

Abstract
Environmental manganese (Mn) toxicity causes an extrapyramidal, parkinsonian-type movement disorder with characteristic magnetic resonance images of Mn accumulation in the basal ganglia. We have recently reported a suspected autosomal recessively inherited syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia in cases without environmental Mn exposure. Whole-genome mapping of two consanguineous families identified SLC30A10 as the affected gene in this inherited type of hypermanganesemia. This gene was subsequently sequenced in eight families, and homozygous sequence changes were identified in all affected individuals. The function of the wild-type protein and the effect of sequence changes were studied in the manganese-sensitive yeast strain Δpmr1. Expressing human wild-type SLC30A10 in the Δpmr1 yeast strain rescued growth in high Mn conditions, confirming its role in Mn transport. The presence of missense (c.266T>C [p.Leu89Pro]) and nonsense (c.585del [p.Thr196Profs∗17]) mutations in SLC30A10 failed to restore Mn resistance. Previously, SLC30A10 had been presumed to be a zinc transporter. However, this work has confirmed that SLC30A10 functions as a Mn transporter in humans that, when defective, causes Mn accumulation in liver and brain. This is an important step toward understanding Mn transport and its role in neurodegenerative processes.
Introduction
Manganese (Mn) is an essential trace metal that plays a critical role as a cofactor for a variety of enzymes involved in amino acid, lipid and carbohydrate metabolism, immune function, bone and connective tissue growth, and blood clotting.1 The main source of Mn in humans is through dietary ingestion. Stable tissue levels are maintained by tight homeostatic control of intestinal absorption and biliary excretion of Mn.2 Although Mn is critical for cell function, overexposure is known to be neurotoxic and causes “manganism.” This distinct syndrome of extrapyramidal movement disorder combined with high signal intensity of the basal ganglia on T1-weighted magnetic resonance images (MRI) of the brain is caused by Mn accumulation in the basal ganglia.3,4
Most frequently, hypermanganesemia occurs because of environmental overexposure. It has been described in workers in mining and welding industries who inhale Mn-laden dust or fumes3,5,6 and in individuals ingesting contaminated drinking water.7 However, cases of Mn intoxication have also been observed in drug addicts who use intravenous methcathinone contaminated with potassium permanganate8,9 and in children and adults receiving parenteral nutrition.10,11 Acquired hepatocerebral degeneration (AHD) can develop in individuals with impaired biliary excretion of Mn including those with advanced hepatic cirrhosis or portosystemic shunts causing a debilitating motor disorder.12–15 Predisposition to Mn toxicity has also been suggested in individuals with idiopathic Parkinson's disease (PD).16,17 Indeed, it has been reported that the use of methylcyclopentadienyl manganese tricarbonyl (an organic derivative of Mn) as an octane enhancer in gasoline might result in earlier presentation of PD.18 Although manganism resembles PD (with symptoms of generalized bradykinesia and rigidity), there are characteristic features distinguishing Mn intoxication from idiopathic PD. Manganism more frequently causes dystonia, shows poor therapeutic response to levodopa, and has normal fluorodopa uptake by PET scan.19,20
Several cases of hypermanganesemia, dystonia, and polycythemia with a variable degree of hepatic dysfunction have been described in children without significant environmental Mn exposure.21–26 Identification of two affected siblings, from a consanguineous family, with this disorder suggested an inherited defect of Mn homeostasis [MIM 613280].21 We have identified a further 13 individuals from seven families with similar symptoms, some of them previously reported,22–24 and performed homozygosity mapping to identify the affected gene. Affected individuals carry mutations in SLC30A10 [MIM 611146], solute carrier family 30, member 10, a previously presumed zinc (Zn) transporter. The effects of sequence changes were investigated in the Mn-sensitive yeast strain Δpmr1, and SLC30A10 was shown to be a potent Mn transporter.
Material and Methods
Subjects
For clinical details see Results and Table 1. The research protocol was approved by the West London Research Ethics Committee. All participants provided written informed consent for participation in the study.
Table 1.
Clinical Characteristics of Individuals with a Syndrome of Hepatic Cirrhosis, Dystonia, Polycythemia, and Hypermanganesemia
| Subject | Gender | Current Age (years) | Ethnicity (Country of Origin) | Consanguinity | Age at Presentation (years) | Whole-Blood Mn (<320 nmol/l) | Dystonia | Spastic Paraparesis | Hepatomegaly | Liver Cirrhosis on Biopsy | Hemoglobin (11.5–15.5 g/dL) | TIBC (50–90 μmol/l) | Iron (5–25 μmol/l) | Ferritin (7–150 μg/l) | Unconjugated Bilirubin (<18 μmol/l) | ALT (15–55 U/l) | Erythropoietin (mIU/ml) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A-II-1 | F | 18 | Punjabi (Pakistan) | + | 3 | 6180 | + | – | – | n/a | 17.3 | 169 | 6.4 | 10.4 | 29 | 156 | n/a | |
| A-II-2 | F | 16 | Punjabi (Pakistan) | + | 3 | 3767 | + | – | – | n/a | 16.2 | 143 | 8.7 | 19 | 32 | 142 | n/a | |
| A-II-4 | M | 11 | Punjabi (Pakistan) | + | 5 | 5096 | + | – | – | n/a | 18.4 | 100 | 13.6 | 20 | n/a | 33 | n/a | |
| A-II-5 | M | 9 | Punjabi (Pakistan) | + | 5 | 6370 | + | – | + | n/a | 16.7 | 148 | 5 | 7.46 | 10 | 85 | 20.4 (3.3–16.6) | |
| B-II-2 | F | 12 | Arabic (Yemen) | + | 2 | 2109 | + | – | – | n/a | 20.5 | n/a | 8.7 | 30 | 22 | 246 | n/a | |
| B-II-5 | F | 8 | Arabic (Yemen) | + | 2 | 1636 | + | – | – | n/a | 19.1 | n/a | n/a | n/a | 26 | 145 | n/a | |
| B-II-7 | F | 4 | Arabic (Yemen) | + | 2 | 1600 | + | – | – | n/a | 18.0 | n/a | n/a | n/a | 17 | 40 | n/a | |
| C-II-2 | F | 12 | Acadian (Canada) | – | 4 | 1145 | + | – | – | – | 19 | 114 | n/a | 2 | 12 | 57 | n/a | Brna et al.23and Sahni et al.24 |
| D-II-3a | M | †18 | Arabic (United Arab Emirates) | + | 2 | n/a | + | – | + | + | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Tuschl21 |
| D-II-6 | F | 21 | Arabic (United Arab Emirates) | + | 11 | 3285 | + | – | + | + | 18 | >120 | 22 | 8 | 45 | 23 | 39.9 (2.5–10.5) | |
| E-II-1 | M | 37 | Caucasian (US) | – | 14 | 3480 | – | + | + | + | 22.5 | 116 | 3.7 | 6 | normal | normal | 473 (0–16) | Gospe22 |
| F-II-1 | F | 12 | Indo-Malay (Philippines) | – | 11 | 3272 | + | – | – | n/a | 21 | n/a | 13.1 | 14 | 23.8 | 72 | 30.2 (3.7–31.5) | |
| G-II-1a | M | †25 | Portuguese/ Amerindian (Brazil) | + | 2 | n/a | + | – | – | + | n/a | n/a | n/a | n/a | n/a | n/a | n/a | |
| G-II-2 | F | 24 | Portuguese/ Amerindian (Brazil) | + | 3 | 3114 | + | – | – | + | 15.9 | 147 | 8.9 | 7.7 | 11.9 | 52 | n/a | |
| H-II-1 | F | 7 | Punjabi (India) | – | 5 | 2366 | + | – | + | n/a | 19.5 | 40 | 10.2 | 17.4 | 23 | 142 | n/a |
The following abbreviations are used: n/a, not available; TIBC, total iron binding capacity; ALT, alanine aminotransferase. Normal reference ranges are given in brackets.
