

Abstract
Degeneration of the cerebrum, cerebellum, and retina in infancy is part of the clinical spectrum of lysosomal storage disorders, mitochondrial respiratory chain defects, carbohydrate glycosylation defects, and infantile neuroaxonal dystrophy. We studied eight individuals from two unrelated families who presented at 2–6 months of age with truncal hypotonia and athetosis, seizure disorder, and ophthalmologic abnormalities. Their course was characterized by failure to acquire developmental milestones and culminated in profound psychomotor retardation and progressive visual loss, including optic nerve and retinal atrophy. Despite their debilitating state, the disease was compatible with survival of up to 18 years. Laboratory investigations were normal, but the oxidation of glutamate by muscle mitochondria was slightly reduced. Serial brain MRI displayed progressive, prominent cerebellar atrophy accompanied by thinning of the corpus callosum, dysmyelination, and frontal and temporal cortical atrophy. Homozygosity mapping followed by whole-exome sequencing disclosed a Ser112Arg mutation in ACO2, encoding mitochondrial aconitase, a component of the Krebs cycle. Specific aconitase activity in the individuals' lymphoblasts was severely reduced. Under restrictive conditions, the mutant human ACO2 failed to complement a yeast ACO1 deletion strain, whereas the wild-type human ACO2 succeeded, indicating that this mutation is pathogenic. Thus, a defect in mitochondrial aconitase is associated with an infantile neurodegenerative disorder affecting mainly the cerebellum and retina. In the absence of noninvasive biomarkers, determination of the ACO2 sequence or of aconitase activity in lymphoblasts are warranted in similarly affected individuals, based on clinical and neuroradiologic grounds.
Main Text
The differential diagnosis of neurodegenerative disorders with atrophy of the cerebral and cerebellar cortex and cerebellar vermis is broad even in young age groups; concomitant occurrence of degeneration of the retina points to lysosomal storage disorders, mitochondrial disorders, defects of carbohydrate glycosylation, and infantile neuroaxonal dystrophy.1–4 We hereby report on the identification of a tricarboxylic acid (TCA) cycle defect manifesting in infancy by the degeneration of the cerebrum, cerebellum, and retina.
The subjects of this study were eight individuals, two males and six females, originating from two unrelated families of Arab Muslim origin (Figure 1). Their clinical data are summarized in Table 1. All were born following an uneventful pregnancy and delivery and had normal birth weights, head circumferences, and Apgar scores. Symptoms were first noted at 2–6 months of age and included variable appearance of truncal hyptonia, head bobbing, athetosis, generalized seizures, and ophthalmologic abnormalities that included strabismus, nystagmus, abnormal eye movements, and abnormal pursuit. The course was characterized by failure to thrive, gradual evolution of severe muscle wasting despite satisfactory oral intake, worsening of the hypotonia, attenuating deep tendon reflexes until complete areflexia was reached, progressive microcephaly, variable forms of seizures that were typically triggered by intercurrent febrile illnesses and were partly responsive to anticonvulsants, and failure to gain developmental milestones, culminating in profound psychomotor retardation. A distinctive feature was the appearance of head bobbing and athetoid movements of the upper limbs along with horizontal nystagmus when the individuals were held at the upright position. Visual tracking, normal at the newborn period, steadily deteriorated with the gradual evolution of bilateral optic atrophy. Visual evoked potentials in early infancy were normal, but the electroretinogram responses became critically reduced over the first two years of life, indicating severe retinal dystrophy. Similarly, auditory brain responses declined steadily, reflecting sensorineural hearing loss of variable severity. EEG disclosed generalized or focal spike and wave activity and slow background compatible with nonspecific encephalopathy. Electrophysiologic assessment of peripheral muscles and nerves performed in individual V-2 at 2 years revealed a normal electromyogram and a mild decrease of peripheral sensory and motor velocity suggesting demyelination. Taken together with the gradual disappearance of tendon reflexes, this finding indicates an evolving peripheral demyelinating neuropathy.
Figure 1.
