

Abstract
Fuchs corneal dystrophy (FCD) is a genetic disorder of the corneal endothelium and is the most common cause of corneal transplantation in the United States. Previously, we mapped a late-onset FCD locus, FCD2, on chromosome 18q. Here, we present next-generation sequencing of all coding exons in the FCD2 critical interval in a multigenerational pedigree in which FCD segregates as an autosomal-dominant trait. We identified a missense change in LOXHD1, a gene causing progressive hearing loss in humans, as the sole variant capable of explaining the phenotype in this pedigree. We observed LOXHD1 mRNA in cultured human corneal endothelial cells, whereas antibody staining of both human and mouse corneas showed staining in the corneal epithelium and endothelium. Corneal sections of the original proband were stained for LOXHD1 and demonstrated a distinct increase in antibody punctate staining in the endothelium and Descemet membrane; punctate staining was absent from both normal corneas and FCD corneas negative for causal LOXHD1 mutations. Subsequent interrogation of a cohort of >200 sporadic affected individuals identified another 15 heterozygous missense mutations that were absent from >800 control chromosomes. Furthermore, in silico analyses predicted that these mutations reside on the surface of the protein and are likely to affect the protein's interface and protein-protein interactions. Finally, expression of the familial LOXHD1 mutant allele as well as two sporadic mutations in cells revealed prominent cytoplasmic aggregates reminiscent of the corneal phenotype. All together, our data implicate rare alleles in LOXHD1 in the pathogenesis of FCD and highlight how different mutations in the same locus can potentially produce diverse phenotypes.
Main Text
Fuchs corneal dystrophy (FCD) is the most common genetic disorder of the corneal endothelium1–3 and accounts for a significant portion of corneal transplantation performed in the United States every year.4,5 Clinically, FCD is marked by the development of guttae, excrescences of the Descemet membrane that typically appear in the fourth or fifth decade of life and increase in number over time.6,7 As the disease progresses, visual acuity decreases secondary to corneal edema and endothelial cell loss, and the last stage of the disease is evidenced by painful epithelial bullae.8
FCD is genetically heterogeneous. A rare early-onset form of an endothelial dystrophy with some of the clinical features of FCD has been ascribed to mutations in COL8A2 (MIM 12052),9,10 whereas rare mutations in SLC4A11 (MIM 610206) and TCF8 (MIM 189909) have been implicated in the pathogenesis of the common form of late-onset FCD.9–14 Additionally, large pedigrees that exhibit dominant inheritance have been used for mapping four loci—FCD1, FCD2, FCD3, and FCD4—on chromosomes 13, 18, 5, and 9, respectively.13,15–17 Finally, rs613872, an intronic SNP at the TCF4 (MIM 602272) locus on chromosome 18q, has also been associated with late-onset FCD.18–20 However, haplotype analyses have suggested that this risk factor is probably independent of FCD2,19 intimating that multiple loci might account for the linkage and association signals on 18q.
Previously, we reported on three large families with multiple affected members, each of which generated LOD scores >3 on 18q. To identify mutations that cause the disorder in these pedigrees, we generated a custom exon-capture library that includes all annotated exons in the most conservative critical region, which is defined as a minimum of two recombination events at each boundary and spans 26 Mb. Given the complexity of the phenotype and the documented presence of both nonpenetrance and phenocopies, we extended our capture target region by 5 MB on each side.
One affected and one unaffected individual per family were sequenced at the Genetic Variation and Gene Discovery Core Facility at Cincinnati Children's Hospital. Exon capture with a Roche/Nimblegen 385k chip and next-generation paired-end sequencing on an Illumina GAIIx instrument generated on average 1.5 GB of data per sample; these data correspond to approximately 25 million 36 bp reads. We captured 98% of the target with an average read depth of 95×, and only a small fraction (0.01%) of the sequences had coverage less than 25×. The raw data were aligned to the reference sequence by Eland (Illumina), and alleles were called by Seqmate software.
We analyzed the coding sequence and intron-exon junctions of 134 genes. For two of the three families, we found no changes that could explain the disorder. This was true not only for rare alleles (minor allele frequency [MAF] <1%), which would have been the expectation from linkage analysis (LOD scores were calculated with a disease allele frequency of 0.01), but also for more common alleles. In addition, we could not observe any overtransmission of common alleles because our study design could not detect association. As such, the causal mutations in these two families could be common, in which case they might act in conjunction with trans alleles at other loci, as shown for FCD4,13 could reside in regions that were not captured (such regions are either the 2% that failed capture or the regulatory elements that were not targeted), or could represent larger CNVs that were undetectable by our tools.
