

Abstract
Manganese is essential for several metabolic pathways but becomes toxic in excessive amounts. Manganese levels in the body are therefore tightly regulated, but the responsible protein(s) remain incompletely known. We studied two consanguineous families with neurologic disorders including juvenile-onset dystonia, adult-onset parkinsonism, severe hypermanganesemia, polycythemia, and chronic hepatic disease, including steatosis and cirrhosis. We localized the genetic defect by homozygosity mapping and then identified two different homozygous frameshift SLC30A10 mutations, segregating with disease. SLC30A10 is highly expressed in the liver and brain, including in the basal ganglia. Its encoded protein belongs to a large family of membrane transporters, mediating the efflux of divalent cations from the cytosol. We show the localization of SLC30A10 in normal human liver and nervous system, and its depletion in liver from one affected individual. Our in silico analyses suggest that SLC30A10 possesses substrate specificity different from its closest (zinc-transporting) homologs. We also show that the expression of SLC30A10 and the levels of the encoded protein are markedly induced by manganese in vitro. The phenotype associated with SLC30A10 mutations is broad, including neurologic, hepatic, and hematologic disturbances. Intrafamilial phenotypic variability is also present. Chelation therapy can normalize the manganesemia, leading to marked clinical improvements. In conclusion, we show that SLC30A10 mutations cause a treatable recessive disease with pleomorphic phenotype, and provide compelling evidence that SLC30A10 plays a pivotal role in manganese transport. This work has broad implications for understanding of the manganese biology and pathophysiology in multiple human organs.
Introduction
Manganese (Mn) is one of the most abundant elements on earth, an essential trace metal in biology, and necessary for several metabolic pathways and cellular homeostasis.1 Manganese becomes toxic when present in excessive amounts. Manganese levels in the body are therefore tightly regulated, but the responsible protein(s) and pathways remain incompletely known.2
In normal conditions, most manganese amounts are absorbed in the intestine from dietary intake and excreted by the liver in the biliary system.1 Manganese intoxication might occur in different contexts, such as professional heavy exposure in miners, smelters, and welders;3 in individuals with chronic liver disease due to the failure of manganese hepatic clearance;4 and in individuals undergoing prolonged total parenteral nutrition, if excessive manganese amounts by-pass the hepatic filter and enter the bloodstream.5 More recently, manganese intoxication has been reported in drug-addicts exposed to this metal as a contaminant of the amphetamine-like drug ephedrone.6,7
During manganese intoxication the metal accumulates in the liver, muscle, and brain, particularly in the basal ganglia (globus pallidus and substantia nigra), and causes a neurologic disorder (manganism) characterized by early behavioral disturbances (also referred to as manganese madness) and a subsequent, prominent extrapyramidal syndrome, bearing similarities to dystonia (MIM 128100) and Parkinson disease (MIM 168600) (manganese-induced parkinsonism).8,9
The familial occurrence of severe hypermanganesemia and hepatic cyrrhosis, in combination with an extrapyramidal motor disorder and polycythemia, was described in 2008 in two young individuals born from consanguineous parents and suggests an autosomal-recessive disorder (MIM 613280).10 At least two other isolated individuals with similar phenotypes have been reported.11,12 Interestingly, in these individuals not only were all the known causes and contexts of manganese intoxication excluded, but the blood manganese levels were also much higher than those occurring in individuals with manganese intoxications due to known causes. These considerations suggest the presence of a primary disorder of the manganese metabolism, leading to hypermanganesemia and accumulation and secondary toxicity in the liver, bone marrow, and the nervous system. Here we report two unrelated families with this syndrome and the identification of recessive mutations in SLC30A10 (MIM 611146) as the disease cause.
Subjects and Methods
We studied two white consanguineous families (one of Italian and one of Dutch descent) with multiple affected siblings; a pattern compatible with autosomal-recessive inheritance (Figure 1). There was no history of neurological, hepatic, or hematological diseases in the previous generations. The parents of the affected individuals could not be personally examined by us because they were dead or unavailable. The study was approved by the appropriate institutional review board, and written informed consent was obtained from all subjects.
Figure 1.

Clinical and Neuroimaging Findings in Individuals with SLC30A10 Mutations
(A and G) Pedigrees of the Italian and the Dutch families. Black symbols denote affected individuals. The proband is indicated with an arrow.
(B–F) Brain MRI images in the individuals from the Italian family. In both brothers marked T1 hyperintensities, typically seen in persons with manganese intoxication, are present in MTC T1-weighted images in the globus pallidus, putamen, caudate nucleus, midbrain, and cerebellum, bilaterally (axial [B–C] and sagittal [E–F] images in individual SIE-03 [B and E] and SIE-02 [C and F]. In (D), a 3DFFET1-weighted, coronal oblique multiplanar reconstruction image shows hyperintensity along the cerebello-rubro-thalamic pathways in individual SIE-03.