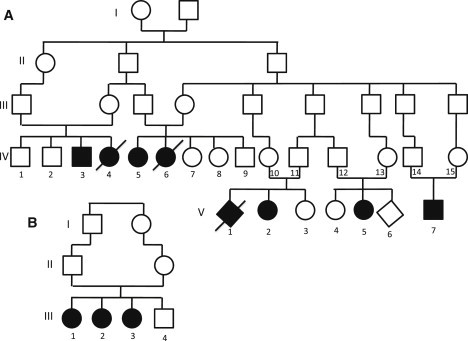
Pedigrees of the Two Families
(A) Family A and (B) Family B. Affected individuals are represented by filled symbols.
Table 1.
Clinical Features of Affected Individuals
|
Family A |
Family B |
|||||||
|---|---|---|---|---|---|---|---|---|
| IV-3 | IV-5 | V-2 | V-5 | V-7 | III-1 | III-2 | III-3 | |
| Sex | m | f | f | f | m | f | f | f |
| Current age (years) | 18 | 13 | 7 | 3 | 0.5 | 9 | 8 | 2 |
| Birth weight (centile) | 2,900 g (10) | 3,700 g (75) | 2,700 g (10) | 2,800 g (15) | 3,700 g (75) | 3,220 g (25) | 3,450 g (50) | 2,750 g (10) |
| Birth head circumference (centile) | 35 cm (40) | 35 cm (50) | 35 cm (50) | 34 cm (25) | 35 cm (40) | 34.5 cm (40) | 35 cm (50) | 34 cm (25) |
| Initial manifestations (age at presentation) | H, A (5 months) | H, A (6 months) | H, A (2 months) | H, E (4 months) | H, E (3 months) | H, S (3 months) | A, H (5 months) | A, E (5 months) |
| Current weight (centile) | 25 kg (−5 SD) | 26 kg (−4 SD) | 11 kg (−5 SD) | 9 kg (−4 SD) | 7.7 kg (25) | 13 kg (−6 SD) | 12 kg (−6 SD) | 8 kg (−4 SD) |
| Current head circumference (centile) | 47 cm (−5 SD) | 49.5 cm (−3 SD) | 46.5 cm (−3.5 SD) | 46.5 cm (5) | 42.7 cm (25) | 48.5 cm (−2.5 SD) | 48 cm (3) | 45.5 cm (5) |
| Global DD/MR | profound | severe | profound | profound | moderate | profound | profound | severe |
| Current motor skills | no intentional movements | crawls, rolls sits with support | does not roll | rolls, does not sit | lift head when supine | does not role | does not role | roles both sides |
| Current cognitive skills | vegetative state, no smile | smile, recognize family | smile, recognize family | smile, recognize family | smile, recognize family | no eye contact, does not recognize family | no eye contact, responds to auditory stimuli | smile, recognize family |
| Ataxia | yes | yes | yes | yes | very mild | yes | yes | yes |
| Seizures | generalized | generalized | focal and generalized | focal and generalized | no | generalized myoclonic and focal | focal and generalized | no |
| Retinal dystrophy (age at diagnosis) | not evaluated | severe (4 years) | severe (1 year) | severe (1 year) | not evaluated | profound (2 years) | profound (1.5 years) | moderate (1.5 years) |
| ophthalmology | optic atrophy, strabismus | optic atrophy, strabismus | optic atrophy, nystagmus | optic atrophy, nystagmus, strabismus | strabismus | optic atrophy, nystagmus | optic atrophy, nystagmus | optic atrophy, nystagmus, strabismus |
| Initial MR scan (age) | not done | normal (11 months) | mild CoA (3 months) | mild CoA (frontal and temporal) (11 months) | normal (in utero, 30 weeks) | CoA (frontal and temporal) mild thinning of CC (7 months) | CoA abnormal WM (16 months) | normal (in utero, 31 weeks) |
| Last MR scan (age) | not done | severe CeA, moderate CoA (frontal and temporal), abnormal periventricular WM signal (12 years) | severe CeA, moderate CoA (frontal and temporal), thin CC abnormal WM signal (4 years) | severe CeA, moderate CoA (frontal and temporal), thin CC, abnormal WM signal (2.5 years) | not done | not done | mild CoA and CeA (1y) | |
| Tendon reflexes | absent | brisk | absent | absent | normal | absent | decreased | absent |
| Other | severe scoliosis | moderate SNHL | severe SNHL (hearing aids) | |||||
The following abbreviations are used: A, ataxia/athetoid movements; H, hypotonia; E, eye abnormalities; S, seizures; HC, head circumference; DD, developmental delay; MR, mental retardation; CoA, cortical atrophy; CeA, cerebellar atrophy; WM, white matter; CC, corpus callosum; SNHL, sensorineural hearing loss.