For the third family (HU: Kindred II in Sundin et al.), however, we identified a C>T transition, c.1639C>T (p.Arg547Cys), in LOXHD1 (MIM 613072; RefSeq accession number NM_144612.6), and it was predicted by PolyPhen2 to be probably pathogenic. The variant was absent from both the 1,000 Genomes Project and the 1,500 sequenced exomes available from the University of Seattle Exome Variant Server. Moreover, the allele was unique to the affected individuals of family HU with the exception of one individual (individual III:1; Figure 1). Our clinical records showed that deceased individual II:2 was never examined, and we therefore cannot rule out the possibility that this individual was affected and subsequently transmitted the causal mutation to his offspring (affected individuals III:1 and III:2). We also did not detect this change in 384 chromosomes of individuals who were older than 60 years of age and whose corneas were phenotypically normal (no guttae) upon slit-lamp examination; this finding is consistent with the allele being pathogenic. Recently, this variation was identified in one individual (Bushman: rs113444922) of African descent in a whole-genome sequencing project; however, the variation was never validated, and, more importantly, there is no information available regarding the phenotype of this individual.
Figure 1.
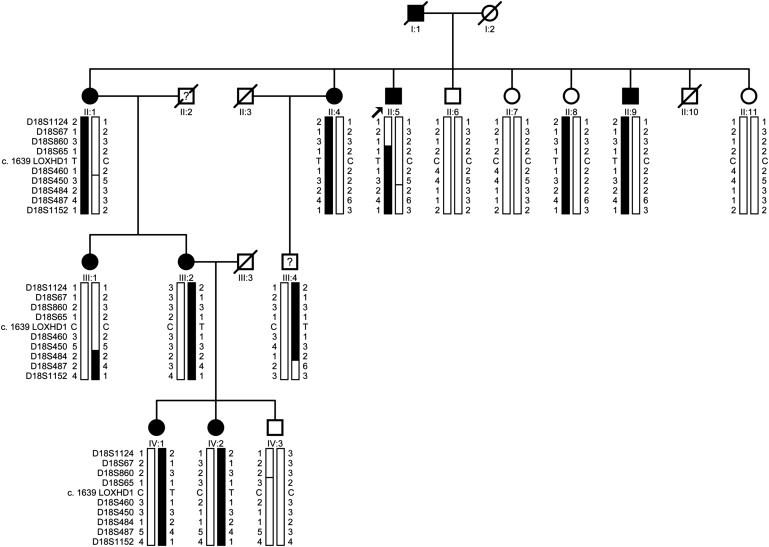
Cyrillic Pedigree of Family HU with the c.1639C>T (p.Arg547Cys) Mutation Segregating with the Disease Phenotype in the Family
The filled symbols represent affected individuals. A diagonal line through a symbol represents a deceased individual. A question mark indicates that the affectation status of an individual is unknown. Haplotypes of 18q microsatellite markers are shown, and the alleles forming the disease-bearing haplotype are shaded black.
The sequencing results were of high quality, and there is no reason to expect that we had missed the allele associated with FCD. Nevertheless, to bolster confidence in our findings, we sequenced the entire exome of the proband (individual II:5) by using Agilent whole-human-exome liquid capture array and sequenced the captured regions on Illumina HighSeq2000. The raw data were aligned to the reference sequence by ELAND, and our results reconfirmed our previous findings. All together, the results from both the target capture and the exome sequencing strongly advocate for the candidacy of LOXHD1.
As a first step toward assessing the candidacy of LOXHD1, we examined the expression of this transcript in the cornea. Previously, Grillet and colleagues reported the expression of Loxhd1 mRNA in cochlear and vestibular hair cells in P4 mice.21 First, we asked whether LOXHD1 expression was present in cultured human corneal endothelial cells. These were grown in cultured FBS medium. Upon confluence, the cells were harvested, and total RNA was extracted with Trizol (Invitrogen, CA). cDNA was synthesized with a commercially available kit (Invitrogen, CA) with polydT and LOXHD1 specific primers. We performed quantitative RT-PCR in triplicate on Applied Biosystems 7900HT by using gene-specific Taqman probes (Applied Biosystems, CA). We analyzed the results with the sequence-detection software (Applied Biosystems, CA). For each group, we calculated the mean cycle threshold (CT) value for each target (LOXHD1) and endogenous reference (GAPDH and β-actin); these CT values represent the PCR cycle at which the ABI 7900 HT Detection System first detects a noticeable increase in reporter florescence above the baseline signal. We observed expression of LOXHD1 in cultured human corneal endothelial cells at levels 5 orders of magnitude lower than that of GAPDH (MIM 138400) or β-actin (MIM 102630) (Table S1, available online).