Died because of complications of cirrhosis.
Homozygosity Mapping
Affected and unaffected siblings of two consanguineous families were included in homozygosity mapping studies. For pedigrees see Figure S1A, available online.
Total genomic DNA was extracted from whole blood of affected individuals with the Gentra Puregene Blood Kit (QIAGEN). Whole-genome mapping was performed with the Illumina HumanCytoSNP-12 DNA Analysis BeadChipKit according to the manufacturer's instructions. SNP data were analyzed with Illumina GenomeStudio Software to identify areas of homozygosity in affected individuals. Corresponding gene lists were generated for each area of homozygosity with the National Center for Biotechnology Information (NCBI) genome viewer build 36.3.
Sequencing of SLC30A10
The four exons and exon-intron boundaries of the coding sequence of SLC30A10 (NM_018713.2) were amplified by PCR from genomic DNA (primers listed in Table S1). The amplicons were purified with shrimp alkaline phosphatase and exonuclease I and subsequently sequenced bidirectionally with the ABI BigDye Terminator 1.1 system (Applied Biosystems) on an ABI DNA sequencer. Sequence data were analyzed with the Sequencer 4.9 software (Gene Codes). Mutations were confirmed by restriction enzyme digest test (enzymes obtained from New England Biolabs). Two hundred control alleles of healthy individuals (who were, where possible ethnicity matched) were screened by restriction enzyme digest test or sequencing for each mutation to exclude common variants.
Sequence Alignment and Phylogeny
Members of the SLC30A10 and Zrc1 family were identified with Ensembl and NCBI. Sequences were aligned with ClustalW2 and the neighbor-joined dendrogram generated with ClustalX.27 For protein accession numbers see Table S2.
Yeast Expression Studies
Expression studies were performed with Gateway technology (Invitrogen). AttB site-introducing primers were designed according to the manufacturer's instructions (Table S3). cDNA was synthesized from human fetal liver total RNA (purchased from Stratagene) with the SuperScript III First-Strand Synthesis System (Invitrogen), and SLC30A10 was amplified with Platinum Pfx DNA Polymerase (Invitrogen). Similarly, ZRC1 was amplified from Saccharomyces cerevisiae genomic DNA (purchased from Novagen). The PCR products were purified with 30% PEG 8000/30mM MgCl2.
Entry clones of SLC30A10 and ZRC1 were produced with the pDONR221 vector and BP Clonase II enzyme mix (Invitrogen). OneShot OmniMAX 2-T1 chemically competent Escherichia coli cells were transformed with the BP recombination reaction and entry clones selected on LB agar plates containing Kanamycin (50 μg/ml). After plasmid extraction with the QIAGEN Plasmid Mini Kit and DNA sequencing of the plasmid insert, a clone with the correct insert sequence of SLC30A10 or ZRC1 was selected and used for site-directed mutagenesis.
Site-directed mutagenesis, with SLC30A10/pDONR221 and ZRC1/pDONR221 as DNA templates, was performed with the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene) according to manufacturer's instructions (primers are listed in Table S4). OneShot OmniMAX 2-T1 chemically competent E. coli cells were transformed with the mutated vectors as described above and mutations confirmed by DNA sequencing after plasmid extraction.
Expression clones of SLC30A10, SLC30A10 (c.266T>C [p.Leu89Pro]), SLC30A10 (c.585del [p.Thr196Profs∗17]), ZRC1, and ZRC1 (c.131A>T [p.Asn44Ile]) were created with the pYES-DEST52 Gateway Vector and LR Clonase II enzyme mix (Invitrogen). OneShot OmniMAX 2-T1 chemically competent E. coli cells were transformed and expression clones selected on LB agar plates containing ampicillin (50 μg/ml). The plasmids were extracted and used for yeast transformation. The correct sequences of the various inserts were confirmed by DNA sequencing.
Wild-type BY4743 S. cerevisiae (Mat a/α his3Δ1/ his3Δ1 leu2Δ0/ leu2Δ0 lys2Δ0/+ met15Δ0/+ ura3Δ0/ ura3Δ0) and Δpmr1::KanMX strains in the BY4743 background were obtained from Open Biosystems. Yeast strains were grown on Yeast Extract Peptone Dextrose (YPD) medium (1% yeast extract, 2% peptone, 2% Dextrose) at 30°C for 72 hr and transformed with the S.f EasyComp Transformation Kit (Invitrogen). Expression clones were selected on SC-Ura medium (0.67% yeast nitrogen base without amino acids, 2% Dextrose and 0.13% amino acid drop-out mixture without Uracil) at 30°C. For gene expression SC-Ura induction medium containing 20% galactose and 10% raffinose was used. High Mn agar plates were prepared by adding MnCl2 to the medium at the required concentrations (1M stock in H2O).