During gestation and throughout the first 6 months, brain MRI was typically normal. Thereafter, repeated MR scans (Figure 2) displayed progressive cerebellar atrophy of both hemispheres and the vermis. In addition, symmetric cortical atrophy gradually evolved, affecting mainly the frontal and temporal lobes along with a striking thinning of the corpus callosum. White matter abnormalities, indicated by T2 and fluid-attenuated inversion recovery (FLAIR) periventricular hyperintense signals, were first noted during the second year of life. Taken together, these serial MR scans revealed generalized progressive brain atrophy affecting primarily the cerebellum and to a lesser extent the white matter and the frontal and parietal lobes of the cerebral cortex.
Figure 2.
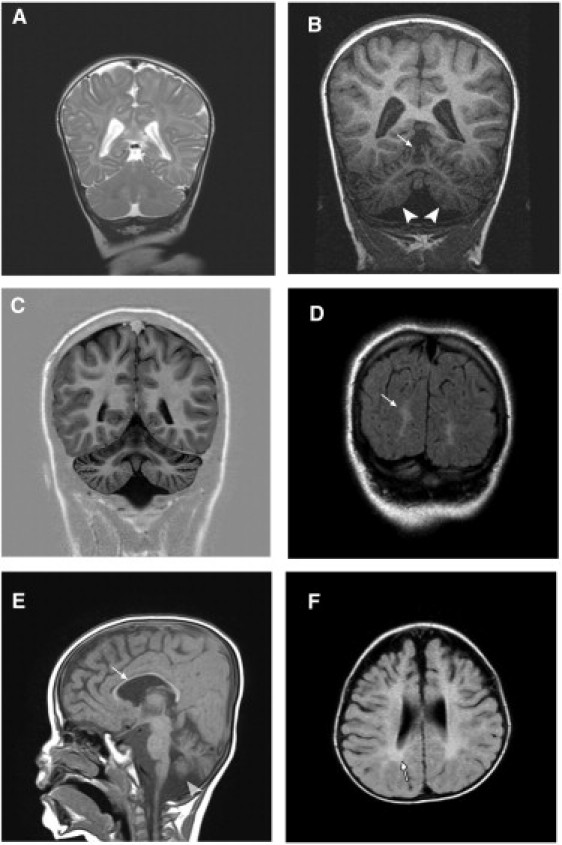
Representative MRI Findings
(A) T2-weighted, coronal section image of individual V-2 at 3 months shows a normal cerebellum and mild cortical atrophy.
(B) T1-weighted, coronal section scan of the same individual at 4 years displays severe atrophy of the cerebellar hemispheres (arrowheads) and the vermis (arrows).
(C) T1-weighted, coronal section image of individual IV-5 at 12 years shows enlarged cerebellar folia and reduced volume indicating generalized (vermian and hemispheral) atrophy. Note also the mild cortical atrophy and visible sulci.
(D) Cortical atrophy and white matter abnormal signals (arrows) demonstrated by FLAIR image of coronal section in the same individual.
(E) Moderate cortical atrophy, very thin corpus callosum (arrow), and cerebellar atrophy (arrowhead) in a T1-weighted, sagital section of individual V-5 at 2.5 years.
(F) FLAIR image, axial section of individual V-5 displaying frontal and temporal cortical atrophy compared with relatively preserved occipital cortex. The abnormal periventricular white matter signal (arrow) is consistent with dysmyelination.
Extensive laboratory investigations, including complete blood count, routine serum chemistry, glucose, lactate, ammonia, thyroid functions, creatine kinase, acylcarnitines, amino acids, very long chain fatty acids, and isoelectric focusing of transferrins, were all normal. The profile of the urinary organic acids and specifically aconitic, citric, and isocitric acids was normal. Cerebrospinal fluid analyses for cells, glucose, protein, lactate, amino acids, and neurotransmitters were normal. Enzymatic assays in white blood cells excluded infantile neuronal lipofuscinosis, GM1 gangliosidosis [MIM 230500], Tay Sachs disease [MIM 272800], Krabbe disease [MIM 245200], and metachromatic leukodystrophy [MIM 250100]. One individual underwent a rectal biopsy that revealed no evidence of storage disease. Muscle biopsies of three individuals disclosed normal histology by light and electron microscopy and by immunohistochemistry. The activities of the five enzymatic complexes of the mitochondrial respiratory chain and the pyruvate dehydrogenase complex were normal in isolated mitochondria from muscle. However, the oxidation of glutamate was slightly reduced to 62.7% (range 57%–66%) of the control mean, normalized to citrate synthase activity, whereas the oxidation of the other mitochondrial substrates, pyruvate, succinate, and ascorbate were 88.3%, 86.7%, and 90.3% of the control mean, normalized to citrate synthase activity, respectively.
