

Abstract
ATR (ataxia telangiectasia and Rad3 related) is an essential regulator of genome integrity. It controls and coordinates DNA-replication origin firing, replication-fork stability, cell-cycle checkpoints, and DNA repair. Previously, autosomal-recessive loss-of-function mutations in ATR have been demonstrated in Seckel syndrome, a developmental disorder. Here, however, we report on a different kind of genetic disorder that is due to functionally compromised ATR activity, which translates into an autosomal-dominant inherited disease. The condition affects 24 individuals in a five-generation pedigree and comprises oropharyngeal cancer, skin telangiectases, and mild developmental anomalies of the hair, teeth, and nails. We mapped the disorder to a ∼16.8 cM interval in chromosomal region 3q22–24, and by sequencing candidate genes, we found that ATR contained a heterozygous missense mutation (c.6431A>G [p.Gln2144Arg]) that segregated with the disease. The mutation occurs within the FAT (FRAP, ATM, and TRRAP) domain—which can activate p53—of ATR. The mutation did not lead to a reduction in ATR expression, but cultured fibroblasts showed lower p53 levels after activation of ATR with hydroxyurea than did normal control fibroblasts. Moreover, loss of heterozygosity for the ATR locus was noted in oropharyngeal-tumor tissue. Collectively, the clinicopathological and molecular findings point to a cancer syndrome and provide evidence implicating a germline mutation in ATR and susceptibility to malignancy in humans.
Main Text
All cells possess complex mechanisms that respond to and repair DNA damage. Integral to regulating the DNA-damage response are the phosphatidylinositol-3-kinase-related protein kinases (PI3KKs), which include ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR).1,2 These kinases can phosphorylate and activate several downstream proteins, including Chk1, Chk2, and p53, that are implicated in cell-cycle arrest, DNA repair, and apoptosis.1,2 ATM and ATR share significant sequence homology as well as several biochemical and functional similarities in promoting cell-cycle arrest and DNA repair. However, whereas ATM is activated after DNA double-strand breakage, ATR is activated by single-stranded regions of DNA generated, for example, in S-phase after replication-fork stalling or during the repair of certain types of DNA damage.2,3 Notably, ATR is vital for replicating cell viability, but ATM is not.4–6 Abnormalities in DNA-damage checkpoint kinases have been implicated in cancer biology, and somatic mutations in ATR (MIM 601215), ATM (MIM 607585), CHK1 (MIM 603078), CHK2 (MIM 604373), BRCA1 (MIM 113705), and BRCA2 (MIM 600185) have all been observed in a variety of malignant human tumors.7–11 Germline mutations in both ATM and ATR, however, have also been reported but are associated with clinically distinct congenital disorders. Autosomal-recessive mutations in ATM underlie ataxia telangiectasia (MIM 208900), a progressive neurodegenerative condition associated with Purkinje neuron loss, cerebellar ataxia, telangiectases, immune dysfunction, and predisposition to B cell lymphoma and T cell leukemia.12 Furthermore, heterozygous carriers of ATM mutations also exhibit increased risk for nonhematologic cancers.13 Mutations in ATR are much rarer. To date, a single homozygous synonymous mutation, which is inherited in an autosomal-recessive manner and which causes mis-splicing of exon 9 of ATR, has been described in two related families afflicted with Seckel syndrome (MIM 210600), which affects five individuals in total.14 Seckel syndrome is characterized by intrauterine-growth retardation, dwarfism, microcephaly with intellectual disability, and a characteristic “bird-headed” facial appearance. Seckel syndrome is, however, genetically heterogeneous, and five recorded loci and three known genes (ATR, CENPJ [MIM 609279], and CEP152 [MIM 613529]) associated with the syndrome contain pathogenic mutations. Cells derived from individuals with Seckel syndrome resulting from ATR mutations exhibit reduced expression of ATR and impaired DNA-damage-induced phosphorylation of important ATR substrates, including H2AX, Chk1, NBS1, and p53, along with defective ATR-dependent G2-M cell-cycle checkpoint activation.14 Considering the fundamental role of ATR in maintaining replication-fork stability and preserving genomic integrity, it is perhaps surprising that, to date, inherited mutations in ATR, i.e., in individuals with Seckel syndrome, have not been implicated in malignancy. In contrast, we now report on a pedigree with an autosomal-dominant disorder that reveals a germline mutation in ATR in a hereditary cancer syndrome.