We next examined the presence of LOXHD1 in mouse corneas. P120 (postnatal day 120) mouse eyes were enucleated and flash frozen in optimal cutting medium. Blocks were cryosectioned and 20 μm sections on slides were fixed with 4% paraformaldehyde in a phosphate buffer for 10 min. The corneal distribution of LOXHD1 was visualized with a rabbit polyclonal LOXHD1 antibody (Santa Cruz Biotechnology, SC85038) at a 1:400 dilution overnight at 4°C, and treatment with goat anti-rabbit IgG conjugated with AlexaFluor 594 (Invitrogen, A11012) at a 1:1000 dilution for 1 hr at 25°C followed. Nuclei were counter stained with DAPI (at a 1:20000 dilution), and images were captured with a Zeiss LSM 710 Meta and digitalized with Zen software. We detected LOXHD1 both in the corneal epithelium and in the endothelium; however, expression was significantly higher in epithelial cells (Figures 2A and 2B). Importantly, LOXHD1 expression in the corneal epithelium and endothelium could be competed away with a LOXHD1 peptide (Santa Cruz Biotechnology, SC85038P), suggesting that the antibody is specific to LOXHD1 (Figures 2C and 2D).
Figure 2.
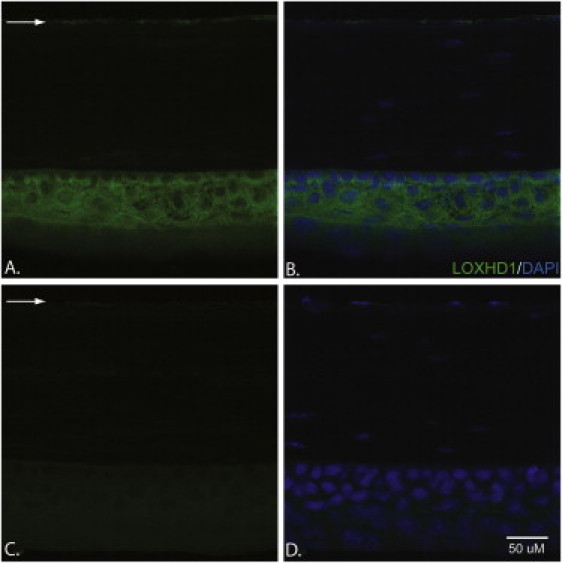
Immunohistochemical Analyses of LOXHD1 in 20 μm Cryosections of Mouse Cornea
(A and B) A slide stained with the LOXHD1 antibody (A) and a slide stained with both DAPI and the LOXHD1 antibody (B). LOXHD1 is present both in the corneal epithelium and in the endothelium; however, the level of LOXHD1 is much higher in the epithelium than in the endothelium.
(C and D) A slide stained with a LOXHD1 antibody +50× peptide (C) and a slide stained with both DAPI and a LOXHD1 antibody +50× peptide (D). The expression in the corneal epithelium and endothelium could be competed away with a commercially available LOXHD1 peptide.
The arrows point to the LOXHD1 present in the corneal endothelium.
We next asked whether we could observe LOXHD1 defects at the site of the pathology in the cornea of the proband. We were able to examine the distribution of LOXHD1 in the cornea, which was obtained at the time of corneal transplantation, of individual II:5 (indicated by an arrow in Figure 1). One control cornea was from a transplanted FCD patient who had been screened and found not to have any coding nonsynonymous variants in LOXHD1. Another control cornea was non-Fuchs tissue obtained from an individual who had been transplanted for keratoconus and who had normal corneal endothelium. Tissue was fixed with formaldehyde and embedded in paraffin. The 10 μm-thick sections were treated with rabbit polyclonal LOXHD1 antibody at a 1:250 dilution for 4 hr at room temperature or overnight at 4°C, and treatment with goat anti-rabbit IgG conjugated with AlexaFluor 594 (Invitrogen) followed. Slides were costained for nuclei with DAPI (in a 1:5000 dilution). Sectioned images were captured with a Zeiss LSM 710 Meta and were digitalized with Zen software.