At the time of writing, all of the individuals were alive; the oldest was 18 years old and already in a vegetative state; six individuals older than 2 years were profoundly intellectually disabled and completely handicapped. Throughout childhood and adolescence, the disease remained CNS exclusive with normal results of serial echocardiograms, abdominal ultrasounds, and routine chemistry. Despite this prolonged survival, the medical history of family A was remarkable in that two individuals (IV-4 and IV-6) with similar phenotype died in their first decade.
In order to localize the mutated gene, we searched for homozygous regions common to three of the individuals (IV-3, V-2, and V-5) by using the GeneChip Human Mapping 250K Nsp array (Affymetrix), as previously described.5 We then used selected short tandem repeat (STR) markers for the genotyping of the remaining family members. All experiments involving DNA of the individuals, their relatives, and healthy controls were approved by the Hadassah and Emek Medical Centers Ethical Review Boards. This analysis resulted in the identification of a single 4 Mb homozygous genomic region on chromosome 22: 38.3–42.3 Mb (corresponding to rs136805–rs6006714, numbering according to NCBI build 36.1/hg18) encompassing 175 SNP markers with identical genotype. Within this region there were 65 protein-coding genes, which included a total of 657 exons. These data were used for prenatal diagnosis of three pregnancies—fetus V-6 was predicted to be heterozygous and fetuses V-1 and V-7 were predicted to be affected. The pregnancy of fetus V-1 was terminated, and postmortem pathological study was unremarkable; in particular, brain macroscopic and microscopic examinations were normal. The parents of fetus V-7 elected to continue the pregnancy, and although normal at birth, at 6 months of age this individual already suffered from hypotonia, developmental delay, strabismus, and athetoid movements.
Because of the large number of genes within the region, we performed exome sequencing. Exonic sequences in DNA from individual V-2 were captured with the Agilent SureSelect All Exon 50 Mb kit and sequenced on 1/4 slide with SOLiD4. Read alignment was performed with Bioscope, and single-nucleotide variants were called with DiBayes and SAMtools.6 The 657 exons within the region of homozygosity had an average read coverage of 66×, and 430 exons were covered more than 6×. We identified 112 homozygous single-nucleotide variants in the region, and 42 variants were located in coding exons and flanking intronic sequencing (±12 nt). Variants were further filtered to remove polymorphisms present in dbSNP132 and the 1000 Genomes Project. Only a single coding homozygous variant remained after filtering. This variant was c.336C>G in ACO2 [MIM 100850] (NM_001098.2), which causes p.Ser112Arg (NP_001089.1). This position is highly conserved and the substitution was scored as “probably damaging” with PolyPhen2 (score of 0.992). All eight individuals were homozygous for the mutation; ten parents and one of the healthy sibs were heterozygous, and three healthy sibs were homozygous for the normal allele. Affected individuals of both unrelated clans shared a similar homozygous haplotype, indicating that Ser112Arg is a founder mutation in this population. None of the 128 anonymous individuals of the same ethnic origin carried the mutation.
