

Abstract
Olmsted syndrome (OS) is a rare congenital disorder characterized by palmoplantar and periorificial keratoderma, alopecia in most cases, and severe itching. The genetic basis for OS remained unidentified. Using whole-exome sequencing of case-parents trios, we have identified a de novo missense mutation in TRPV3 that produces p.Gly573Ser in an individual with OS. Nucleotide sequencing of five additional affected individuals also revealed missense mutations in TRPV3 (which produced p.Gly573Ser in three cases and p.Gly573Cys and p.Trp692Gly in one case each). Encoding a transient receptor potential vanilloid-3 cation channel, TRPV3 is primarily expressed in the skin, hair follicles, brain, and spinal cord. In transfected HEK293 cells expressing TRPV3 mutants, much larger inward currents were recorded, probably because of the constitutive opening of the mutants. These gain-of-function mutations might lead to elevated apoptosis of keratinocytes and consequent skin hyperkeratosis in the affected individuals. Our findings suggest that TRPV3 plays essential roles in skin keratinization, hair growth, and possibly itching sensation in humans and selectively targeting TRPV3 could provide therapeutic potential for keratinization or itching-related skin disorders.
Main Text
Olmsted syndrome (OS) is a rare congenital disorder characterized by bilateral mutilating palmoplantar keratoderma (PPK) and periorificial keratotic plaques with severe itching at all lesions.1,2 Diffused alopecia, constriction of digits, and onychodystrophy have also been reported.2 Infections and squamous cell carcinomas can arise on the keratotic areas.3 The diagnosis of OS is based on its characteristic clinical picture without any specific biologic markers or histopathological patterns. It should be differentiated from several genodermatoses, such as Vohwinkel syndrome (MIM 124500) and acrodermatitis enteropathica (MIM 201100).2 Until recently, only 46 individuals with OS had been reported,4,5 including 36 sporadic cases and four families containing ten affected individuals. The definite mode of inheritance was still uncertain, and autosomal-dominant,6 X-linked-dominant7 and X-linked-recessive8 modes of inheritance had been proposed. Treatment of OS includes surgical removal of keratotic palmoplantar mass and oral retinoids. However, the relief is often temporary with a high rate of recurrence.2 No pathogenic mutations in any of the four genes (KRT1 [MIM 139350], GJB2 [MIM 121011], SLURP1 [MIM 606119], LOR [MIM 152445]), which were previously implicated in the pathogenesis of several hereditary diseases with mutilating PPK, were identified in a single individual with OS.2 Here, we demonstrate that gain-of-function mutations within TRPV3 (MIM 607066) on chromosomal region 17p13, which encodes a transient receptor potential vanilloid-3 cation channel, give rise to the OS phenotype.
We investigated six cases of OS in China (Table 1, Figures 1A–1C, and Figure S1, available online). Our study was approved by the Clinical Research Ethics Committee of Peking University First Hospital, Beijing China (institutional review board number 2011[360]). Informed consent was obtained following the rules from the institutional review board. All the cases are sporadic except for individual 3, whose affected daughter died of infection at 2 years; no DNA sample from the daughter was available (Figure 1F). In all affected individuals the symptoms developed in the first year of life. Keratotic lesions were yellowish brown and had a sharp erythematous border. Periorificial keratotic plaques were present around the mouth, nostrils, ear meatus, anus, and perigenital region. They might appear to be mild and restricted or extend to involve neck, upper thorax, lower abdomen, inguinal folds, and upper inner thighs. PPK aggravated gradually to become mutilating, including flexion deformity of the fingers, constriction of the digits, that is pseudoainhum, and even spontaneous amputation of the digits or the hands in the severely affected individuals. Painful fissures on the soles interfered with walking in most of the affected individuals. Hair involvement varied, ranging from alopecia universalis with follicular papules to merely sparse curly hair. All individuals complained of severe itching in the lesions, resulting in frequent scratching and sleep disturbances. Thermosensation was normal in all individuals, even on affected areas. No additional neurologic or sensory anomalies were detected. No atopic history was reported, and the serum zinc level was within normal range in all the individuals. Consanguinity was denied in all the families. Individuals 1, 2, and 6 were treated with oral acitretin. Significant improvement in skin lesions and itching sensation were noted in the three individuals after 4 weeks, though changes in hair growth seemed limited after 1 year of treatment. The histopathological findings of skin biopsies from keratotic lesions of individuals 1, 2, and 6 demonstrated psoriasiform hyperplasia, orthohyperkeratosis, and parakeratosis with profound mast cell infiltration in the upper dermis (Figures 1D and 1E). Hair follicles from nonpalmoplantar skin biopsy were not examined in this study.