Examination of corneal sections in which the genotype was masked showed a distinct increase in staining in the corneal endothelium and Descemet membrane of the proband with the c.1639C>T (p.Arg547Cys) LOXHD1 mutation (Figures 3A, 3D, and 3G)when compared to the non-LOXHD1 Fuchs (Figures 3B, 3E, and 3H) and keratoconus controls (Figures 3C, 3F, and 3I). Furthermore, we observed multiple aggregate staining in the corneal endothelium of HU-II-5 and a clearly distinguishable increase in protein abundance in the Descemet membrane, as well as increased thickness of the membrane—a pathognomonic hallmark of FCD. Together with our mutational data, these studies indicate that the LOXHD1 mutation has a causal role, most likely through a mechanism of protein aggregation in the endothelium and Descemet membrane, in the HU family.
Figure 3.
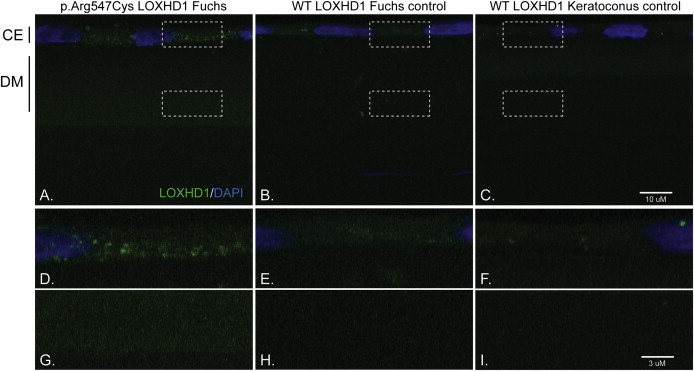
Immunohistochemical Analyses of LOXHD1 in Human Corneal Endothelium and Descemet Membrane
(A) Fuchs-affected individual harboring the c.1639C>T (p.Arg547Cys) LOXHD1 mutation.
(B) Fuchs-affected individual negative for any causal LOXHD1 mutation.
(C) Keratoconus-affected individual negative for Fuchs corneal dystrophy (note: panels D, E, F, G, H, and I are enlarged images of the boxed areas in panels A, B, and C). Aggregates of LOXHD1 are noted in the corneal endothelium and throughout the thickened Descemet membrane of a Fuchs patient harboring the c.1639C>T (p.Arg547Cys) LOXHD1 mutation. Few protein deposits in the corneal endothelium (“CE”) and Descemet membrane (“DM”) are seen in a Fuchs patient negative for any causal LOXHD1 mutation and in the patient transplanted for keratoconus. Note: The gain (exposure) is an order of magnitude higher in (C) than in (A) and (B).
To further investigate the contribution of LOXHD1 to the total genetic load of late-onset FCD, we interrogated our entire cohort of small nuclear families and sporadic cases. Recruitment, examination, and procedures for DNA-sample collection were approved by the institutional review boards for human subjects research at the Johns Hopkins University School of Medicine and were in accordance with the Declaration of Helsinki. All participating individuals gave informed written consent and underwent detailed ophthalmic examination, including slit-lamp biomicroscopy. Severity was graded on a modified scale according to Krachmer and colleagues; >12 central guttae in one eye was indicative of disease (grade 1) in individuals over 40 years of age.6
We sequenced all coding exons of LOXHD1 with Big Dye Terminator Ready Reaction Mix according to the manufacturer's (Applied Biosystems) instructions as described.13, 14 Primer pairs are available upon request. We identified 14 additional nonsynonymous coding variants (one variant, c.5272A>T [p.Thr1758Ser], was found in two sporadic affected individuals) and a missense variant, c.1904T>C (p.Leu635Pro), in the LOXHD1 splice isoform (RefSeq accession number NM_001145472) in our cohort of 207 unrelated subjects (Figure 4A and Table S2A). None of these alleles were present in 384 ethnically matched control chromosomes, and they were also not reported in the 1,000 Genome Project or the 1,500 exome project. Additionally, we found the mutated residues to be highly conserved in other LOXHD1 orthologs, and, with the exception of one variant, c.1945G>A (p.Asp649Asn), all are predicted to be damaging (Table S2A).