We investigated a Caucasian family containing 24 affected individuals spanning five generations originating from Indiana, USA (Figure 1A). All those with the disorder had telangiectases (Figures 1B, 1C, and 1D) that appeared during infancy (<18 months) in both sun-exposed and sun-protected sites; <18 months is somewhat earlier than in most individuals with ataxia telangiectasia due to recessive mutations in ATM. Other clinical features included thinning of the lateral part of the eyebrows and patchy alopecia in areas of skin with prominent telangiectases. Thin dental enamel and dental caries in both primary and secondary dentition were also noted. With regard to malignancy, 10 of the 24 cases developed oropharyngeal cancer (Figures 1A, 1E, 1F, and 1G), typically in the third decade of life or thereafter. Other malignancies reported included nonmelanoma skin cancer (basal cell carcinoma, squamous cell carcinoma, and sebaceous carcinoma) in three individuals, breast cancer in one individual, and cervical cancer in one other family member. No further cutaneous or developmental anomalies were noted (for additional clinical details, see Tables S1 and S2, available online).
Figure 1.
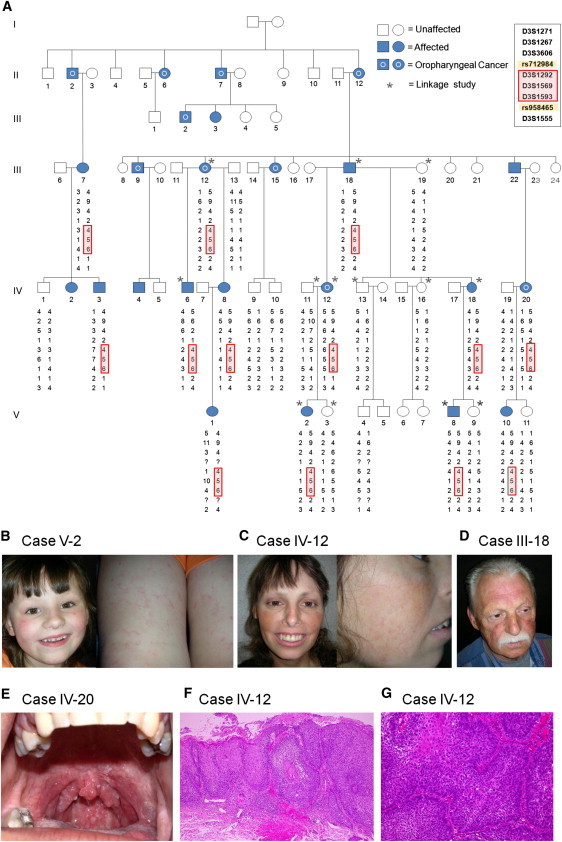
Pedigree, Linkage Analysis, and Clinicopathological Features of This Autosomal-Dominant Disorder Associated with a Missense Mutation in ATR
(A) The family pedigree. Squares denote male family members, and circles denote female family members. A 17.3 Mb critical region is defined by recombination events in individual IV:6 for rs712984 and in individuals IV:3 and IV:12 for SNP rs958465.
(B, C, and D) Carriers are a 6-year-old female (B), a 35-year-old female (C), and a 56-year-old male (D) with telangiectases on the face as well as thin outer eyebrows.
(E) A 25-year old female carrier with the additional feature of a papillomatous tumor in the upper-pharynx (tonsillar) region with histological features of squamous cell carcinoma.
(F and G) Histopathological feature of squamous cell carcinoma in the upper pharynx (tonsillar) region of affected individual IV-12. The epidermis shows full-thickness dysplasia with hyperkeratosis and parakeratosis (hematoxylin and eosin stain; original magnification is ×100) (F). Nests of carcinoma cells are infiltrating into the submucosal tissue (hematoxylin and eosin stain; original magnification is ×200) (G).
Additional pedigree data are detailed in Table S1 and S2.