Table 1.
Phenotypic Characteristics and Amino Acid Changes in TRPV3 in Subjects with OS
| Patient | Age (Years) | Sex | Family History | Palmoplantar Keratosis | Periorificial Keratosis | Alopecia | Amino Acid Change in TRPV3 |
|---|---|---|---|---|---|---|---|
| 1 | 9 | female | − | +++, CDB | ++++ | ++++ | Gly573Ser |
| 2 | 14 | female | − | ++ | ++ | +++ | Gly573Cys |
| 3 | 25 | female | + | ++++, SDA | ++++ | ++++ | Gly573Ser |
| 4 | 12 | female | − | ++++, CDB, SDA | +++ | +++ | Gly573Ser |
| 5 | 23 | male | − | +++, CDB | +++ | ++ | Trp692Gly |
| 6 | 27 | female | − | ++, CDB | ++ | + (dry curly hair) | Gly573Ser |
The following abbreviations are used: CDB, Constricting digit bands; SDA, Spontaneous digit amputation. The following symbols are used: −, absent; +, present; ++, mild; +++, moderate; ++++, severe.
Figure 1.
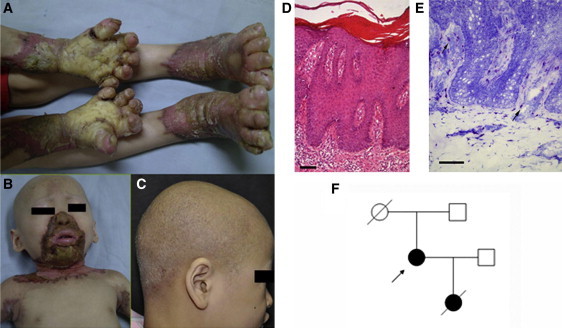
Phenotypic Characteristics of OS
(A) Bilateral mutilating palmoplantar keratoderma, flexion deformities and constriction of digits in individual 1.
(B) Keratotic plaques involving periorificial, neck and axillary areas in individual 1.
(C) Diffused alopecia with follicular papules in individual 2.
(D) Skin biopsy section demonstrating psoriasiform hyperplasia, orthohyperkeratosis and parakeratosis in individual 1 (hematoxylin-eosin staining; the scale bar represents 100 μm).
(E) Individual 1, skin biopsy section showing profound mast cell infiltration in upper dermis (toluidine blue staining; the scale bar represents 100 μm).
(F) The pedigree of individual 3 (arrow indicates individual 3).
On the basis of the familial case (individual 3) we enrolled and previous reports,6,7,9 an autosomal-dominant trait of OS was postulated. In this way, a sporadic case was supposed to carry a de novo pathogenic heterozygous mutation in the causative gene. We then applied a genome-wide approach and sequenced the exomes of individual 1 and her parents (Beijing Genomic Institute, Shenzhen, China). Exome capture was carried out with SureSelect Human All Exon Kit (Agilent, Santa Clara, CA, USA), guided by the manufacturer's protocols and then sequenced by Hiseq2000 platform (Illumina, San Diego, CA, USA). We obtained about 2.4 to 3.7 Gb of mappable sequence data per sample. Single-nucleotide variations of individual 1 were filtered by dbSNP131, the 1000 Genomes Project, and HapMap8 databases. To detect de novo variations, we further filtered out heterozygous variations from her parents. Forty-five apparent de novo damaging variations were found in the individual through SIFT software10 prediction. Given that the expected rarity of true de novo events in the targeted exome is less than one per trio,11 we sequenced all of these candidate variations by using Sanger sequencing. Only one de novo heterozygous point mutation, c.1717G>A in TRPV3, was identified in individual 1 (Table S1). This transition substitution causes an amino acid alteration of p.Gly573Ser (Figure 2A). We then screened the coding exons and their flanking intron sequences (primers are listed in Table S2) in TRPV3 in the five additional individuals and found heterozygous missense mutations in all of them (Table 1 and Figure 2A). No same variations were found in the parents of individuals 2, 4, 5, and 6, indicating that they are de novo mutations. The mother of individual 3 passed away, so the origin of the mutation in individual 3 cannot be identified. We also sequenced these sites in TRPV3 in 216 ethnically matched normal controls, and these three mutations were not detected.