Figure 4.
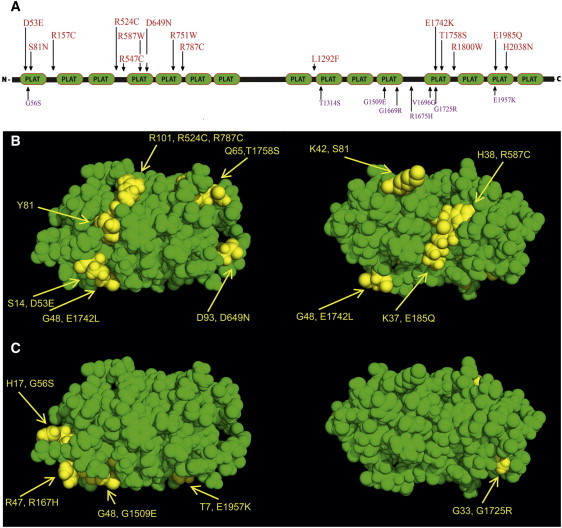
LOXHD1 Variants Identified in Late-Onset FCD Cases and Phenotypically Normal Control Samples
(A) Schematic representation of LOXHD1 variations identified in FCD cases (red) and phenotypically normal controls (purple). Also shown is the localization profile of the variations identified in FCD cases (B) and normal controls (C) in the homology model that was constructed with the crystal coordinates of human lipoxygenase. This model suggests that most of these residues that mutated in late-onset FCD lie on the surface of the protein. In addition to 15 variations, a missense variant, c.1904T>C (p.Leu635Pro), was identified in the LOXHD1 splice isoform (NM_001145472), and it was not present in 384 ethnically matched control chromosomes.
Despite this apparent enrichment of pathogenic alleles, sequencing data from the 1,000 Genomes Project and Seattle exomes suggested that multiple LOXHD1 variants that are absent from dbSNP are present in the general population; this suggestion raises the possibility that the alleles discovered in our sporadic cases represent chance events. To investigate this possibility, we sequenced all coding exons of LOXHD1 in 288 unaffected and unrelated control samples (576 chromosomes); we identified eight variants that were not present in any database. Nevertheless, the variants were predicted to be pathogenic by PolyPhen2 algorithms (Figure 4A and Table S2B). Furthermore, the 1,500 exome project had previously identified in 550 samples of European descent 12 variants that were predicted to be damaging by PolyPhen2. We therefore asked whether there were more damaging alleles in our cases than in our controls. Analysis of these data showed a significant enrichment (p = 0.003), which, together with the absence of these damaging alleles from ∼2,000 control chromosomes, suggests that the observed mutational load of this locus is relevant to FCD.
Cognizant of the imperfect predictive nature of PolyPhen and the relatively modest number of FCD cases sequenced, both of which could lead to false-positive results, we asked whether the three-dimensional structure of LOXHD1 displays nonrandom distribution of the private variants found in our cases. We therefore constructed a three-dimensional structure of LOXHD1 by using the coordinates of human lipoxygenase (Figures 4B and 4C). The degree of amino acid identity ranged between 26% and 39%, which is sufficient to ensure tertiary-structure alignment with human lipoxygenase.22 The model predicts that most of the mutations identified in our cases reside on the surface of the protein, making them more likely to affect the way in which the protein interacts with other proteins. This homology model further suggests that conservatively, 5 out of the 15 variants identified in our cohort, including the allele identified in family HU, reside in the interdomain linker regions. In contrast, most of the variants (seven out of eight) identified in our phenotypically normal controls are present in structured regions. These data predict that substituting amino acids that have aromatic or cyclic side chains for positively charged amino acids is bound to increase hydrophobicity of these linker regions and result in conformational changes, whereas the arginine to cysteine changes potentially allow for disulfide formation.