After obtaining approval from the ethics committee and informed consent from all subjects, we extracted genomic DNA from peripheral blood samples from 26 individuals (13 subjects with clinical features of the disorder and 13 unaffected individuals) in compliance with the Declaration of Helsinki Principles. The subjects were genotyped with the Human Mapping 10K Array version 2 (Affymetrix, High Wycombe, UK) according to the manufacturer's protocol. To generate appropriate input files for the linkage-analysis program Merlin,15 we utilized ALOHOMORA software.16 Assuming autosomal-dominant inheritance with complete penetrance, we then carried out parametric linkage analysis by using the genotyping data of 13 individuals (7 affected individuals and 6 unaffected individuals; indicated by asterisks in Figure 1A). Linkage analysis identified a single significant locus—with a LOD score of 2.7—in chromosomal region 3q22–24 (Figure 2A). The peak on chromosome 3 extends from SNP rs722813 to rs952032 (116–155 cM) (Figure 2B). Additional microsatellite markers were genotyped for fine mapping of this interval. The primers were obtained from the ABI PRISM Linkage Mapping Set version 2.5 (Applied Biosystems, Warrington, UK). The PCR products were analyzed on an ABI 3730 DNA sequencer with GeneMapper software version 3.0 (Applied Biosystems). Fine mapping with microsatellite markers and SNP array data narrowed the candidate region to a ∼16.8 cM interval in region 3q22–24 between rs712984 and rs951465 (Figure 1A). This interval contains ∼90 genes, including those predicted in silico (Figure 2C). Candidate-gene analysis was then performed. Potential candidate genes (which were chosen on the basis of their associated phenotype) included the following: AMOTL2 (related to angiogenesis), MRAS and RASA2 (related to oncogenesis), TOPBP1, CEP63, STAG1, NCK1, PIK3CB, and ATR (which play a role in DNA-damage response and DNA replication), and NEK11, ANAPC13, and TFDP2 (which control the cell cycle). All of the candidate genes that were sequenced are listed in Table S3. For sequencing, DNA samples were amplified with published primers sited in introns flanking individual exons of each gene.17 However, sequencing the genomic DNA of clinically affected individuals for all genes in the interval only disclosed a single heterozygous point mutation (c.6431A>G in ATR [NCBI RefSeq accession number NM_001184.3]) that segregated with the disease. This mutation, which converts a highly evolutionarily conserved glutamine to arginine at amino acid 2144 (p.Gln2144Arg) (Figure 3C), was present in all the affected individuals but not in any of the unaffected individuals (Figures 3A and 3B). The nucleotide substitution was not found in the screening of 220 ethnically matched control chromosomes. This mutation occurs within the FAT (FRAP, ATM and TRRAP) domain of ATR and is positioned immediately adjacent to a potential phosphorylation site (SQ/TQ motif) at Ser2143 (Figure 3C). Within the FAT domain of ATR, phosphorylation of Thr1989 has previously been implicated in the activation of ATR by the ATR stimulator TopBP1.18,19 Nevertheless, the function of the FAT domain of PI3KKs, including ATR specifically, has not been unequivocally demonstrated experimentally, although it has been suggested to have some structural role, possibly in mediating protein-protein interactions.20 Consistent with this suggestion, the FAT domain of ATM and ATR has been shown to bind to AIMP3 (aminoacyl-tRNA synthetase-interacting multifunctional protein 3, previously known as p18), a haploinsufficient tumor suppressor that can activate p53.21 Another possibility, extrapolated from studies on ATM, is that the FAT domain normally loops to have a direct physical interaction with the kinase domain, thereby stabilizing the whole carboxy end of the protein and permitting optimal downstream regulatory activities.22 However, this has yet to be proven for the FAT domain of ATR. To evaluate the functional consequences of the mutation, we first used RT-PCR and immunoblotting to assess ATR-mRNA expression in skin samples and in cultured fibroblasts and to assess protein levels in cultured fibroblasts from two affected subjects, two control skin samples, and one individual with Seckel syndrome. In our p.Gln2144Arg mutant samples, there was no reduction in ATR mRNA (Figures S1 and S2) or ATR levels (c.f. reduced expression in Seckel syndrome, Figure 3D). Next, on the basis of the clinical feature of malignancy and existing data implicating AIMP3 in the normal upregulation of p53 in response to DNA damage, we examined expression of p53 by immunoblotting in ATR-mutated fibroblasts; we noted both constitutively reduced p53 and also reduced p53 levels after ATR activation with hydroxyurea (Figure 3E). Furthermore, under similar experimental conditions, hydroxyurea-induced Chk1 and H2AX phosphorylation did not show any clear reduction in these ATR-mutated cells, suggesting differential substrate activation (Figures S3 and S4).