Figure 2.
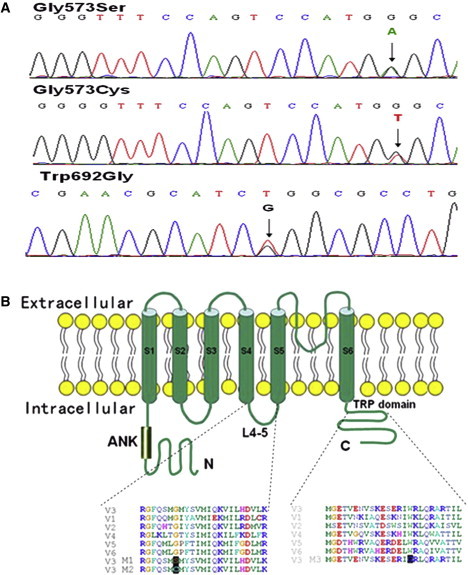
Mutations and Schematic Structure of TRPV3
(A)Sequencing results demonstrating heterozygous TRPV3 mutation of c.1717G>A (amino acid change of p.Gly573Ser) in individuals 1, 3, 4, and 6; c.1717G>T (p.Gly573Cys) in individual 2; and c.2074T>G (p.Trp692Gly) in individual 5.
(B) Schematic structure of TRPV3 protein and multiple alignment of the linker region between S4 and S5 and the TRP domain, with the alignments of corresponding segments of human TRPV1-2 and 4-6 proteins. Gly573 and Trp692 are highly conserved residues. The mutation sites that produce p.Gly573Ser (M1), p.Gly573Cys (M2), and p.Trp692Gly (M3) are also aligned with the substitution positions (black highlighted).
Because TRPV3 is a transmembrane ion channel protein, we further investigated the electrophysiological effect of TRPV3 mutations via the patch-clamp technique. cDNA clones of the whole coding region of human TRPV3 in the pCMV6-AC-GFP vector was obtained from Origene (Rockville, MD). With this plasmid as the template, site-directed mutagenesis (Fast Mutagenesis System, Transgen Biotech, Beijing, China) was performed to obtain three clones containing the point mutations found in the individuals with OS (the primers used are listed in Table S3). HEK293 cells were transiently transfected with plasmids expressing wild-type (WT), p.Gly573Ser, p.Gly573Cys, or p.Trp692Gly TRPV3 proteins with 1 μl lipofectamine 2000 (Invitrogen). After 24 hr of transfection, HEK293 cells with green fluorescence located primarily in the cellular membrane were chosen for patch-clamp recordings under an inverted fluorescence microscope (IX71, Olympus, Tokyo, Japan). Both whole-cell and inside-out patch were performed with an EPC10 amplifier (HEKA, Lambrecht, Germany) driven by PatchMaster software (HEKA). Both bath and pipette solutions contained 130 mM NaCl, 3 mM HEPES, and 0.3 mM EGTA (pH 7.4). All measurements were carried out at room temperature (22–25°C). We evaluated the voltage-dependent properties for each mutant. After breaking into the whole-cell configuration, we found that patches from the HEK293 cells expressing p.Gly573Ser, p.Gly573Cys, or p.Trp692Gly TRPV3 altered channels were leaky and showed large inward leakage-like currents with almost linear current-voltage relationship and secondary conducting phase of TRPV3 current.12 This indicates that the mutant channels are fully open (Figures 3A–3D). To rule out the possibility that these large currents were caused by leakage due to poor cell conditions, we evaluated these mutants by inside-out patch recordings. It is notable that all the three mutants exhibited fast inactivation currents at a more depolarization potential (+220 mV) (Figure S2). For outwardly rectifying WT TRPV3 channels, the inactivation can only be observed when the channels are fully activated by saturating concentrations of ligands,13,14 indicating these mutants resembling an overactive WT TRPV3 channel. To further test the pharmacology of the mutants in response to ligand stimulation, we determined agonist-evoked currents from inside-out patches by applying 300 μM 2-aminoethoxydiphenyl borate (2-APB), and nonspecific leak currents were assessed by superfusing Ba2+ from intracellular side to completely block TRPV3 currents.15 Unlike WT TRPV3, the saturating concentrations of 2-APB had no significant effects on or even partially inhibited the basal currents from the cells expressing the mutant channels (Figures 3E–3H). Indeed, 2-APB not only activated TRPV3 but also slightly inhibited the channel activity when the channel was fully open.14,16 Collectively, our data indicate that p.Gly573Ser, p.Gly573Cys, and p.Trp692Gly TRPV3 are constitutively active and act in a gain-of-function manner. Similarly, in a previous study that used Xenopus oocytes expressing murine p.Gly573Ser or p.Gly573Cys, larger basal currents and lower temperature thresholds were observed.17
Figure 3.
