or palate have not been reported as a feature of patients with MCTO.
Studies in mice have suggested that MAFB plays important roles in pancreatic β cell development and in hindbrain development. MCTO is not known to be associated with pancreatic or central nervous system abnormalities, although these have not been investigated in detail.
Of the ten separate mutations observed, all were predicted to be probably or possibly damaging (Table 2). The protein structure of MafB is only established for the fragment of protein interacting with its DNA recognition motif, Cmare (amino acids 238–264).35 The mutations we observed cluster in a narrow region of the transitional activation domain where the protein structure is not yet established, although of note the protein domains in this regions are highly evolutionarily conserved (Figure 2). The function prediction algorithm PolyPhen predicts that all the variants are damaging or possibly damaging. We tested two additional algorithms Pmut and SIFT (Table 2). Pmut predicts four of the variants to be neutral; however, three of these calls have low reliability according to their own scoring criteria (all 0/10 except for S70A scored at 7/10).
Skeletal dysplasias highlight important pathways in bone of relevance to the general population and to common bone diseases such as osteoporosis. For example, the identification of SOST [MIM 605740] as the gene underlying sclerosteosis [MIM 269500]36 and van Buchem's disease [MIM 239100]37 has lead to the development of anti-sclerostin antibodies as a treatment for osteoporosis.38 For patients with MCTO, bisphosphonates have not proven useful in treatment to date. Our data suggest that it might be worth targeting MafB signaling instead in these patients. Anti-RANKL antibody therapy is now available for the treatment of osteoporosis39 and could be considered for treatment of at least the bone disease that characterizes the MCTO phenotype.
In conclusion, we have identified previously unreported missense mutations clustering in a narrow region of MAFB in 11 unrelated simplex cases and in two families with MCTO. MAFB is known to play a critical role in regulation of osteoclastogenesis and in normal renal development, making this a highly plausible gene underlying this skeletal dysplasia. This study opens up the possibility of new therapeutic options in these patients and other patients with excessive osteoclastic activity.
Acknowledgments
The affected individuals and their families who contributed clinical and genetic material are gratefully acknowledged. The assistance of Jordan Young, Linda Bradbury, and Aideen McInerney-Leo in patient recruitment and Jessica Harris and Catherine Cremin in laboratory work is gratefully acknowledged. This work was supported by a research grant from the Royal Children's Hospital Foundation, Brisbane, Australia. M.A.B. was supported by an National Health and Medical Research Council Principal Research Fellowship.
Contributor Information
Emma L. Duncan, Email: e.duncan@uq.edu.au.
Matthew A. Brown, Email: matt.brown@uq.edu.au.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
Complete Genomics, http://www.completegenomics.com/sequence-data/download-data/
Ensembl, http://www.ensembl.org/index.html
GATK, http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit
NCBI dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/
Novalign alignment tool, www.Novocraft.com
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen, http://genetics.bwh.harvard.edu/pph/
SIFT, http://sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu/
UniProt, http://www.uniprot.org/uniprot/
References
- 1.Glazov E.A., Zankl A., Donskoi M., Kenna T.J., Thomas G.P., Clark G.R., Duncan E.L., Brown M.A. Whole-exome re-sequencing in a family quartet identifies POP1 mutations as the cause of a novel skeletal dysplasia. PLoS Genet. 2011;7:e1002027. doi: 10.1371/journal.pgen.1002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simpson M.A., Irving M.D., Asilmaz E., Gray M.J., Dafou D., Elmslie F.V., Mansour S., Holder S.E., Brain C.E., Burton B.K. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 2011;43:303–305. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- 3.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shurtleff D.B., Sparkes R.S., Clawson D.K., Guntheroth W.G., Mottet N.K. Hereditary Osteolysis with Hypertension and Nephropathy. JAMA. 1964;188:363–368. doi: 10.1001/jama.1964.03060300025005. [DOI] [PubMed] [Google Scholar]