ACO2 consists of 18 exons that encode an 805 amino acid TCA-cycle protein, the mitochondrial aconitate hydratase (mAH). Cellular aconitase activity, measured in lymphoblasts of two individuals by monitoring the decrease in the absorbance of the substrate cis-aconitate at 240 nm,7 was reduced at 37°C to 11.9% ± 9.2% of the control. In contrast, fumarase activity in individuals' lymphoblasts, assayed at 250 nm with L-malic acid as the substrate,8 was comparable to that of the control (Figure 3). The serine at codon 112 is highly conserved throughout evolution, and we have used this attribute to study the effect of the mutation in a yeast strain in which the aconitase gene was deleted (Saccharomyces cerevisiae ACO1). In yeast, a small fraction of the mAH resides in the cytosol and has been shown to participate in the glyoxylate shunt that is required for growth on two-carbon substrates such as ethanol.7 Because the human and yeast mAHs are highly conserved, we cloned the human mAH into a yeast expression vector and shuffled it into a yeast strain harboring a chromosomal deletion of the yeast mAH gene (Δaco1, ΔYLR304C, as previously described7,8). We found that both the nonmutated and the Ser112Arg mutated human cDNA fully complement the yeast mAH deletion strain under a variety of conditions (not shown). We then examined whether the Ser112Arg mutated human gene can complement the yeast mAH deletion under high-requirement conditions such as respiration and growth on two-carbon substrates. We discovered that under growth conditions requiring the TCA cycle and the glyoxylate shunt (growth on ethanol medium) and growth at 37°C, yeast strains expressing a Ser112Arg mutated human gene show a very clear growth defect (Figure 4, second row), whereas the same strain harboring the nonmutated human ACO2 cDNA grows similarly to the wild-type (third row). These results indicate functional significance of the ACO2 Ser112Arg mutation, which is revealed under specific physiological conditions. It is expected that a more deleterious mutation in the human ACO2 gene would not be compatible with life because yeast lacking the aconitase gene exhibit very severe growth defects (Figure 4, fourth row).
Figure 3.
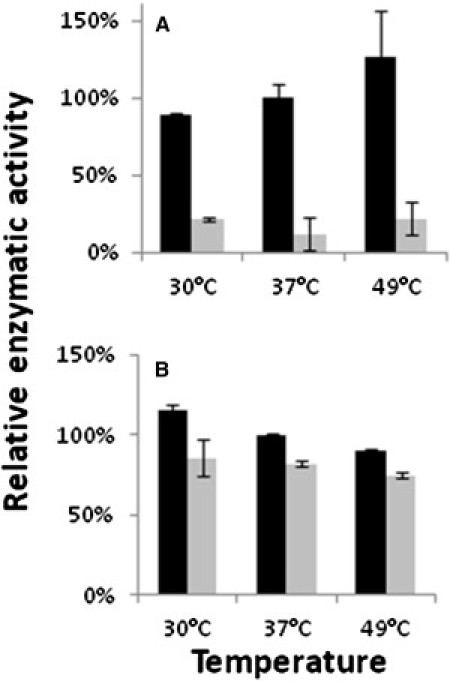
Aconitase-Specific Activity
Lymphoblasts from individuals harboring either a wild-type ACO2 (black bars) or a Ser112Arg-ACO2 mutated gene (grey bars) were lysed and cell extracts were assayed for aconitase (A) and fumarase (B) activity at the indicated temperatures. The relative specific enzymatic activity is presented with reference to the wild-type extract at 37°C (100%). Error bars indicate the standard deviation of two independent experiments.
Figure 4.
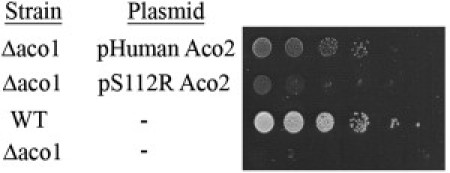
Expression of S112R mutant human mitochondrial aconitase, ACO2, in yeast, only partially complements the depletion of the endogenous aconitase, S. cerevisiae ACO1
Wild-type (WT) yeast and a mutant deleted for the aconitase gene (Δaco1) were transformed with the indicated plasmids. Cultures were serially diluted at 10-fold intervals, and 10 μl of each was spotted onto ethanol plates, which were incubated at 37°C for 5 days.