The structural model of the protein predictions is consistent with the detection of apparent aggregates in the cornea of the individual from family HU. To probe this possibility further, we expressed the HU mutation and two other randomly chosen alleles in ARPE-19, a cell line derived from the human retinal pigment epithelium, and we used expression of wild-type LOXHD1 as a negative control. We grew ARPE-19 cells in Dulbecco's Modified Eagle Medium and Ham's F-12 Nutrient 1:1 mixture (DMEM/F-12, Invitrogen) with 10% FBS and 2 mM L-glutamine. We used FuGene6 Transfection Reagent (Roche) to transfect the cells with plasmids harboring the wild-type Loxhd1 tagged at the N-terminus with GFP, plasmids harboring the HU mutation, or plasmids harboring two other alleles randomly picked from our cohort. Strikingly, analysis of ∼100 transfected cells masked to the transfection cocktail showed distinct cytoplasmic puncta in cells expressing each of the three mutants; cytoplasmic puncta were seen rarely in cells expressing the wild-type protein (Figure 5). Although we are cautious in interpreting data gathered from a heterologous in vitro system, these observations are consistent with both the endogenous findings in the affected cornea and our three-dimensional protein model.
Figure 5.
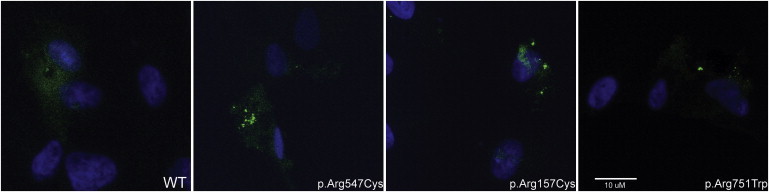
Localization of GFP-Tagged Wild-Type and Mutant LOXHD1
ARPE-19 cells were transfected with plasmids harboring (A) wild-type, (B) c.469C>T (p.Arg157Cys), (C) c.1639C>T (p.Arg547Cys), and (D) c.2251C>T (p.Arg751Trp) Loxhd1 tagged at the N-terminus with GFP. The wild-type LOXHD1 is present throughout the cytoplasm, whereas the p.Arg157Cys, p.Arg547Cys, and p.Arg751Trp mutant proteins show puncta in transfected cells.
LOXHD1 is an evolutionarily conserved protein predicted to consist of 15 PLAT (polycystin-1, lipoxygenase, alpha-toxin) domains. The biological function of PLAT domains is not well established, but it is posited that they target proteins to the plasma membrane.21 Although the exact mechanism of late-onset-FCD causality associated with LOXHD1 mutations identified in our cohort is not yet clear, the presence of marked precipitates in the corneas of our index family suggests that some fraction of FCD might be caused by aggregation defects similar to other late-onset degenerative disorders, such as limb-girdle muscular dystrophy and Parkinson disease.23,24 In that context, determining the composition of these aggregates might reveal important clues regarding pathomechanism. Furthermore, LOXHD1 appears to be expressed at low abundance in the cornea, whereas our results suggest that high levels of protein in cells induced aggregate formation, exacerbated further by the presence of mutations. These results raise the possibility that increased LOXHD1 concentrations might have long-term cytotoxic effects. Therefore, we speculate that transcriptional upregulation independent of coding-sequence mutations might also predispose individuals to late-onset FCD.
Finally, LOXHD1 is the second FCD locus involved in both corneal dystrophy and deafness; SLC4A11, a sodium-borate transporter, has been associated with both congenital hereditary endothelial dystrophy with progressive deafness (Harboyan syndrome: MIM 217400) and late-onset FCD.12,14,25 To date, two causal mutations, c.2008C>T (p.Arg670∗) and c.4714C>T (p.Arg1572∗), in LOXHD1 have been associated with hearing loss.21,26 Many of the affected individuals in our Fuchs cohort harboring rare LOXHD1 variants did self report hearing impairment, and recently, hearing disability was reported in FCD cases, suggesting an association between FCD and hearing disability.27 Nonetheless, we are cautious when inferring causal links from these observations, given that (1) FCD is a disorder typically diagnosed in people in their 5th or 6th decade of life (or older), and assessing comorbidities between two common traits is challenging, and (2) although we have observed significantly more coding LOXHD1 changes in our cases than in our controls, interpreting with confidence the causal potential of any single allele within that series is not possible. Exploring the possible links between common hearing deficits and FCD will require significant replication of our studies.