Results
Clinical Characteristics of Affected Individuals
Affected individuals present at the age of 2 to 15 years with gait disturbance and hypertonia of the limbs with some becoming wheelchair bound. Although most cases show pure four-limb dystonia leading to a characteristic high-stepping gait (a “cock-walk” gait) and fine motor impairment sometimes accompanied by dysarthria, fine tremor, and bradykinesia, one affected individual has pure spastic paraparesis without extrapyramidal dysfunction (Table 1).22 Childhood developmental milestones are age-appropriate, and intellectual development appears normal in affected individuals; however, no formal cognitive testing was performed. All cases have pathognomonic MRI brain appearances with hyperintensity of globus pallidus, putamen, caudate, subthalamic and dentate nucleus and sparing of the thalamus and ventral pons on T1-weighted images (Figure S2). When the disease is extensive, this can be accompanied by white matter and anterior pituitary involvement. T2-weighted images also show changes, however, to a much lesser extent, and are often reported as normal.
Most affected individuals have liver impairment suggested by raised transaminases and unconjugated hyperbilirubinemia, and two individuals have died following complications of cirrhosis (Table 1). Liver biopsies show micronodular cirrhosis or fibrosis with positive rhodanine staining and elevated Mn content.21,22
Whole-blood Mn levels are significantly raised in all affected individuals (on average more than 2,000 nmol/l; normal is less than 320 nmol/l) (Table 1), mostly to a much higher extent than observed previously in environmental Mn exposure,3,7 parenteral nutrition,10 or chronic liver disease21 where blood levels are less than 2,000 nmol/l. Parental blood Mn levels were mildly elevated in three families (380 to 649 nmol/l), consistent with carrier status. These parents and heterozygous siblings were asymptomatic and were therefore not subjected to detailed evaluation of neurological and hepatic function.
Further features generally observed in this form of inherited hypermanganesemia are polycythemia associated with high erythropoietin levels and depleted iron stores indicated by low ferritin and iron levels and increased total iron binding capacity (Table 1).
Chelation therapy with disodium calcium edetate as recommended for environmental Mn intoxication28 has been initiated in some individuals with improvement of neurological symptoms. Supplementation of iron, a competitive inhibitor of intestinal Mn uptake29,30 in individuals with low iron stores, has also proven effective to lower blood Mn levels. A significant reduction in blood Mn levels, normalization of the erythrocyte count, and improvement of neurological symptoms was observed in individual D-II-621 and G-II-2 (Figure S3). Long-term follow-up data of manganese blood levels is currently only available for individual D-II-6. Combined disodium calcium edetate and iron supplementation has resulted in significant improvement of gait and fine motor movements, resolution of polycythemia, and normalization of liver Mn content with no further progression of cirrhosis.21
Homozygosity Mapping Identifies a Candidate Gene for a Syndrome of Hepatic Cirrhosis, Dystonia, Polycythemia, and Hypermanganesemia
In order to identify shared regions of homozygosity and potential candidate genes, DNA from the siblings of the consanguineous families A and D were analyzed by homozygosity mapping (Figure S1A). Analysis of SNP data for family A revealed a large area of homozygosity on chromosomal region 1q (198.403.205 to 219.513.863 bp) shared between all four affected siblings with the unaffected sibling being heterozygous. Within this area there was a region of no-call SNPs (218.057.426 to 218.158.564 bp) consistent with a deletion. The affected sibling (D-II-6) of family D was also homozygous in this area (217.537.040 to 219.914.224 bp) (Figure S1B and Table S5).
This area contained only one gene, SLC30A10, and this was considered a strong candidate gene. Analysis of coordinate data suggested that exons 1 and 2 of this gene were affected by the deletion in family A.
Sanger Sequencing of SLC30A10
Sequencing of SLC30A10 in individual D-II-6 revealed a homozygous nine base deletion in exon 1 (c.314_322del) resulting in a deletion of three amino acids (p.Ala105_Pro107del) (Figure S4). Both parents and three unaffected siblings of family D were heterozygous for the mutation. All other healthy siblings did not carry this mutation. The sequencing data were confirmed by a restriction enzyme digest test that used MspI (data not shown). This mutation was absent in 200 control alleles of healthy individuals.
Subsequent mutation analysis of SLC30A10 in a further six families revealed homozygous sequence changes in all affected individuals (Figure 1). Where DNA of parents was available, they were found to be heterozygous carriers. None of the unaffected siblings were homozygous for the familial mutation and mutations were absent in 200 control chromosomes of healthy unaffected individuals. Amplification of SLC30A10 from blood, lymphoblastoid, and fibroblast cDNA from healthy control subjects was unsuccessful suggesting that SLC30A10 is not expressed in these cell lines. This is consistent with previous studies in rats showing tissue specific expression in liver, brain, and intestine.31 Unfortunately, such tissue samples in a form suitable for RNA analysis were not available from affected individuals.
Figure 1.
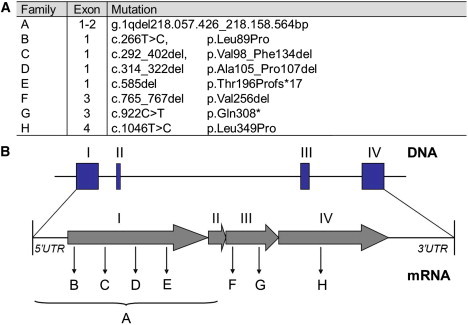
SLC30A10 Mutations in Affected Families with a Syndrome of Hepatic Cirrhosis, Dystonia, Polycythemia, and Hypermanganesemia
(A) Mutations in SLC30A10 identified by DNA sequencing. (For families D and G no DNA was available for analysis of deceased siblings D-II-3 and G-II-1).
(B) Genomic structure of the exons encoding SLC30A10 and positions of identified mutations. The large deletion spanning exon 1 and 2 in family A is indicated by a bracket.