- 6.Tyler T., Rosenbaum H.D. Idiopathic multicentric osteolysis. AJR Am. J. Roentgenol. 1976;126:23–31. doi: 10.2214/ajr.126.1.23. [DOI] [PubMed] [Google Scholar]
- 7.Bennett W.M., Houghton D.C., Beals R.C. Nephropathy of idiopathic multicentric osteolysis. Nephron. 1980;25:134–138. doi: 10.1159/000181769. [DOI] [PubMed] [Google Scholar]
- 8.Fryns J.P. Essential osteolysis with carpal and tarsal onset. J. Genet. Hum. 1982;30(Suppl 5):423–428. [PubMed] [Google Scholar]
- 9.Addor M.C., Pescia G., Egloff D., Quéloz J. Hereditary multicentric osteolysis. J. Genet. Hum. 1986;34:293–303. [PubMed] [Google Scholar]
- 10.Carnevale A., Canún S., Mendoza L., del Castillo V. Idiopathic multicentric osteolysis with facial anomalies and nephropathy. Am. J. Med. Genet. 1987;26:877–886. doi: 10.1002/ajmg.1320260415. [DOI] [PubMed] [Google Scholar]
- 11.Pai G.S., Macpherson R.I. Idiopathic multicentric osteolysis: Report of two new cases and a review of the literature. Am. J. Med. Genet. 1988;29:929–936. doi: 10.1002/ajmg.1320290425. [DOI] [PubMed] [Google Scholar]
- 12.De Ravel T.J., Matthijs G., Holvoet M., Wouters C., Legius E., Fryns J.P. Idiopathic multicentric osteolysis presents early and is not linked to chromosome 18q21.1. J. Med. Genet. 2000;37:E34. doi: 10.1136/jmg.37.11.e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faber M.R., Verlaak R., Fiselier T.J., Hamel B.C., Franssen M.J., Gerrits G.P. Inherited multicentric osteolysis with carpal-tarsal localisation mimicking juvenile idiopathic arthritis. Eur. J. Pediatr. 2004;163:612–618. doi: 10.1007/s00431-004-1502-1. [DOI] [PubMed] [Google Scholar]
- 14.Singhal R., Salim J., Walker P. Idiopathic multicentric osteolysis: A case report and literature review. Acta Orthop. Belg. 2005;71:328–333. [PubMed] [Google Scholar]
- 15.Thomas C.W., Bisset A.J., Sampson M.A., Armstrong R.D. Case report: Multicentric carpal/tarsal osteolysis: Imaging review and 25-year follow-up. Clin. Radiol. 2006;61:892–895. doi: 10.1016/j.crad.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 16.Connor A., Highton J., Hung N.A., Dunbar J., MacGinley R., Walker R. Multicentric carpal-tarsal osteolysis with nephropathy treated successfully with cyclosporine A: A case report and literature review. Am. J. Kidney Dis. 2007;50:649–654. doi: 10.1053/j.ajkd.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 17.Martignetti J.A., Aqeel A.A., Sewairi W.A., Boumah C.E., Kambouris M., Mayouf S.A., Sheth K.V., Eid W.A., Dowling O., Harris J. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat. Genet. 2001;28:261–265. doi: 10.1038/90100. [DOI] [PubMed] [Google Scholar]
- 18.Zankl A., Bonafé L., Calcaterra V., Di Rocco M., Superti-Furga A. Winchester syndrome caused by a homozygous mutation affecting the active site of matrix metalloproteinase 2. Clin. Genet. 2005;67:261–266. doi: 10.1111/j.1399-0004.2004.00402.x. [DOI] [PubMed] [Google Scholar]
- 19.Zankl A., Pachman L., Poznanski A., Bonafé L., Wang F., Shusterman Y., Fishman D.A., Superti-Furga A. Torg syndrome is caused by inactivating mutations in MMP2 and is allelic to NAO and Winchester syndrome. J. Bone Miner. Res. 2007;22:329–333. doi: 10.1359/jbmr.061013. [DOI] [PubMed] [Google Scholar]
- 20.Wenkert D., Mumm S., Wiegand S.M., McAlister W.H., Whyte M.P. Absence of MMP2 mutation in idiopathic multicentric osteolysis with nephropathy. Clin. Orthop. Relat. Res. 2007;462:80–86. doi: 10.1097/BLO.0b013e3180d09db8. [DOI] [PubMed] [Google Scholar]
- 21.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang P.W., Eisenbart J.D., Cordes S.P., Barsh G.S., Stoffel M., Le Beau M.M. Human KRML (MAFB): cDNA cloning, genomic structure, and evaluation as a candidate tumor suppressor gene in myeloid leukemias. Genomics. 1999;59:275–281. doi: 10.1006/geno.1999.5884. [DOI] [PubMed] [Google Scholar]