Acknowledgments
We thank all family members for their enthusiastic participation in this study. None of the contributing authors have any financial interest related to this work. We are thankful to Ulrich Mueller for providing the Loxhd1 construct. This study was supported in part by the National Eye Institute grant R01EY016835 (J.D.G.) and the Kwok Research Fund (J.D.G.). J.D.G. is a distinguished Margaret C. Mosher Professor of Ophthalmology. N.K. is a distinguished Brumley Professor.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Primer3, http://primer3.sourceforge.net
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Fuchs E. Dystrophia epithelialis corneae. Graefes Arch. Clin. Exp. Ophthalmol. 1910;76:478–508. [Google Scholar]
- 2.Wilson S.E., Bourne W.M. Fuchs' dystrophy. Cornea. 1988;7:2–18. [PubMed] [Google Scholar]
- 3.Klintworth G.K. The molecular genetics of the corneal dystrophies—current status. Front. Biosci. 2003;8:d687–d713. doi: 10.2741/1018. [DOI] [PubMed] [Google Scholar]
- 4.Lorenzetti D.W., Uotila M.H., Parikh N., Kaufman H.E. Central cornea guttata. Incidence in the general population. Am. J. Ophthalmol. 1967;64:1155–1158. [PubMed] [Google Scholar]
- 5.Mannis M.J., Holland E.J., Beck R.W., Belin M.W., Goldberg M.A., Gal R.L., Kalajian A.D., Kenyon K.R., Kollman C., Ruedy K.J., Cornea Donor Study Group Clinical profile and early surgical complications in the Cornea Donor Study. Cornea. 2006;25:164–170. doi: 10.1097/01.ico.0000164832.69668.4b. [DOI] [PubMed] [Google Scholar]
- 6.Krachmer J.H., Purcell J.J., Jr., Young C.W., Bucher K.D. Corneal endothelial dystrophy. A study of 64 families. Arch. Ophthalmol. 1978;96:2036–2039. doi: 10.1001/archopht.1978.03910060424004. [DOI] [PubMed] [Google Scholar]
- 7.Gottsch J.D., Sundin O.H., Rencs E.V., Emmert D.G., Stark W.J., Cheng C.J., Schmidt G.W. Analysis and documentation of progression of Fuchs corneal dystrophy with retroillumination photography. Cornea. 2006;25:485–489. doi: 10.1097/01.ico.0000178726.11693.14. [DOI] [PubMed] [Google Scholar]
- 8.Goar E.L. Dystrophy of the Corneal Endothelium (Cornea Guttata), with Report of a Histologic Examination. Trans. Am. Ophthalmol. Soc. 1933;31:48–59. [PMC free article] [PubMed] [Google Scholar]
- 9.Biswas S., Munier F.L., Yardley J., Hart-Holden N., Perveen R., Cousin P., Sutphin J.E., Noble B., Batterbury M., Kielty C. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum. Mol. Genet. 2001;10:2415–2423. doi: 10.1093/hmg/10.21.2415. [DOI] [PubMed] [Google Scholar]
- 10.Gottsch J.D., Sundin O.H., Liu S.H., Jun A.S., Broman K.W., Stark W.J., Vito E.C., Narang A.K., Thompson J.M., Magovern M. Inheritance of a novel COL8A2 mutation defines a distinct early-onset subtype of fuchs corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 2005;46:1934–1939. doi: 10.1167/iovs.04-0937. [DOI] [PubMed] [Google Scholar]
- 11.Gottsch J.D., Zhang C., Sundin O.H., Bell W.R., Stark W.J., Green W.R. Fuchs corneal dystrophy: Aberrant collagen distribution in an L450W mutant of the COL8A2 gene. Invest. Ophthalmol. Vis. Sci. 2005;46:4504–4511. doi: 10.1167/iovs.05-0497. [DOI] [PubMed] [Google Scholar]
- 12.Vithana E.N., Morgan P.E., Ramprasad V., Tan D.T., Yong V.H., Venkataraman D., Venkatraman A., Yam G.H., Nagasamy S., Law R.W. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum. Mol. Genet. 2008;17:656–666. doi: 10.1093/hmg/ddm337. [DOI] [PubMed] [Google Scholar]