Sequence Alignment and Phylogeny
SLC30A10 sequence alignment of various species with its ortholog in fungi, Zrc1, confirmed that all identified amino acid substitutions affect evolutionary highly conserved areas of the protein (Figure 2). The phylogeny tree shows the two distinct evolutionary groups of SLC30A10 and Zrc1 (Figure S5).
Figure 2.

Evolutionary Conservation Data for SLC30A10
Putative transmembrane domains marked I–VI in gray. Positions with single, fully conserved residues are marked with an asterisk (∗). Conservation between groups of strongly and weakly similar properties is indicated by a colon (;) and a period (.), respectively. Amino acid substitutions are marked in color: yellow, missense mutation (c.266T>C [p.Leu89Pro]); black arrows, deletion (c.298_402del, [p.Val98_Phe134del]); blue, deletion (c.314_322del [p.Ala105_Pro107del]); green, single base deletion causing frame shift and premature stop codon (c.585del [p.Thr196Profs∗17]); pink, deletion (c.765_767del [p.Val256 del]); orange, nonsense mutation (c.922C>T [p.Gln308∗]); red, missense mutation (c.1046T>C [p.Leu349Pro]).
Yeast Expression Studies
The function of human wild-type SLC30A10 and the effect of mutations were investigated in the Mn-sensitive Δpmr1 yeast strain. PMR1 (plasma membrane ATPase-related) encodes a P-type Ca2+/Mn2+ ATPase located at the Golgi membrane that is involved in the transport of Mn2+ from the cytosol into the Golgi. Thereby, it plays an important role in the regulation of cellular Mn2+ homeostasis in yeast. Deletion of PMR1 leads to accumulation of Mn2+ in the cytosol and increased sensitivity of cells to high concentrations of Mn2+ in the growth medium.32–34
Out of the eight identified mutations, we chose to investigate two different types of sequence changes: one missense mutation (c.266T>C), which is located in a highly conserved transmembrane region, and one nonsense mutation (c.585del) predicted to lead to the production of a significantly truncated protein of 213 amino acids (p.Thr196Profs∗17; Figure 2). Both mutations were created with site-directed mutagenesis. ZRC1, the yeast ortholog of SLC30A10, and ZRC1 with its previously described Mn resistant mutation (c.131A>T) were used as controls.32 The Δpmr1 yeast strain failed to grow on SC-Ura medium supplemented with a high Mn concentration (1.5 mM MnCl2) (Figure 3). Overexpression of the wild-type human SLC30A10 inserted into the yeast expression vector pYES-DEST52 under the control of the GAL1 promoter restored growth under high Mn concentrations suggesting its crucial role in Mn transport. The presence of the missense (c.266T>C) and nonsense (c.585del) mutation in SLC30A10 abolished growth (Figure 3).
Figure 3.

Yeast Expression Studies
Wild-type cells BY4743 and Δpmr1 cells were transformed with empty vector pYES-Dest52, SLC30A10, SLC30A10 (c.266T>C [p.Leu89Pro]), SLC30A10 (c.585del [p.Thr196Profs∗17]), ZRC1, and ZRC1 (c.131A>T [p.Asn44Ile]). 105, 5 × 104, 104, 5 × 103 and 103 cells (a–e) were spotted onto SC-Ura plates supplemented with or without 1.5 mM MnCl2 and incubated at 30°C for 6 days. Human SLC30A10 rescues growth of the manganese-sensitive Δpmr1 yeast strain on SC-Ura medium supplemented with high manganese concentration. Introducing the missense mutation (c.266T>C [p.Leu89Pro]) or nonsense mutation (c.585del [p.Thr196Profs∗17]) by site-directed mutagenesis abolishes the effect.
Discussion
We have identified the genetic basis of an inborn error of Mn homeostasis with accumulation of Mn in the liver and basal ganglia; it is caused by mutations in SLC30A10.
Human SLC30A10 is on chromosome 1 and has four exons that encode a protein of 485 amino acids (NP_061183.2) (Figure 1). This gene was recently identified by bioinformatics methods and was classified as a member of the SLC30 solute carrier subfamily of the cation diffusion facilitator (CDF) family.35 To date the SLC30 family has been thought to be a group of mammalian Zn transporters.35,36 Proteins of this family have a conserved structure of six transmembrane helices (TMD I–VI) with both N and C termini located on the cytoplasmic side of the plasma membrane.35,37 Solute carriers are secondary active transporters allowing movement of molecules across a cell membrane against a concentration gradient without the direct use of ATP. Members of this family are found in all biological kingdoms and transfer metals out of the cytosol, either extracellularly or into organelles, thereby regulating intracellular metal concentrations.31,35–38
Mutation analysis of SLC30A10 revealed homozygous sequence changes in all affected individuals described. These are predicted to either cause a significantly truncated protein because of a frameshift and premature stop codon or are deletions or affect an evolutionary highly conserved area of the protein and are therefore likely to have a detrimental effect on protein function (Figure 2). All identified sequence changes were absent in 200 control chromosomes of healthy individuals and are not listed as SNPs on dbSNP and Ensembl databases. Analysis of variants in the general population reported on dbSNP and Ensembl identified two SNPs in the coding region of SLC30A10 that are predicted by the bioinformatic tools PolyPhen and SIFT to be damaging to protein function (Table S6). However, both are rare in the general population, 1:4,551 and 1:4,548, respectively. It will be of interest to study these variants and establish whether they are pathological mutations.
The deleterious effects of mutations in this gene were confirmed by expression studies in the Mn-sensitive Δpmr1 yeast strain. Although wild-type SLC30A10 restored growth in high Mn concentrations, SLC30A10 carrying a missense (c.266T>C) or nonsense (c.585del) mutation did not (Figure 3). Not very surprisingly, the effect of human wild-type SLC30A10 in yeast was less pronounced than that of ZRC1 carrying the Mn resistance mutation (c.131A>T). SLC30A10 and Zrc1 (p.Asn44Ile) probably both contribute to Mn uptake into vacuolar vesicles; how this actually occurs and whether there are different mechanisms involved has yet to be determined.
Our studies demonstrate that human wild-type SLC30A10 functions as a Mn transporter that protects cells from Mn toxicity. Amino acid substitutions in the SLC30A10 yeast ortholog Zrc1 have recently been reported to alter substrate specificity from Zn to iron and Mn.33 Evolutionary changes in the amino acid sequence might be responsible for the altered metal affinity in man.