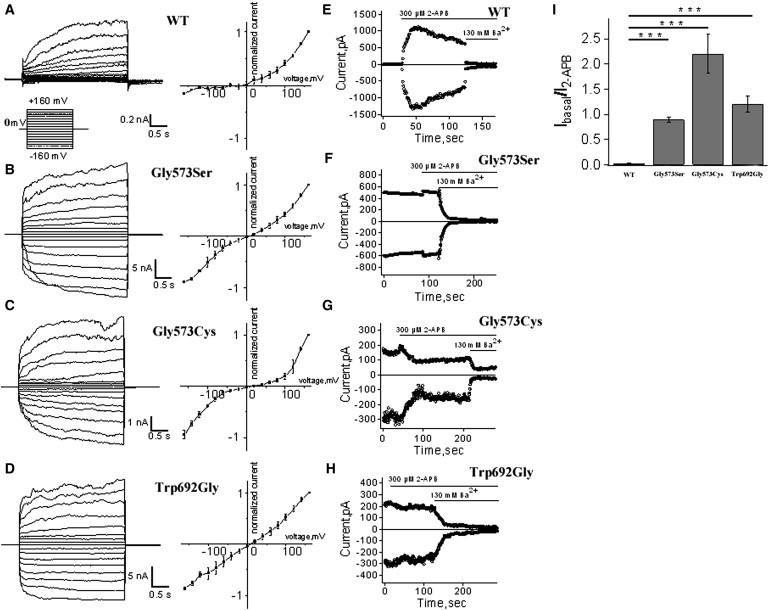
Whole-Cell and Inside-Out Recordings of TRPV3 Currents from Transfected HEK293 Cells
(A–D) Representative whole-cell current traces in HEK293 cells expressing TRPV3 or mutant channels in response to the voltage step protocol (left panels), and respective current-voltage plots from the steady-state currents (right panels).
(E–H) Representative currents from inside-out patches in HEK293 cells expressing TRPV3 or mutant channels in response to stimulation by 300 μM 2-APB or inhibition by 130 mM Ba2+ to assess the level of leak currents, at −80 mV and +80 mV. WT TRPV3 current is activated by 2-APB and followed by a characteristic decay. Note that the mutants Gly573Ser, Gly573Cys, and Trp692Gly show little activation, then inhibition in the presence of 2-APB, and a robust block by Ba2+.
(I) Comparison of ratios of currents evoked by 2-APB (300 μM) over basal currents between the TRPV3 WT and mutant channels. (t test, n = 3–7. p = 0.00001).
Error bars represent SEM.
Because activation of TRPV3 was previously shown to induce apoptosis,18 we next evaluated whether these gain-of-function mutations in TRPV3 could lead to increased cell death of transfected HEK293 cells and keratinocytes in our individuals. Transfected HEK293 cells (detailed above) were cultured for 24 hr and then stained by Hoechst 33342 and propidium iodide (PI) for 15 min before morphological cell death assessment. Cells were observed by inverted fluorescence microscope (IX71, Olympus, Japan) with UV/488 nm dual excitation. Dead cells, shown as PI positive, were counted in five randomly selected high-power fields (400×, more than 1,000 cells total per specimen) with an ImageJ cell counter. Three independent experiments were conducted. For statistical analyses, the experimental data were analyzed by one-way analysis of variance (ANOVA). Comparison of mutant groups with the WT group was carried out by Fisher's least significant difference (LSD) test. Compared to WT, cells expressing the mutant TRPV3 tended to result in a significantly higher cell death rate at 24 hr after transfection (Figures 4A and 4B). The cell death could be partly rescued by a TRP channel inhibitor ruthenium red (Figure S3).