Figure 2.
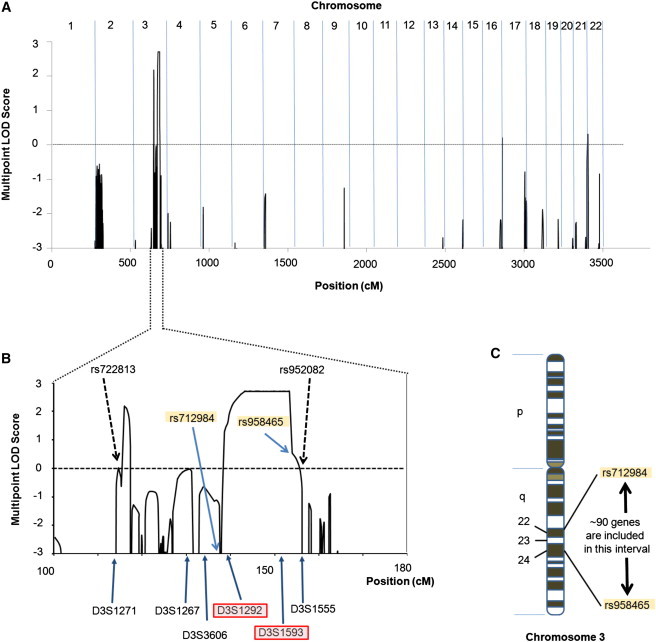
Genome-Wide Linkage Analysis
(A) Genome-wide parametric linkage analysis reveals a LOD score of >2.5 on chromosome 3.
(B) Magnification of the LOD-score diagram of the chromosome 3 region. The chromosome 3 peak extends from SNP rs722813 to rs952032 (116–155 cM), and the max LOD is 2.7. Fine mapping with microsatellite markers and SNP array data (Figure 1A) narrowed the candidate region to a ∼16.8 cM interval in region 3q22–24 between rs712984 and rs951465.
(C) This interval contains ∼90 genes, including ATR.
Figure 3.
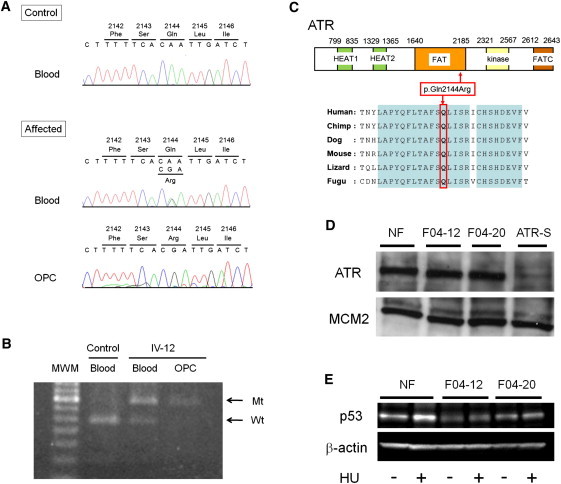
Identification of a Pathogenic Mutation in ATR
(A) Nucleotide sequencing of genomic DNA extracted from the blood of an affected individual reveals a heterozygous missense mutation, c.6431A>G (p.Gln2144Arg), in ATR (“affected blood”) but a homozygous or hemizygous mutation in an oropharyngeal-cancer lesion (“affected OPC”) from carrier individual IV-12.
(B) Analysis of the mutation c.6431A>G in ATR with restriction enzyme MfeI. PCR products from the wild-type (“Wt”) allele were digested, whereas those from the mutant (“Mt”) allele were not. Control-blood DNA shows only a digested band (“control blood”). DNA extracted from blood shows both an undigested band and a digested band (“IV-12 blood”) but only an undigested band from the oropharyngeal-cancer lesion (“IV-12 OPC”) in carrier individual IV-12.
(C) A schematic representation of ATR (NP_001175.2). The mutation occurs within the FAT domain of ATR. The substituted amino acid (p.Gln2144) is well conserved throughout various evolutionary lineages.