CDF transporters share a high degree of sequence conservation in the charged residues of TMD II and V that are thought to function as specific metal binding sites. Zn binding in YiiP, an E. coli CDF member, is facilitated by two highly conserved aspartic acid residues (position 45 and 49) in TMD II and a histidine and aspartic acid (position 153 and 157) in TMD V.39,40 Several residues within TMD II, III, and IV of Zrc1 are also known to be critical for zinc specificity33,41 Alignment of the protein sequences of Zrc1 and SLC30A10 from different species revealed that during evolution Leu33, Asn44, and Ala52 in TMD II and Ser272 and Ile275 in TMD IV have been replaced by Ile, Ser, Gly, The, and Met in SLC30A10, respectively (Table 2).41 These amino acids appear to be highly conserved throughout the SLC30A10 family. Lack of conservation of these amino acids between Zrc1 and SLC30A10 suggests that they have evolved independently and functionally as two different transporters. The Zrc1 subfamily contains mostly prokaryotic members from both eubacterial and archaeal sources, whereas the SLC30A10 subfamily contains eukaryotic members (Figure S5). Therefore, we conclude that evolutionary changes in the amino acid sequence of the protein have altered the substrate specificity of the transporter from Zn in yeast to Mn in mammalian cells. This is consistent with the finding that affected individuals carrying a mutation in SLC30A10 have high blood Mn levels, whereas Zn levels are normal. Although we have not excluded the possibility that SLC30A10 might be involved in the transport of other divalent cations, the mutations that we have encountered in this gene only result in a hypermanganesemia phenotype and other blood cation levels are within the normal range. Further studies are required to investigate the properties of SLC30A10 and determine how SLC30A10 is involved in the regulation of Mn transport. Mn homeostasis in higher organisms is not well understood. Various cellular uptake mechanisms for Mn have been suggested, including divalent metal transporter (DMT1), ZIP-8, the transferrin (Tf)/Tf receptor (TfR) system, voltage and store operated calcium channels, and the glutamate receptor channel; however, none of these transporters are specific for Mn.1,42,43 Given that mutations in SLC30A10 in humans have a detrimental effect on Mn homeostasis, we expect that SLC30A10 is a key player in Mn transport.
Table 2.
Comparison of Conserved Amino Acid Residues in Zrc1, which When Substituted Confer Resistance to High Manganese Toxicity, with Conserved Residues Present in SLC30A10
| Amino Acid Positiona |
Zrc1 (n = 5) |
SLC30A10 (n = 43) |
|---|---|---|
| 33 | Leucine | Isoleucineb |
| 40 | Phenylalanine | Phenylalanine |
| 44 | Asparagine | Serine |
| 52 | Alanine | Glycine |
| 79 | Glycine | Glycine |
| 86 | Phenylalanine | Phenylalanine |
| 87 | Leucine | Leucine |
| 101 | Arginine | Arginine |
| 272 | Serine | Threonine |
| 275 | Isoleucine | Methioninec |
n is used as an abbreviation for the number of genes used for alignment. Highlighted residues are conserved in Zrc1 and SLC30A10 but are not conserved between the two groups.
Residues compared are those reported to be gain-of-function mutations in ZRC1 that determine metal specificity.33,41 Numbering relative to S. cerevisiae Zrc1.
Valine in zebrafish.
Phenylalanine in zebrafish.
Neurological presentation of individuals with a syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia resembles environmental Mn toxicity and AHD, both presenting with atypical parkinsonism that is poorly responsive to L-Dopa treatment.3,6,14,15 Although dystonia and rigidity are more prominent in inherited hypermanganesemia and environmental manganism, AHD is more likely to cause ataxia and chorea.14 Dysarthria can be a feature in all three forms of hypermanganesemia. Occupational manganism can present with psychosis and compulsive behaviors, so-called “Mn madness.”5 Cognitive and behavioral changes have not been observed in inherited hypermanganesemia. MRI brain findings of affected individuals are similar to those seen in environmental Mn toxicity and AHD (Figure S2).4,14,15 The most characteristic finding in inherited hypermanganesemia is increased signal intensity of the globus pallidus on T1-weighted sequences with changes extending into adjacent basal ganglia in most cases. When the disease is extensive, white matter involvement can be observed. More frequently than seen in occupational manganism and AHD, T2 changes of low signal return from the globus pallidus are also present in inherited hypermanganesemia. However, there are no specific MRI features that can distinguish SLC30A10 mutations from acquired causes of hypermanganesemia.
One affected individual has a different neurological phenotype of spastic paraparesis without extrapyramidal motor impairment.22 This presentation is similar to hepatic myelopathy, a rare complication of hepatic failure that has also been attributed to Mn accumulation.44 In both cases, MRI of the spinal cord is normal.
Although cirrhosis has not been observed in environmental Mn exposure, it is the life-limiting factor in this form of inherited hypermanganesemia. The liver is the major organ involved in the excretion of Mn via the bile. In animal models increased exposure to Mn results in hepatocellular necrosis and elevated transaminases.45,46
Polycythemia and depleted iron stores are hallmarks of this disease and should prompt SLC30A10 analysis. Both features have not been observed in cases of environmental Mn toxicity and AHD. Individuals with a diagnosis of cryptogenic liver cirrhosis and AHD should be carefully assessed for these features to allow correct diagnosis. Mn has been suggested to induce erythropoietin gene expression,47 and this is the likely mechanism behind the development of polycythemia in affected individuals. Homeostatic regulation of Mn and iron is closely linked and explains the depleted iron stores in affected individuals.1,29,42,48 Mn has a direct effect on the availability of the bioactive iron pool and induces an iron-starved phenotype with reduced ferritin expression and increased TfR expression. With increasing exposure to Mn, release of iron from intracellular stores, enhanced iron uptake, and decreased iron utilization is favored.49,50 Iron and Mn are chemically and structurally similar and compete for the same serum binding protein (Tf) and membranous transporter protein (DMT1). Therefore, dietary iron status has a direct effect on Mn uptake.29,30 This explains the beneficial effect of iron supplementation in affected individuals.