Figure 4.
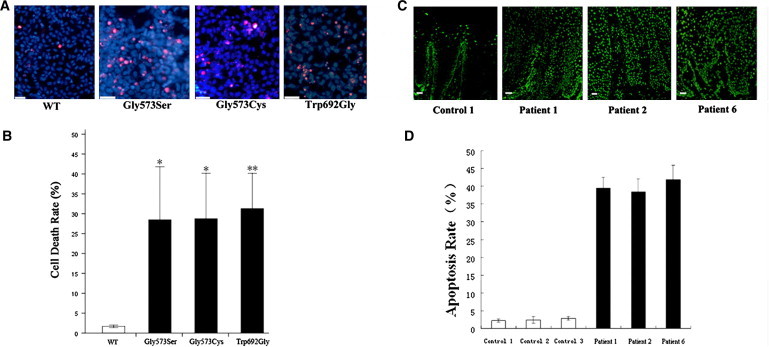
Cell Death of Transfected HEK293 Cells and Apoptosis of Affected Individuals' Keratinocytes
(A) Merged images of transfected HEK293 cells stained with PI (red) and Hoechst 33342 (blue). Mutants show significantly more PI-positive cells compared to wild-type TRPV3.
(B) Quantification of cell death rates in transfected HEK293 cells (red nuclei/blue nuclei × 100%). Data are averaged from three independent experiments. ∗p < 0.05, ∗∗p < 0.01.
(C) Fluorescence microscope images (TUNEL method) of palm skin biopsy sections from an unrelated control, individuals 1, 2, and 6. Large amount of apoptotic cells (with fluorescent nuclei) are seen in individuals with OS. The scale bars represent 50 μm.
(D) Quantification of apoptotic keratinocytes in the skin sections. The mean proportion of apoptotic cells are significantly higher in the group of OS patients than in the group of normal controls (t test, n = 3, p = 0.000005).
Error bars represent SEM.
In situ apoptosis examination of the keratinocytes in keratotic lesions in individuals 1, 2, and 6 was conducted with the TUNEL method (In Situ Cell Death Detection Kit, Roche). Skin sections from the normal edge of surgically excised palm pigmented nevus in three unrelated healthy subjects were used as the controls. Images above the basal layer of the epidermis were taken randomly with a fluorescence microscope with the same photograph parameters. Total cells and apoptotic cells (with fluorescent nucleus) in five random high-power fields (400×) were counted, and the percentage of apoptotic cells was calculated (apoptotic cells/total cells × 100%). Apoptotic cell rates in the affected individuals were significantly higher compared to the normal controls (Figures 4C and 4D).
Here we performed exome sequencing in a single family and have identified mutations in TRPV3 as one of the genetic bases for OS. For sporadic cases of rare autosomal-dominant Mendelian disorders, exome sequencing of case-parents trios has proven to be a cost-effective and promising strategy to unravel causative genes. Our findings suggest OS is inherited in an autosomal-dominant pattern. However, only a few familial cases were reported, possibly because of the severe skin lesions. Other suggested modes of inheritance in OS, including X-linked dominant7 and X-linked recessive,8 are probably due to genetic heterogeneity or incomplete penetrance of this disease.