- 13.Riazuddin S.A., Zaghloul N.A., Al-Saif A., Davey L., Diplas B.H., Meadows D.N., Eghrari A.O., Minear M.A., Li Y.J., Klintworth G.K. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am. J. Hum. Genet. 2010;86:45–53. doi: 10.1016/j.ajhg.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riazuddin S.A., Vithana E.N., Seet L.F., Liu Y., Al-Saif A., Koh L.W., Heng Y.M., Aung T., Meadows D.N., Eghrari A.O. Missense mutations in the sodium borate cotransporter SLC4A11 cause late-onset Fuchs corneal dystrophy. Hum. Mutat. 2010;31:1261–1268. doi: 10.1002/humu.21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sundin O.H., Jun A.S., Broman K.W., Liu S.H., Sheehan S.E., Vito E.C., Stark W.J., Gottsch J.D. Linkage of late-onset Fuchs corneal dystrophy to a novel locus at 13pTel-13q12.13. Invest. Ophthalmol. Vis. Sci. 2006;47:140–145. doi: 10.1167/iovs.05-0578. [DOI] [PubMed] [Google Scholar]
- 16.Sundin O.H., Broman K.W., Chang H.H., Vito E.C., Stark W.J., Gottsch J.D. A common locus for late-onset Fuchs corneal dystrophy maps to 18q21.2-q21.32. Invest. Ophthalmol. Vis. Sci. 2006;47:3919–3926. doi: 10.1167/iovs.05-1619. [DOI] [PubMed] [Google Scholar]
- 17.Riazuddin S.A., Eghrari A.O., Al-Saif A., Davey L., Meadows D.N., Katsanis N., Gottsch J.D. Linkage of a mild late-onset phenotype of Fuchs corneal dystrophy to a novel locus at 5q33.1-q35.2. Invest. Ophthalmol. Vis. Sci. 2009;50:5667–5671. doi: 10.1167/iovs.09-3764. [DOI] [PubMed] [Google Scholar]
- 18.Baratz K.H., Tosakulwong N., Ryu E., Brown W.L., Branham K., Chen W., Tran K.D., Schmid-Kubista K.E., Heckenlively J.R., Swaroop A. E2-2 protein and Fuchs's corneal dystrophy. N. Engl. J. Med. 2010;363:1016–1024. doi: 10.1056/NEJMoa1007064. [DOI] [PubMed] [Google Scholar]
- 19.Riazuddin S.A., McGlumphy E.J., Yeo W.S., Wang J., Katsanis N., Gottsch J.D. Replication of the TCF4 intronic variant in late-onset Fuchs corneal dystrophy and evidence of independence from the FCD2 locus. Invest. Ophthalmol. Vis. Sci. 2011;52:2825–2829. doi: 10.1167/iovs.10-6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y.J., Minear M.A., Rimmler J., Zhao B., Balajonda E., Hauser M.A., Allingham R.R., Eghrari A.O., Riazuddin S.A., Katsanis N. Replication of TCF4 through association and linkage studies in late-onset Fuchs endothelial corneal dystrophy. PLoS ONE. 2011;6:e18044. doi: 10.1371/journal.pone.0018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grillet N., Schwander M., Hildebrand M.S., Sczaniecka A., Kolatkar A., Velasco J., Webster J.A., Kahrizi K., Najmabadi H., Kimberling W.J. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am. J. Hum. Genet. 2009;85:328–337. doi: 10.1016/j.ajhg.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999;12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- 23.Xiong H., Wang D., Chen L., Choo Y.S., Ma H., Tang C., Xia K., Jiang W., Ronai Z., Zhuang X., Zhang Z. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Invest. 2009;119:650–660. doi: 10.1172/JCI37617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schröder R., Schoser B. Myofibrillar myopathies: A clinical and myopathological guide. Brain Pathol. 2009;19:483–492. doi: 10.1111/j.1750-3639.2009.00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vithana E.N., Morgan P., Sundaresan P., Ebenezer N.D., Tan D.T., Mohamed M.D., Anand S., Khine K.O., Venkataraman D., Yong V.H. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2) Nat. Genet. 2006;38:755–757. doi: 10.1038/ng1824. [DOI] [PubMed] [Google Scholar]
- 26.Edvardson S., Jalas C., Shaag A., Zenvirt S., Landau C., Lerer I., Elpeleg O. A deleterious mutation in the LOXHD1 gene causes autosomal recessive hearing loss in Ashkenazi Jews. Am. J. Med. Genet. A. 2011;155A:1170–1172. doi: 10.1002/ajmg.a.33972. [DOI] [PubMed] [Google Scholar]
- 27.Stehouwer M., Bijlsma W.R., Van der Lelij A. Hearing disability in patients with Fuchs' endothelial corneal dystrophy: Unrecognized co-pathology? Clin Ophthalmol. 2011;5:1297–1301. doi: 10.2147/OPTH.S23091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.