Chelation therapy to enhance urinary Mn excretion has been attempted with various chelation agents. Disodium calcium edetate as recommended for environmental Mn intoxication28 has significantly improved neurological symptoms in some subjects, led to reduction of blood Mn, and halted the progression of cirrhosis.21 It carries the disadvantage of intravenous application over 5 days every 4 weeks and the requirement of strict monitoring of other essential heavy metals such as zinc, copper, and selenium.51 Several of the affected individuals are from countries with poor medical resources where intravenous disodium calcium edetate is either not available or the current setting would not allow for frequent application and monitoring of chelation therapy. Other chelating agents have therefore been used. Two siblings (B-II-2 and B-II-5) have improved clinically on oral dimercaptosuccinic acid (DMSA). However, because both siblings also receive iron supplementation, it is difficult to attribute the effect to one or the other. Blood and urine Mn levels and neurological symptoms of two men with occupational Mn intoxication did not respond significantly to DMSA treatment.52 Para-aminosalicylic acid has also been reported not to be beneficial.23 Further research is required to identify specific and efficacious treatment agents for hypermanganesemia that can be administered conveniently, preferably by mouth.
Chronic exposure to Mn has been suggested to be involved in the pathogenesis of PD.53–57 Whole-blood Mn levels in a PD cohort have been found to be significantly higher than in healthy control subjects.55 Also, individuals affected by early onset forms of idiopathic PD with mutations in PARK2 [MIM 602544] (Juvenile Parkinson's Disease type 2 [MIM 600116]), and ATP13A2 [MIM 610513] (Kufor Rakeb Syndrome [MIM 606693]) seem to be at higher risk of Mn toxicity.56,57 Therefore, the importance of understanding Mn homeostasis in more detail is becoming increasingly evident.
In summary, by studying a rare syndrome of inherited Mn accumulation, we have identified a unique Mn transporter in man. SLC30A10 has been shown to function as a specific Mn transporter in higher organisms and when mutated causes a syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia characterized by accumulation of Mn in liver and brain.
Acknowledgments
Karin Tuschl and Philippa Mills are funded by the National Institute for Health Research. Peter Clayton is funded by the Great Ormond Street Hospital Children's charity. Research at the University College London Institute of Child Health and Great Ormond Street Hospital for Children National Health Service (NHS) Trust benefits from research and development funding received from the NHS Executive. We are grateful to K. Pearce for her technical assistance and to Prof G. Moore for provision of control samples.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM). http://www.omim.org/
Entrez Gene. http://www.ncbi.nlm.nih.gov/gene
NCBI Nucleotide database. http://www.ncbi.nlm.nih.gov/nuccore
NCBI Protein database. http://www.ncbi.nlm.nih.gov/protein
NCBI genome viewer. http://www.ncbi.nlm.nih.gov/projects/mapview/
Ensembl. http://www.ensembl.org/index.html
ClustalW2. http://www.ebi.ac.uk/Tools/msa/clustalw2/
NCBI dbSNP. http://www.ncbi.nlm.nih.gov/projects/SNP/
SIFT. http://sift.jcvi.org/
PolyPhen. http://genetics.bwh.harvard.edu/pph/
References
- 1.Aschner M., Guilarte T.R., Schneider J.S., Zheng W. Manganese: Recent advances in understanding its transport and neurotoxicity. Toxicol. Appl. Pharmacol. 2007;221:131–147. doi: 10.1016/j.taap.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Au C., Benedetto A., Aschner M. Manganese transport in eukaryotes: The role of DMT1. Neurotoxicology. 2008;29:569–576. doi: 10.1016/j.neuro.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crossgrove J., Zheng W. Manganese toxicity upon overexposure. NMR Biomed. 2004;17:544–553. doi: 10.1002/nbm.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uchino A., Noguchi T., Nomiyama K., Takase Y., Nakazono T., Nojiri J., Kudo S. Manganese accumulation in the brain: MR imaging. Neuroradiology. 2007;49:715–720. doi: 10.1007/s00234-007-0243-z. [DOI] [PubMed] [Google Scholar]
- 5.Rivera-Mancía S., Ríos C., Montes S. Manganese accumulation in the CNS and associated pathologies. Biometals. 2011;24:811–825. doi: 10.1007/s10534-011-9454-1. [DOI] [PubMed] [Google Scholar]
- 6.Furbee B. Welding and parkinsonism. Neurol. Clin. 2011;29:623–640. doi: 10.1016/j.ncl.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Wasserman G.A., Liu X., Parvez F., Factor-Litvak P., Ahsan H., Levy D., Kline J., van Geen A., Mey J., Slavkovich V. Arsenic and manganese exposure and children's intellectual function. Neurotoxicology. 2011;32:450–457. doi: 10.1016/j.neuro.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stepens A., Logina I., Liguts V., Aldins P., Eksteina I., Platkājis A., Mārtinsone I., Tērauds E., Rozentāle B., Donaghy M. A Parkinsonian syndrome in methcathinone users and the role of manganese. N. Engl. J. Med. 2008;358:1009–1017. doi: 10.1056/NEJMoa072488. [DOI] [PubMed] [Google Scholar]
- 9.Sikk K., Haldre S., Aquilonius S.M., Taba P. Manganese-Induced Parkinsonism due to Ephedrone Abuse. Parkinsons Dis. 2011;2011:865319. doi: 10.4061/2011/865319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quaghebeur G., Taylor W.J., Kingsley D.P., Fell J.M., Reynolds A.P., Milla P.J. MRI in children receiving total parenteral nutrition. Neuroradiology. 1996;38:680–683. doi: 10.1007/s002340050333. [DOI] [PubMed] [Google Scholar]