TRPV3 was cloned in 2002. It belongs to a superfamily of transient receptor potential (TRP) cation channels. It is mainly expressed in keratinocytes, hair follicles, and the brain and spinal cord.19–21 As a thermosensitive channel, TRPV3 is activated at 33°C, and therefore, it was supposed to play a role in warm sensation.22 Notably, in spite of an intolerant itching sensation, the individuals in our study did not experience any other abnormal sensation, including thermosensation. Recent data also suggest that TRPV3 is not a major contributor to mouse heat sensation.23 TRPV3 is also expressed in the central nervous system, but intriguingly, we did not observe any obvious neurological or psychiatric clinical symptoms other than itching in anyone of the individuals we studied. However, we cannot rule out the possibility that some related clinical manifestations were very subtle. Activation of TRPV3 can elevate intracellular Ca2+ concentration, induce apoptosis, and inhibit human hair growth,18 which explains the manifestation of alopecia in these individuals. Recent studies also show that TRPV3 is a Ca2+ entry pathway tightly associated with the TGF-α/EGFR signaling complex orchestrating keratinocyte terminal differentiation.24 TRPV3 knockout mice present abnormal hair morphology and skin barrier function.25 Spontaneous mutant rodent strains, DS-Nh mice (carrying the TRPV3 mutation that produces p.Gly573Ser) and WBN/Kob-Ht rats (carrying the TRPV3 mutation that produces p.Gly573Cys), exhibit hairlessness and dermatitis.26 However, it is still poorly understood how TRPV3 functions in the keratinization of the skin in humans.
Alignment of the TRPV paralogs demonstrates that Gly573 and Trp692, the amino acids that we find altered in the six individuals in this study, have been completely conserved in this region of the protein. (Figure 2B). Notably, all the female individuals we studied carry mutations located in the site that produces Gly573. Individual 5, carrying p.Trp692Gly, has less severe alopecia (Figure S1). Individual 5 is a male, and whether male gender and/or the characteristic mutation relate to his mild hair involvement is yet unknown. Because of the limited number of cases reported here, the genotype-phenotype correlation of OS remains to be defined. Interestingly, spontaneous mutant rodent strains DS-Nh mice and WBN/Kob-Ht rats carry p.Gly573Ser and p.Gly573Cys, respectively, identical to the altered proteins in individuals 1, 2, 3, 4, and 6. These mutant rodents develop the phenotype of whole-body hairless and pruritic dermatitis on their faces without keratosis on their paws. They were considered as an animal model of atopic dermatitis.26 Although all the individuals denied a history of atopy, the itching was obvious in the skin lesions.
TRPV3 channel contains six transmembrane domains with a pore region between the fifth (S5) and sixth (S6) segments, and both C and N termini are intracellularly located (Figure 2B). The two sites we detected, which produce Gly573 and Trp692, are located in the linker region between S4 and S5 and in the TRP box (amino acid sequence of IWRLQR) of TRP domain (Figure 2B), respectively. Based on the high-resolution structure of voltage-gated potassium channel Kv1.2, the movement of the S4 voltage sensor is coupled to pore opening via interactions between residues in the S4-S5 linker and the C-terminal end of S6.27 If the TRPV channels share a similar architecture with the Kv1.2 channel, then the TRP domain is located at the C-terminal immediately after S6, potentially placing it in proximity to the S4-S5 linker. We proposed that similar to voltage-gated potassium channels,28–30 the S4-S5 linker of TRPV channel might interact with the TRP domain to maintain the channel in closed state, and contributes to transferring activating mechanical energy to open the intracellular S6 gate. Therefore, the mutations we detected could disrupt the coupling and lock the channel in the open conformation.
Ion channels serve many functions, including the transport of ions and water, the control of electrical excitability, and the regulation of ionic homeostasis. Proliferation and differentiation of keratinocytes and hair follicles, as well as sensation of pain and itching, are precisely controlled by ionic signals. So far, several kinds of channelopathies mainly involved in skin have been identified. Gap junction proteins, such as GJB2, GJB3, GJB4 and GJB6, are defective in several kinds of keratoderma. Alterations in voltage-gated sodium channel type IX alpha subunit are responsible for primary erythromelalgia (MIM 133020), an autosomal-dominant disorder characterized by burning pain and skin redness in extremities.31 Here, we show a skin channelopathy caused by TRPV3 mutations. The mutant TRPV3 might function in a dominant-positive manner to increase the constitutive TRPV3 activity and elevate Ca2+ in keratinocytes, leading to severe keratoderma and intolerant itching sensation. The nature of the mutations we described here also indicates that modulation of TRPV3 activity could be an alternative approach to the treatment of skin keratinization, hair, and itching disorders.
Acknowledgments
We are grateful to the patients and their family members for participation in this study. We appreciate the helpful discussions with Yan Shen, Jie Ding, Yi Rao, and Stephen G. Waxman. This work was supported in part by National Natural Science Foundation of China (81071289 for Y.Y. and 30970919 for K.W.), and the Program for New Century Excellent Talents (NCET06-0015 to Y.Y.).