(D) Immunoblot of ATR in fibroblast extracts. “NF” indicates control fibroblasts from a healthy donor; “F04-12” and “F04-20” are fibroblasts from carriers IV-12 and IV-20, respectively, in Figure 1A; “ATR-S” denotes fibroblasts derived from an individual with ATR Seckel syndrome; MCM2 (minichromosome maintenance complex component 2) was used as a loading control. The antibodies sc-1887 (for ATR) and sc-9839 (for MCM2) were purchased from Santa Cruz (Santa Cruz, CA).
(E) Immunoblot of p53 in fibroblast extracts (of both carriers and controls) before and after the activation of ATR by hydroxyurea (“HU”) exposure for two hours; β-actin was used as a loading control. The antibodies #9282 (for p53) and #4967 (for β-actin) were purchased from Cell Signaling (Beverly, MA).
Cancer predisposition might be associated with tumor-suppressor inactivation resulting from both an inherited mutation (i.e., a mutant allele) in the germline and a somatic loss of the wild-type allele (such a loss leaves a single copy of the mutant allele).23 For loss of heterozygosity (LOH) analysis in the cancer tissue of this syndrome, we sequenced DNA extracted from paraffin sections of an oropharyngeal-cancer lesion from affected individual IV-12 (Figures 3A and 3B). Sequencing revealed a homozygous or hemizygous change, c.6431A>G in ATR, in the cancer lesion. This LOH event, with the allelic loss of the wild-type ATR allele, does not affect the expression of ATR by immunohistochemistry (Figure S5) but implicates ATR in the pathophysiology of the oropharyngeal cancer and therefore indicates a tumor-suppressing role of ATR. Nevertheless, this interpretation can only be supported after further exploration of the occurrence of LOH in more squamous-cell-carcinoma samples from this pedigree. We also assessed the tumor tissue for the presence of human papillomavirus (HPV), given that HPV is a known risk factor for oropharyngeal cancer.24 However, PCR amplification of tumor DNA via the PCR Human Papillomavirus Detection Set (Takara, Osaka, Japan) failed to identify HPV in two oropharyngeal-cancer samples obtained from subjects IV-12 and IV-20 (data not shown).
Germline mutations in ATR have hitherto only been reported in the autosomal-recessive disorder Seckel syndrome. Moreover, a humanized mouse model (incorporating the Seckel-patient-specific mutation) of the disorder shows similar features—proportionate dwarfism and microcephaly, along with an overt progeroid phenotype coupled with attenuation of stem-cell niches—to those of the human syndrome.4,25 Furthermore, conditional partial deletion of Atr in a postnatal mouse leads to a similar age-related phenotype—hair graying, alopecia, and thymic involution—consistent with the loss of stem cell regenerative capacity.4,26 Nevertheless, neither individuals with Seckel syndrome nor either mouse model display an elevated incidence of malignancy. This might be related to the magnitude of ATR deficiency in this context because heterozygous mutations and haploinsufficiency of ATR have been implicated in some acquired cancers in certain circumstances.27 Notably, somatic mutations in exon 10 of ATR have been detected in some endometrial, colon, and stomach tumors.27 These mutations, which occur upstream of the FAT domain, have been shown to inhibit ATR-dependent responses to DNA damage. A further study also showed that ATR haploinsufficiency leads to high-level genetic instability and accelerated tumorigenesis in a mismatch repair-deficient host.28 Notably, monoallelic ATR targeting in MLH1 (MutL homolog 1)-deficient HCT 116 carcinoma cells resulted in hypersensitivity to genotoxic stress accompanied by an increase in fragile-site instability, as well as chromosomal amplifications and rearrangements.28 Mice bearing the Atr+/− Mlh1−/− genotype were highly prone to both embryonic lethality and early tumor development.28 Furthermore, Chk1, an important effector kinase of ATR, can also act as a haploinsufficient tumor suppressor.29
In contrast, this autosomal-dominant pedigree clearly provides clinicopathologic data linking an inherited abnormality in ATR to malignancy. Collectively, our observations implicate the FAT domain of ATR in the suppression of tumorigenesis. Why an oropharyngeal-cancer tumor occurs instead of a hematologic or solid-organ tumor still remains unclear, and further observation of this single pedigree will be necessary for establishing whether there is a more extensive risk of other cancers. Alternatively, known risk factors for oropharyngeal cancer include alcohol, tobacco, and viral infections.30 Of these factors, viral infection might be the most likely cause of the tumor seen in our pedigree. Notably, the ATR pathway can be targeted by certain viruses during infection,31 and viral proteins are ATR targets.32 Regarding the tumor location, it has been shown previously that Fanconi anemia—an autosomal-recessive syndrome caused by defects in at least 13 proteins that are involved in the recognition and repair of DNA in a DNA-damage response network and that are downstream of ATR signaling and are phosphorylated by ATR—is also associated with oropharyngeal cancer.33,34 In conclusion, our report identifies an autosomal-dominant disorder and also provides further clinical and genetic data associating ATR with human disease.