Aconitate hydratase (AH) catalyzes the reversible isomerization of citrate to isocitrate. The reaction consists of dehydration followed by hydration and the formation of an intermediate product, cis-aconitate, which normally does not separate from AH. The two existing AH forms, cytoplasmic (cAH) and mitochondrial (mAH), are only 30% homologous and differ from each other in physicochemical and structural properties. In humans, the two forms are ubiquitously expressed and are most active in the heart, kidney, and liver.9 Both proteins have four iron-sulfur (Fe-S) clusters, but the fourth iron ion is loosely bound, allowing its dissociation from the cluster thereby rendering the protein inactive (3Fe-4S). The active (4Fe-4S) cluster was shown to be extremely sensitive to superoxide-mediated inactivation10 and a decrease in AH activity was observed in several neurodegenerative diseases associated with the development of oxidative stress, in particular Friedreich ataxia [MIM 229300], Parkinson [MIM 168600], and Alzheimer disease [MIM 104300],11 as well as in mice lacking mitochondrial superoxide dismutase.12 The reduced AH activity in endomyocardial biopsies of individuals with Friedreich ataxia was attributed not only to oxidative stress but also to the importance of Frataxin in the Fe-S cluster assembly.13 The secondary inhibition of mAH in individuals with Friedreich ataxia might thus underlie the clinical similarities with the individuals we studied, in particular the progressive cerebellar atrophy and the optic atrophy.14
The TCA cycle has a crucial role in energy metabolism, generating reducing power in the form of NADH and FADH2. Several TCA cycle enzymopaties, including deficiency of succinate dehydrogenase [MIM 600857], succinyl-CoA synthase [MIM 603921] and fumarate hydratase [MIM 606812] are known to be associated with autosomal-recessive encephalopathies of infancy,15–18 whereas mutations in IDH3 encoding the beta-subunit of NAD-specific isocitrate dehydrogenase have been reported in individuals with retinitis pigmentosa.19 The mAH defect thus joins the growing list of neurodegenerative diseases associated with primary TCA cycle impairment but is distinctive in the prominent involvement of the retina and the cerebellum. Similar to the other TCA cycle defects, individuals in our study hardly had any mitochondrial disease stigmata, such as elevated lactate, pyruvate, and alanine levels in plasma or TCA cycle metabolite levels in urine. Enzymatic activities of the mitochondrial respiratory chain complexes in muscle mitochondria were intact and only glutamate oxidation was slightly reduced, indicating that in clinical settings mAH deficiency would be easily overlooked.
TCA cycle defects are not only associated with mitochondrial encephalopathies but also with benign and malignant tumors; this suggests that TCA cycle metabolites are in fact oncometabolites, and the enzymes themselves are oncogene suppressors. Mutations in the isocitrate dehydrogenase genes were found in individuals with glioblastoma multiforme, glioma, and acute myeloid leukemias,20 pheochromocytoma and paraganglioma were associated with succinate dehydrogenase mutations21 and germline mutations in the fumarate hydratase gene were discovered in individuals with uterine fibroids, skin leiomyomata and papillary renal cell carcinoma.22 Following the identification of the ACO2 mutation in the individuals in our study, we scrutinized the medical files of the obligatory carriers but could not identify an increased prevalence of cancer.
Our approach, combining homozygosity mapping with exome sequencing, enabled the identification of a pathogenic mutation in ACO2 in eight individuals who suffered from a neurodegenerative disorder affecting mainly the cerebellum and the retina. As biomarkers are not available, we recommend measurement of aconitase activity in lymphoblasts or determination of the sequence of ACO2 in similarly affected individuals, on the basis of clinical and neuroradiologic grounds.
Acknowledgements
We thank Rachel Dahan, Lital Sheva, and Noa Cohen for excellent technical assistance.
Contributor Information
Ophry Pines, Email: ophryp@ekmd.huji.ac.il.
Orly Elpeleg, Email: elpeleg@hadassah.org.il.