Contributor Information
Ruoyu Li, Email: lry0660@gmail.com.
Yong Yang, Email: dryongyang@bjmu.edu.cn.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes project, http://www.1000genomes.org/
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man, http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Olmsted H.C. Keratodermia palmaris et plantaris congenitalis: Report of a case showing associated lesions of unusual location. Am. J. Dis. Child. 1927;33:757–764. [Google Scholar]
- 2.Mevorah B., Goldberg I., Sprecher E., Bergman R., Metzker A., Luria R., Gat A., Brenner S. Olmsted syndrome: mutilating palmoplantar keratoderma with periorificial keratotic plaques. J. Am. Acad. Dermatol. 2005;53(5, Suppl 1):S266–S272. doi: 10.1016/j.jaad.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 3.Ogawa F., Udono M., Murota H., Shimizu K., Takahashi H., Ishida-Yamamoto A., Iizuka H., Katayama I. Olmsted syndrome with squamous cell carcinoma of extremities and adenocarcinoma of the lung: failure to detect loricrin gene mutation. Eur. J. Dermatol. 2003;13:524–528. [PubMed] [Google Scholar]
- 4.Vosynioti V., Kosmadaki M., Tagka A., Katsarou A. A case of Olmsted syndrome. Eur. J. Dermatol. 2010;20:837–838. doi: 10.1684/ejd.2010.1088. [DOI] [PubMed] [Google Scholar]
- 5.Tharini G.K., Hema N., Jayakumar S., Parveen B. Olmsted syndrome: report of two cases. Indian J. Dermatol. 2011;56:591–593. doi: 10.4103/0019-5154.87166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rivers J.K., Duke E.E., Justus D.W. Etretinate: management of keratoma hereditaria mutilans in four family members. J. Am. Acad. Dermatol. 1985;13:43–49. doi: 10.1016/s0190-9622(85)70141-7. [DOI] [PubMed] [Google Scholar]
- 7.Cambiaghi S., Tadini G., Barbareschi M., Caputo R. Olmsted syndrome in twins. Arch. Dermatol. 1995;131:738–739. [PubMed] [Google Scholar]
- 8.Larrègue M., Callot V., Kanitakis J., Suau A.M., Foret M. Olmsted syndrome: report of two new cases and literature review. J. Dermatol. 2000;27:557–568. doi: 10.1111/j.1346-8138.2000.tb02229.x. [DOI] [PubMed] [Google Scholar]
- 9.Armstrong A.P., Percival N. Olmsted's syndrome. J. R. Soc. Med. 1997;90:81–82. doi: 10.1177/014107689709000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 11.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA. 2010;107:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grandl J., Hu H., Bandell M., Bursulaya B., Schmidt M., Petrus M., Patapoutian A. Pore region of TRPV3 ion channel is specifically required for heat activation. Nat. Neurosci. 2008;11:1007–1013. doi: 10.1038/nn.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bang S., Yoo S., Yang T.J., Cho H., Hwang S.W. Farnesyl pyrophosphate is a novel pain-producing molecule via specific activation of TRPV3. J. Biol. Chem. 2010;285:19362–19371. doi: 10.1074/jbc.M109.087742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung M.K., Lee H., Mizuno A., Suzuki M., Caterina M.J. 2-aminoethoxydiphenyl borate activates and sensitizes the heat-gated ion channel TRPV3. J. Neurosci. 2004;24:5177–5182. doi: 10.1523/JNEUROSCI.0934-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang F., Cui Y., Wang K., Zheng J. Thermosensitive TRP channel pore turret is part of the temperature activation pathway. Proc. Natl. Acad. Sci. USA. 2010;107:7083–7088. doi: 10.1073/pnas.1000357107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu H., Grandl J., Bandell M., Petrus M., Patapoutian A. Two amino acid residues determine 2-APB sensitivity of the ion channels TRPV3 and TRPV4. Proc. Natl. Acad. Sci. USA. 2009;106:1626–1631. doi: 10.1073/pnas.0812209106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao R., Tian J., Tang J., Zhu M.X. The TRPV3 mutation associated with the hairless phenotype in rodents is constitutively active. Cell Calcium. 2008;43:334–343. doi: 10.1016/j.ceca.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borbíró I., Lisztes E., Tóth B.I., Czifra G., Oláh A., Szöllosi A.G., Szentandrássy N., Nánási P.P., Péter Z., Paus R. Activation of transient receptor potential vanilloid-3 inhibits human hair growth. J. Invest. Dermatol. 2011;131:1605–1614. doi: 10.1038/jid.2011.122. [DOI] [PubMed] [Google Scholar]