- 25.Kim K., Kim J.H., Lee J., Jin H.M., Kook H., Kim K.K., Lee S.Y., Kim N. MafB negatively regulates RANKL-mediated osteoclast differentiation. Blood. 2007;109:3253–3259. doi: 10.1182/blood-2006-09-048249. [DOI] [PubMed] [Google Scholar]
- 26.Moriguchi T., Hamada M., Morito N., Terunuma T., Hasegawa K., Zhang C., Yokomizo T., Esaki R., Kuroda E., Yoh K. MafB is essential for renal development and F4/80 expression in macrophages. Mol. Cell. Biol. 2006;26:5715–5727. doi: 10.1128/MCB.00001-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly L.M., Englmeier U., Lafon I., Sieweke M.H., Graf T. MafB is an inducer of monocytic differentiation. EMBO J. 2000;19:1987–1997. doi: 10.1093/emboj/19.9.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smink J.J., Bégay V., Schoenmaker T., Sterneck E., de Vries T.J., Leutz A. Transcription factor C/EBPbeta isoform ratio regulates osteoclastogenesis through MafB. EMBO J. 2009;28:1769–1781. doi: 10.1038/emboj.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wethmar K., Bégay V., Smink J.J., Zaragoza K., Wiesenthal V., Dörken B., Calkhoven C.F., Leutz A. C/EBPbetaDeltauORF mice—a genetic model for uORF-mediated translational control in mammals. Genes Dev. 2010;24:15–20. doi: 10.1101/gad.557910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rocques N., Abou Zeid N., Sii-Felice K., Lecoin L., Felder-Schmittbuhl M.P., Eychène A., Pouponnot C. GSK-3-mediated phosphorylation enhances Maf-transforming activity. Mol. Cell. 2007;28:584–597. doi: 10.1016/j.molcel.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Duncan E.L., Danoy P., Kemp J.P., Leo P.J., McCloskey E., Nicholson G.C., Eastell R., Prince R.L., Eisman J.A., Jones G. Genome-wide association study using extreme truncate selection identifies novel genes affecting bone mineral density and fracture risk. PLoS Genet. 2011;7:e1001372. doi: 10.1371/journal.pgen.1001372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bakker S.J., Vos G.D., Verschure P.D., Mulder A.H., Tiebosch A.T. Abnormal glomerular basement membrane in idiopathic multicentric osteolysis. Pediatr. Nephrol. 1996;10:200–202. doi: 10.1007/BF00862077. [DOI] [PubMed] [Google Scholar]
- 33.Sadl V., Jin F., Yu J., Cui S., Holmyard D., Quaggin S., Barsh G., Cordes S. The mouse Kreisler (Krml1/MafB) segmentation gene is required for differentiation of glomerular visceral epithelial cells. Dev. Biol. 2002;249:16–29. doi: 10.1006/dbio.2002.0751. [DOI] [PubMed] [Google Scholar]
- 34.Beaty T.H., Murray J.C., Marazita M.L., Munger R.G., Ruczinski I., Hetmanski J.B., Liang K.Y., Wu T., Murray T., Fallin M.D. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat. Genet. 2010;42:525–529. doi: 10.1038/ng.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Textor L.C., Wilmanns M., Holton S.J. Expression, purification, crystallization and preliminary crystallographic analysis of the mouse transcription factor MafB in complex with its DNA-recognition motif Cmare. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007;63:657–661. doi: 10.1107/S1744309107031260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brunkow M.E., Gardner J.C., Van Ness J., Paeper B.W., Kovacevich B.R., Proll S., Skonier J.E., Zhao L., Sabo P.J., Fu Y. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 2001;68:577–589. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balemans W., Patel N., Ebeling M., Van Hul E., Wuyts W., Lacza C., Dioszegi M., Dikkers F.G., Hildering P., Willems P.J. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 2002;39:91–97. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X., Ominsky M.S., Warmington K.S., Morony S., Gong J., Cao J., Gao Y., Shalhoub V., Tipton B., Haldankar R. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J. Bone Miner. Res. 2009;24:578–588. doi: 10.1359/jbmr.081206. [DOI] [PubMed] [Google Scholar]
- 39.Cummings S.R., San Martin J., McClung M.R., Siris E.S., Eastell R., Reid I.R., Delmas P., Zoog H.B., Austin M., Wang A., FREEDOM Trial Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N. Engl. J. Med. 2009;361:756–765. doi: 10.1056/NEJMoa0809493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.