Acknowledgments
We thank P. Green, T. Techanukul, and C. Mathew for discussions. We also thank S. Clements and N. Almaani for technical assistance. M.O. is a Cancer Research UK (CRUK) Senior Cancer Research fellow, supported by CRUK and the Medical Research Council. We acknowledge financial support from the UK Department of Health via the National Institute for Health Research comprehensive Biomedical Research Centre award to Guy's and St. Thomas' National Health Services (NHS) Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BLAST Assembled RefSeq Genomes, http://blast.ncbi.nlm.nih.gov/Blast.cgi
Ensembl Genome Browser, http://www.ensembl.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Primer3, http://frodo.wi.mit.edu/primer3/
The National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Yang J., Yu Y., Hamrick H.E., Duerksen-Hughes P.J. ATM, ATR and DNA-PK: Initiators of the cellular genotoxic stress responses. Carcinogenesis. 2003;24:1571–1580. doi: 10.1093/carcin/bgg137. [DOI] [PubMed] [Google Scholar]
- 2.Abraham R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 3.O'Driscoll M., Jeggo P.A. The role of double-strand break repair - insights from human genetics. Nat. Rev. Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- 4.O'Driscoll M. Mouse models for ATR deficiency. DNA Repair (Amst.) 2009;8:1333–1337. doi: 10.1016/j.dnarep.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 5.de Klein A., Muijtjens M., van Os R., Verhoeven Y., Smit B., Carr A.M., Lehmann A.R., Hoeijmakers J.H. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 2000;10:479–482. doi: 10.1016/s0960-9822(00)00447-4. [DOI] [PubMed] [Google Scholar]
- 6.Brown E.J., Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 7.Menoyo A., Alazzouzi H., Espín E., Armengol M., Yamamoto H., Schwartz S., Jr. Somatic mutations in the DNA damage-response genes ATR and CHK1 in sporadic stomach tumors with microsatellite instability. Cancer Res. 2001;61:7727–7730. [PubMed] [Google Scholar]
- 8.Ahmed M., Rahman N. ATM and breast cancer susceptibility. Oncogene. 2006;25:5906–5911. doi: 10.1038/sj.onc.1209873. [DOI] [PubMed] [Google Scholar]
- 9.Meijers-Heijboer H., Wijnen J., Vasen H., Wasielewski M., Wagner A., Hollestelle A., Elstrodt F., van den Bos R., de Snoo A., Fat G.T. The CHEK2 1100delC mutation identifies families with a hereditary breast and colorectal cancer phenotype. Am. J. Hum. Genet. 2003;72:1308–1314. doi: 10.1086/375121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antoniou A., Pharoah P.D., Narod S., Risch H.A., Eyfjord J.E., Hopper J.L., Loman N., Olsson H., Johannsson O., Borg A. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003;72:1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poehlmann A., Roessner A. Importance of DNA damage checkpoints in the pathogenesis of human cancers. Pathol. Res. Pract. 2010;206:591–601. doi: 10.1016/j.prp.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 12.Savitsky K., Bar-Shira A., Gilad S., Rotman G., Ziv Y., Vanagaite L., Tagle D.A., Smith S., Uziel T., Sfez S. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 13.Renwick A., Thompson D., Seal S., Kelly P., Chagtai T., Ahmed M., North B., Jayatilake H., Barfoot R., Spanova K., Breast Cancer Susceptibility Collaboration (UK) ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006;38:873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 14.O'Driscoll M., Ruiz-Perez V.L., Woods C.G., Jeggo P.A., Goodship J.A. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 15.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 16.Rüschendorf F., Nürnberg P. ALOHOMORA: A tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–2125. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- 17.Sjöblom T., Jones S., Wood L.D., Parsons D.W., Lin J., Barber T.D., Mandelker D., Leary R.J., Ptak J., Silliman N. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 18.Nam E.A., Zhao R., Glick G.G., Bansbach C.E., Friedman D.B., Cortez D. Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase. J. Biol. Chem. 2011;286:28707–28714. doi: 10.1074/jbc.M111.248914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S., Shiotani B., Lahiri M., Maréchal A., Tse A., Leung C.C., Glover J.N., Yang X.H., Zou L. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol. Cell. 2011;43:192–202. doi: 10.1016/j.molcel.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosotti R., Isacchi A., Sonnhammer E.L. FAT: A novel domain in PIK-related kinases. Trends Biochem. Sci. 2000;25:225–227. doi: 10.1016/s0968-0004(00)01563-2. [DOI] [PubMed] [Google Scholar]
- 21.Park B.J., Kang J.W., Lee S.W., Choi S.J., Shin Y.K., Ahn Y.H., Choi Y.H., Choi D., Lee K.S., Kim S. The haploinsufficient tumor suppressor p18 upregulates p53 via interactions with ATM/ATR. Cell. 2005;120:209–221. doi: 10.1016/j.cell.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 22.Lempiäinen H., Halazonetis T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009;28:3067–3073. doi: 10.1038/emboj.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Assié G., LaFramboise T., Platzer P., Eng C. Frequency of germline genomic homozygosity associated with cancer cases. JAMA. 2008;299:1437–1445. doi: 10.1001/jama.299.12.1437. [DOI] [PubMed] [Google Scholar]
- 24.Syrjänen S. Human papillomavirus (HPV) in head and neck cancer. J. Clin. Virol. 2005;32(Suppl 1):S59–S66. doi: 10.1016/j.jcv.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 25.Murga M., Bunting S., Montaña M.F., Soria R., Mulero F., Cañamero M., Lee Y., McKinnon P.J., Nussenzweig A., Fernandez-Capetillo O. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet. 2009;41:891–898. doi: 10.1038/ng.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruzankina Y., Pinzon-Guzman C., Asare A., Ong T., Pontano L., Cotsarelis G., Zediak V.P., Velez M., Bhandoola A., Brown E.J. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis K.A., Mullany S., Thomas B., Chien J., Loewen R., Shridhar V., Cliby W.A. Heterozygous ATR mutations in mismatch repair-deficient cancer cells have functional significance. Cancer Res. 2005;65:7091–7095. doi: 10.1158/0008-5472.CAN-05-1019. [DOI] [PubMed] [Google Scholar]
- 28.Fang Y., Tsao C.C., Goodman B.K., Furumai R., Tirado C.A., Abraham R.T., Wang X.F. ATR functions as a gene dosage-dependent tumor suppressor on a mismatch repair-deficient background. EMBO J. 2004;23:3164–3174. doi: 10.1038/sj.emboj.7600315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lam M.H., Liu Q., Elledge S.J., Rosen J.M. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell. 2004;6:45–59. doi: 10.1016/j.ccr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 30.Gillison M.L. Current topics in the epidemiology of oral cavity and oropharyngeal cancers. Head Neck. 2007;29:779–792. doi: 10.1002/hed.20573. [DOI] [PubMed] [Google Scholar]
- 31.Chaurushiya M.S., Weitzman M.D. Viral manipulation of DNA repair and cell cycle checkpoints. DNA Repair (Amst.) 2009;8:1166–1176. doi: 10.1016/j.dnarep.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blackford A.N., Bruton R.K., Dirlik O., Stewart G.S., Taylor A.M., Dobner T., Grand R.J., Turnell A.S. A role for E1B-AP5 in ATR signaling pathways during adenovirus infection. J. Virol. 2008;82:7640–7652. doi: 10.1128/JVI.00170-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson L.H. Unraveling the Fanconi anemia-DNA repair connection. Nat. Genet. 2005;37:921–922. doi: 10.1038/ng0905-921. [DOI] [PubMed] [Google Scholar]
- 34.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.