- 19.Smith G.D., Gunthorpe M.J., Kelsell R.E., Hayes P.D., Reilly P., Facer P., Wright J.E., Jerman J.C., Walhin J.P., Ooi L. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature. 2002;418:186–190. doi: 10.1038/nature00894. [DOI] [PubMed] [Google Scholar]
- 20.Peier A.M., Reeve A.J., Andersson D.A., Moqrich A., Earley T.J., Hergarden A.C., Story G.M., Colley S., Hogenesch J.B., McIntyre P. A heat-sensitive TRP channel expressed in keratinocytes. Science. 2002;296:2046–2049. doi: 10.1126/science.1073140. [DOI] [PubMed] [Google Scholar]
- 21.Xu H., Ramsey I.S., Kotecha S.A., Moran M.M., Chong J.A., Lawson D., Ge P., Lilly J., Silos-Santiago I., Xie Y. TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature. 2002;418:181–186. doi: 10.1038/nature00882. [DOI] [PubMed] [Google Scholar]
- 22.Moqrich A., Hwang S.W., Earley T.J., Petrus M.J., Murray A.N., Spencer K.S., Andahazy M., Story G.M., Patapoutian A. Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science. 2005;307:1468–1472. doi: 10.1126/science.1108609. [DOI] [PubMed] [Google Scholar]
- 23.Huang S.M., Li X., Yu Y., Wang J., Caterina M.J. TRPV3 and TRPV4 ion channels are not major contributors to mouse heat sensation. Mol. Pain. 2011;7:37. doi: 10.1186/1744-8069-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montell C. Preventing a Perm with TRPV3. Cell. 2010;141:218–220. doi: 10.1016/j.cell.2010.03.043. [DOI] [PubMed] [Google Scholar]
- 25.Cheng X., Jin J., Hu L., Shen D., Dong X.P., Samie M.A., Knoff J., Eisinger B., Liu M.L., Huang S.M. TRP channel regulates EGFR signaling in hair morphogenesis and skin barrier formation. Cell. 2010;141:331–343. doi: 10.1016/j.cell.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asakawa M., Yoshioka T., Matsutani T., Hikita I., Suzuki M., Oshima I., Tsukahara K., Arimura A., Horikawa T., Hirasawa T., Sakata T. Association of a mutation in TRPV3 with defective hair growth in rodents. J. Invest. Dermatol. 2006;126:2664–2672. doi: 10.1038/sj.jid.5700468. [DOI] [PubMed] [Google Scholar]
- 27.Long S.B., Tao X., Campbell E.B., MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 28.Tristani-Firouzi M., Chen J., Sanguinetti M.C. Interactions between S4-S5 linker and S6 transmembrane domain modulate gating of HERG K+ channels. J. Biol. Chem. 2002;277:18994–19000. doi: 10.1074/jbc.M200410200. [DOI] [PubMed] [Google Scholar]
- 29.Labro A.J., Boulet I.R., Choveau F.S., Mayeur E., Bruyns T., Loussouarn G., Raes A.L., Snyders D.J. The S4-S5 linker of KCNQ1 channels forms a structural scaffold with the S6 segment controlling gate closure. J. Biol. Chem. 2011;286:717–725. doi: 10.1074/jbc.M110.146977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choveau F.S., Rodriguez N., Abderemane Ali F., Labro A.J., Rose T., Dahimène S., Boudin H., Le Hénaff C., Escande D., Snyders D.J. KCNQ1 channels voltage dependence through a voltage-dependent binding of the S4-S5 linker to the pore domain. J. Biol. Chem. 2011;286:707–716. doi: 10.1074/jbc.M110.146324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y., Wang Y., Li S., Xu Z., Li H., Ma L., Fan J., Bu D., Liu B., Fan Z. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 2004;41:171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.