- 11.Chalela J.A., Bonillha L., Neyens R., Hays A. Manganese encephalopathy: An under-recognized condition in the intensive care unit. Neurocrit. Care. 2011;14:456–458. doi: 10.1007/s12028-010-9476-5. [DOI] [PubMed] [Google Scholar]
- 12.Klos K.J., Ahlskog J.E., Kumar N., Cambern S., Butz J., Burritt M., Fealey R.D., Cowl C.T., Parisi J.E., Josephs K.A. Brain metal concentrations in chronic liver failure patients with pallidal T1 MRI hyperintensity. Neurology. 2006;67:1984–1989. doi: 10.1212/01.wnl.0000247037.37807.76. [DOI] [PubMed] [Google Scholar]
- 13.Uchino A., Aoki T., Ohno M. Portal-systemic encephalopathy: Report of a case with unusual MR findings. Radiat. Med. 1993;11:21–23. [PubMed] [Google Scholar]
- 14.Ferrara J., Jankovic J. Acquired hepatocerebral degeneration. J. Neurol. 2009;256:320–332. doi: 10.1007/s00415-009-0144-7. [DOI] [PubMed] [Google Scholar]
- 15.Fernández-Rodriguez R., Contreras A., De Villoria J.G., Grandas F. Acquired hepatocerebral degeneration: Clinical characteristics and MRI findings. Eur. J. Neurol. 2010;17:1463–1470. doi: 10.1111/j.1468-1331.2010.03076.x. [DOI] [PubMed] [Google Scholar]
- 16.Gorell J.M., Johnson C.C., Rybicki B.A., Peterson E.L., Kortsha G.X., Brown G.G., Richardson R.J. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson's disease. Neurotoxicology. 1999;20:239–247. [PubMed] [Google Scholar]
- 17.Aschner M., Erikson K.M., Herrero Hernández E., Tjalkens R. Manganese and its role in Parkinson's disease: From transport to neuropathology. Neuromolecular Med. 2009;11:252–266. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dobson A.W., Erikson K.M., Aschner M. Manganese neurotoxicity. Ann. N Y Acad. Sci. 2004;1012:115–128. doi: 10.1196/annals.1306.009. [DOI] [PubMed] [Google Scholar]
- 19.Calne D.B., Chu N.S., Huang C.C., Lu C.S., Olanow W. Manganism and idiopathic parkinsonism: Similarities and differences. Neurology. 1994;44:1583–1586. doi: 10.1212/wnl.44.9.1583. [DOI] [PubMed] [Google Scholar]
- 20.Olanow C.W., Good P.F., Shinotoh H., Hewitt K.A., Vingerhoets F., Snow B.J., Beal M.F., Calne D.B., Perl D.P. Manganese intoxication in the rhesus monkey: A clinical, imaging, pathologic, and biochemical study. Neurology. 1996;46:492–498. doi: 10.1212/wnl.46.2.492. [DOI] [PubMed] [Google Scholar]
- 21.Tuschl K., Mills P.B., Parsons H., Malone M., Fowler D., Bitner-Glindzicz M., Clayton P.T. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia-A new metabolic disorder. J. Inherit. Metab. Dis. 2008;31:151–163. doi: 10.1007/s10545-008-0813-1. [DOI] [PubMed] [Google Scholar]
- 22.Gospe S.M., Jr., Caruso R.D., Clegg M.S., Keen C.L., Pimstone N.R., Ducore J.M., Gettner S.S., Kreutzer R.A. Paraparesis, hypermanganesaemia, and polycythaemia: A novel presentation of cirrhosis. Arch. Dis. Child. 2000;83:439–442. doi: 10.1136/adc.83.5.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brna P., Gordon K., Dooley J.M., Price V. Manganese toxicity in a child with iron deficiency and polycythemia. J. Child Neurol. 2011;26:891–894. doi: 10.1177/0883073810393962. [DOI] [PubMed] [Google Scholar]
- 24.Sahni V., Léger Y., Panaro L., Allen M., Giffin S., Fury D., Hamm N. Case report: A metabolic disorder presenting as pediatric manganism. Environ. Health Perspect. 2007;115:1776–1779. doi: 10.1289/ehp.10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sue W.C., Chen C.Y., Chen C.C. Dyskinesia from manganism in a hepatic dysfunction patient. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi. 1996;37:59–64. [PubMed] [Google Scholar]
- 26.Gruber Gikovate C., Zirretta J.C., Ferreira Bezerra J.M., Canedo de Magalhães G., Barkovich A.J. Transient globus pallidus T1 shortening associated with polycythaemia and dystonia. Neuroradiology. 1999;41:288–291. doi: 10.1007/s002340050750. [DOI] [PubMed] [Google Scholar]
- 27.Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herrero Hernandez E., Discalzi G., Valentini C., Venturi F., Chiò A., Carmellino C., Rossi L., Sacchetti A., Pira E. Follow-up of patients affected by manganese-induced Parkinsonism after treatment with CaNa2EDTA. Neurotoxicology. 2006;27:333–339. doi: 10.1016/j.neuro.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 29.Fitsanakis V.A., Zhang N., Garcia S., Aschner M. Manganese (Mn) and iron (Fe): Interdependency of transport and regulation. Neurotox. Res. 2010;18:124–131. doi: 10.1007/s12640-009-9130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen S.L., Ashwell M.S., Moeser A.J., Fry R.S., Knutson M.D., Spears J.W. High dietary iron reduces transporters involved in iron and manganese metabolism and increases intestinal permeability in calves. J. Dairy Sci. 2010;93:656–665. doi: 10.3168/jds.2009-2341. [DOI] [PubMed] [Google Scholar]
- 31.Sreedharan S., Stephansson O., Schiöth H.B., Fredriksson R. Long evolutionary conservation and considerable tissue specificity of several atypical solute carrier transporters. Gene. 2011;478:11–18. doi: 10.1016/j.gene.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 32.Ton V.K., Mandal D., Vahadji C., Rao R. Functional expression in yeast of the human secretory pathway Ca(2+), Mn(2+)-ATPase defective in Hailey-Hailey disease. J. Biol. Chem. 2002;277:6422–6427. doi: 10.1074/jbc.M110612200. [DOI] [PubMed] [Google Scholar]