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
References
- 1.Nardocci N., Verga M.L., Binelli S., Zorzi G., Angelini L., Bugiani O. Neuronal ceroid-lipofuscinosis: a clinical and morphological study of 19 patients. Am. J. Med. Genet. 1995;57:137–141. doi: 10.1002/ajmg.1320570205. [DOI] [PubMed] [Google Scholar]
- 2.Uziel G., Moroni I., Lamantea E., Fratta G.M., Ciceri E., Carrara F., Zeviani M. Mitochondrial disease associated with the T8993G mutation of the mitochondrial ATPase 6 gene: a clinical, biochemical, and molecular study in six families. J. Neurol. Neurosurg. Psychiatry. 1997;63:16–22. doi: 10.1136/jnnp.63.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaeken J., Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu. Rev. Genomics Hum. Genet. 2007;8:261–278. doi: 10.1146/annurev.genom.8.080706.092327. [DOI] [PubMed] [Google Scholar]
- 4.Gregory A., Westaway S.K., Holm I.E., Kotzbauer P.T., Hogarth P., Sonek S., Coryell J.C., Nguyen T.M., Nardocci N., Zorzi G. Neurodegeneration associated with genetic defects in phospholipase A(2) Neurology. 2008;71:1402–1409. doi: 10.1212/01.wnl.0000327094.67726.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Regev-Rudzki N., Karniely S., Ben-Haim N.N., Pines O. Yeast aconitase in two locations and two metabolic pathways: seeing small amounts is believing. Mol. Biol. Cell. 2005;16:4163–4171. doi: 10.1091/mbc.E04-11-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Regev-Rudzki N., Yogev O., Pines O. The mitochondrial targeting sequence tilts the balance between mitochondrial and cytosolic dual localization. J. Cell Sci. 2008;121:2423–2431. doi: 10.1242/jcs.029207. [DOI] [PubMed] [Google Scholar]
- 9.Slaughter C.A., Hopkinson D.A., Harris H. The distribution and properties of aconitase isozymes in man. Ann. Hum. Genet. 1977;40:385–401. [PubMed] [Google Scholar]
- 10.Gardner P.R., Raineri I., Epstein L.B., White C.W. Superoxide radical and iron modulate aconitase activity in mammalian cells. J. Biol. Chem. 1995;270:13399–13405. doi: 10.1074/jbc.270.22.13399. [DOI] [PubMed] [Google Scholar]
- 11.Hinerfeld D., Traini M.D., Weinberger R.P., Cochran B., Doctrow S.R., Harry J., Melov S. Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. J. Neurochem. 2004;88:657–667. doi: 10.1046/j.1471-4159.2003.02195.x. [DOI] [PubMed] [Google Scholar]
- 12.Kil I.S., Park J.W. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J. Biol. Chem. 2005;280:10846–10854. doi: 10.1074/jbc.M411306200. [DOI] [PubMed] [Google Scholar]
- 13.Bulteau A.L., O'Neill H.A., Kennedy M.C., Ikeda-Saito M., Isaya G., Szweda L.I. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science. 2004;305:242–245. doi: 10.1126/science.1098991. [DOI] [PubMed] [Google Scholar]
- 14.Patel M. Mitochondrial dysfunction and oxidative stress: cause and consequence of epileptic seizures. Free Radic. Biol. Med. 2004;37:1951–1962. doi: 10.1016/j.freeradbiomed.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 15.Elpeleg O., Miller C., Hershkovitz E., Bitner-Glindzicz M., Bondi-Rubinstein G., Rahman S., Pagnamenta A., Eshhar S., Saada A. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am. J. Hum. Genet. 2005;76:1081–1086. doi: 10.1086/430843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ottolenghi C., Hubert L., Allanore Y., Brassier A., Altuzarra C., Mellot-Draznieks C., Bekri S., Goldenberg A., Veyrieres S., Boddaert N. Clinical and biochemical heterogeneity associated with fumarase deficiency. Hum. Mutat. 2011;32:1046–1052. doi: 10.1002/humu.21534. [DOI] [PubMed] [Google Scholar]
- 17.Bourgeron T., Rustin P., Chretien D., Birch-Machin M., Bourgeois M., Viegas-Péquignot E., Munnich A., Rötig A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat. Genet. 1995;11:144–149. doi: 10.1038/ng1095-144. [DOI] [PubMed] [Google Scholar]
- 18.Ostergaard E., Christensen E., Kristensen E., Mogensen B., Duno M., Shoubridge E.A., Wibrand F. Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am. J. Hum. Genet. 2007;81:383–387. doi: 10.1086/519222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartong D.T., Dange M., McGee T.L., Berson E.L., Dryja T.P., Colman R.F. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat. Genet. 2008;40:1230–1234. doi: 10.1038/ng.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prensner J.R., Chinnaiyan A.M. Metabolism unhinged: IDH mutations in cancer. Nat. Med. 2011;17:291–293. doi: 10.1038/nm0311-291. [DOI] [PubMed] [Google Scholar]
- 21.Astuti D., Latif F., Dallol A., Dahia P.L., Douglas F., George E., Sköldberg F., Husebye E.S., Eng C., Maher E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomlinson I.P., Alam N.A., Rowan A.J., Barclay E., Jaeger E.E., Kelsell D., Leigh I., Gorman P., Lamlum H., Rahman S., Multiple Leiomyoma Consortium Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002;30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]