- 33.Lin H., Kumánovics A., Nelson J.M., Warner D.E., Ward D.M., Kaplan J. A single amino acid change in the yeast vacuolar metal transporters ZRC1 and COT1 alters their substrate specificity. J. Biol. Chem. 2008;283:33865–33873. doi: 10.1074/jbc.M804377200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lapinskas P.J., Cunningham K.W., Liu X.F., Fink G.R., Culotta V.C. Mutations in PMR1 suppress oxidative damage in yeast cells lacking superoxide dismutase. Mol. Cell. Biol. 1995;15:1382–1388. doi: 10.1128/mcb.15.3.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seve M., Chimienti F., Devergnas S., Favier A. In silico identification and expression of SLC30 family genes: An expressed sequence tag data mining strategy for the characterization of zinc transporters' tissue expression. BMC Genomics. 2004;5:32. doi: 10.1186/1471-2164-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lichten L.A., Cousins R.J. Mammalian zinc transporters: Nutritional and physiologic regulation. Annu. Rev. Nutr. 2009;29:153–176. doi: 10.1146/annurev-nutr-033009-083312. [DOI] [PubMed] [Google Scholar]
- 37.Palmiter R.D., Findley S.D. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 1995;14:639–649. doi: 10.1002/j.1460-2075.1995.tb07042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacDiarmid C.W., Milanick M.A., Eide D.J. Biochemical properties of vacuolar zinc transport systems of Saccharomyces cerevisiae. J. Biol. Chem. 2002;277:39187–39194. doi: 10.1074/jbc.M205052200. [DOI] [PubMed] [Google Scholar]
- 39.Wei Y., Fu D. Binding and transport of metal ions at the dimer interface of the Escherichia coli metal transporter YiiP. J. Biol. Chem. 2006;281:23492–23502. doi: 10.1074/jbc.M602254200. [DOI] [PubMed] [Google Scholar]
- 40.Lu M., Fu D. Structure of the zinc transporter YiiP. Science. 2007;317:1746–1748. doi: 10.1126/science.1143748. [DOI] [PubMed] [Google Scholar]
- 41.Lin H., Burton D., Li L., Warner D.E., Phillips J.D., Ward D.M., Kaplan J. Gain-of-function mutations identify amino acids within transmembrane domains of the yeast vacuolar transporter Zrc1 that determine metal specificity. Biochem. J. 2009;422:273–283. doi: 10.1042/BJ20090853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roth J.A., Garrick M.D. Iron interactions and other biological reactions mediating the physiological and toxic actions of manganese. Biochem. Pharmacol. 2003;66:1–13. doi: 10.1016/s0006-2952(03)00145-x. [DOI] [PubMed] [Google Scholar]
- 43.Yokel R.A. Manganese flux across the blood-brain barrier. Neuromolecular Med. 2009;11:297–310. doi: 10.1007/s12017-009-8101-2. [DOI] [PubMed] [Google Scholar]
- 44.Campellone J.V., Lacomis D., Giuliani M.J., Kroboth F.J. Hepatic myelopathy. Case report with review of the literature. Clin. Neurol. Neurosurg. 1996;98:242–246. doi: 10.1016/0303-8467(96)00018-2. [DOI] [PubMed] [Google Scholar]
- 45.Huang P., Li G., Chen C., Wang H., Han Y., Zhang S., Xiao Y., Zhang M., Liu N., Chu J. Differential toxicity of Mn(2+) and Mn(3+) to rat liver tissues: Oxidative damage, membrane fluidity and histopathological changes. Exp. Toxicol. Pathol. 2010 doi: 10.1016/j.etp.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 46.Witzleben C.L., Boyer J.L., Ng O.C. Manganese-bilirubin cholestasis. Further studies in pathogenesis. Lab. Invest. 1987;56:151–154. [PubMed] [Google Scholar]
- 47.Ebert B.L., Bunn H.F. Regulation of the erythropoietin gene. Blood. 1999;94:1864–1877. [PubMed] [Google Scholar]
- 48.Yin Z., Jiang H., Lee E.S., Ni M., Erikson K.M., Milatovic D., Bowman A.B., Aschner M. Ferroportin is a manganese-responsive protein that decreases manganese cytotoxicity and accumulation. J. Neurochem. 2010;112:1190–1198. doi: 10.1111/j.1471-4159.2009.06534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crooks D.R., Ghosh M.C., Braun-Sommargren M., Rouault T.A., Smith D.R. Manganese targets m-aconitase and activates iron regulatory protein 2 in AF5 GABAergic cells. J. Neurosci. Res. 2007;85:1797–1809. doi: 10.1002/jnr.21321. [DOI] [PubMed] [Google Scholar]
- 50.Kwik-Uribe C.L., Reaney S., Zhu Z., Smith D. Alterations in cellular IRP-dependent iron regulation by in vitro manganese exposure in undifferentiated PC12 cells. Brain Res. 2003;973:1–15. doi: 10.1016/s0006-8993(03)02457-0. [DOI] [PubMed] [Google Scholar]
- 51.Sata F., Araki S., Murata K., Aono H. Behavior of heavy metals in human urine and blood following calcium disodium ethylenediamine tetraacetate injection: Observations in metal workers. J. Toxicol. Environ. Health A. 1998;54:167–178. doi: 10.1080/009841098158881. [DOI] [PubMed] [Google Scholar]
- 52.Angle C.R. Dimercaptosuccinic acid (DMSA): Negligible effect on manganese in urine and blood. Occup. Environ. Med. 1995;52:846. doi: 10.1136/oem.52.12.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lucchini R.G., Martin C.J., Doney B.C. From manganism to manganese-induced parkinsonism: A conceptual model based on the evolution of exposure. Neuromolecular Med. 2009;11:311–321. doi: 10.1007/s12017-009-8108-8. [DOI] [PubMed] [Google Scholar]
- 54.Roth J.A. Are there common biochemical and molecular mechanisms controlling manganism and parkisonism. Neuromolecular Med. 2009;11:281–296. doi: 10.1007/s12017-009-8088-8. [DOI] [PubMed] [Google Scholar]
- 55.Fukushima T., Tan X., Luo Y., Kanda H. Relationship between blood levels of heavy metals and Parkinson's disease in China. Neuroepidemiology. 2010;34:18–24. doi: 10.1159/000255462. [DOI] [PubMed] [Google Scholar]
- 56.Roth J.A., Singleton S., Feng J., Garrick M., Paradkar P.N. Parkin regulates metal transport via proteasomal degradation of the 1B isoforms of divalent metal transporter 1. J. Neurochem. 2010;113:454–464. doi: 10.1111/j.1471-4159.2010.06607.x. [DOI] [PubMed] [Google Scholar]
- 57.Tan J., Zhang T., Jiang L., Chi J., Hu D., Pan Q., Wang D., Zhang Z. Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. J. Biol. Chem. 2011;286:29654–29662. doi: 10.1074/jbc.M111.233874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.