(H and I) Brain MRI images in the individuals from the Dutch family. In individual NIJ005, marked hyperintensities are present in T1-weighted images in the globus pallidus, bilaterally (coronal image [H]); in individual NIJ010, T1-weighted images obtained around the time the manganese blood levels had returned at almost normal levels showed no obvious abnormalities (axial image shown in [I]).
DNA Studies
Genomic DNA was isolated from peripheral blood with standard protocols. We performed genome-wide homozygosity mapping in three affected individuals (two from the Italian family and one from the Dutch family). Genome-wide genotyping was performed with the Illumina HumanOmniExpress BeadChip arrays (730,525 SNPs at a median distance of 2.1 kb). Homozygosity mapping was performed with GenomeStudio 2011,V2011.1 (Illumina, San Diego, CA) and Nexus Copy Number, Discovery Edition, ver. 5.1 (BioDiscovery, El Segundo, CA).
We then performed exome sequencing in one Dutch affected individual (NIJ005, IV-3 in Figure 1G) by using in-solution capturing (Agilent SureSelect Human All Exon 50Mb Kit, Agilent Technologies, Santa Clara, CA) and Illumina HiSeq, paired-end sequencing, at an average of 30× coverage. Reads were aligned to the human reference genome (hg19 assembly) with the Burrows-Wheeler-Aligner (BWA, version 0.5.9c). Single-nucleotide variants and small insertions and deletions (indels) were annotated with the Genome Analysis Tool Kit (GATK, version 1.0.5777).
Last, we sequenced all the exons and exon-intron boundaries of one positional candidate (SLC30A10) by using Sanger protocols. Primers and PCR protocols are reported in Table S1, available online. The variants detected were tested in ethnically matched healthy controls by Sanger sequencing. The SLC30A10 variants are named according to the GenBank accession numbers (NM_018713.2 and NP_061183.2) and numbered at nucleotide level from the “A” of the ATG-translation initiation codon.
Immunohistochemistry
Human brain and spinal cord tissues were obtained from The Netherlands Brain Bank, Netherlands Institute for Neuroscience, Amsterdam, The Netherlands. Human normal liver tissue was obtained from the Department of Pathology, Erasmus MC, Rotterdam. All material has been collected from donors from whom a written informed consent for autopsy and the use of the material and clinical information for research purposes had been obtained. We also studied specimens from a liver biopsy performed in 1981 in one affected individual from the Dutch family with SLC30A10 mutations (NIJ012, IV-2 in Figure 1G).
For immunohistochemistry, paraffin-embedded sections (7 μm) were analyzed from the cerebral cortex, globus pallidus, cerebellum, and spinal cord of two normal male donors (their ages at death were 76 and 81 years). Briefly, dewaxed sections were pretreated for antigen retrieval by pressure cooking in 0.1 M sodium citrate buffer (pH 6) for 5 min. Subsequently, an indirect immunoperoxidase labeling was performed with goat polyclonal antibody against SLC30A10 (Santa Cruz, 1/50) as a primary antibody and anti-goat conjugated with HRP (1/100; DAKO) as a secondary antibody. Sections were counterstained with heamatoxylin and mounted with entellan.
Cell Culture and Immunoblotting
HepG2 (hepatocellular carcinoma) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Lonza) supplemented with 10% fetal calf serum, 50 U/ml penicillin and 50 mg/ml streptomycin, in a humidified 5% CO2 incubator. Twenty-four hours after seeding, the cells were treated with sublethal concentrations of MnCl2 (100μM) or H2O (blank) for 5 hr. They were then suspended in HES buffer (20 mM HEPES [pH 7.4], 1 mM EDTA, 250 mM sucrose, protease inhibitor cocktail [Roche Molecular Biochemicals]) and homogenated by sonication. After spindown at 100,000 g for 20 min at 4°C, the pellet was resuspended in HES buffer, and the protein concentration was determined by using the bicinchoninic acid (BCA) protein assay kit (Pierce) according to the manufacturer's instructions. The protein samples (80 μg) were separated by 6%–12% Criterion XT, 4%–12% Bis-Tris Gel (Bio-Rad) and then transferred to nitrocellulose membranes. Membranes were blocked with 5% low-fat milk powder (Fluka) in 1× PBS containing 0.1% Tween 20 (PBST) for 1 hr at room temperature and incubated overnight at 4°C with goat polyclonal antibody against SLC30A10 (Santa Cruz, 1/500) and mouse monoclonal against actin (Sigma, 1/2,000). After washing three times with PBST, the membranes were incubated in the dark for 1 hr with PBST containing donkey anti-goat or anti-mouse secondary antibodies (700 nm or 800 nm, LI-COR Biosciences Lincoln, 1/5,000 dilution). After washing, the membranes were scanned with the Odyssey Infrared Imager (Li-COR Biosciences).
RT-qPCR Studies
HepG2 cells were cultured as mentioned before. Twenty-four hours after seeding, HepG2 cells were treated with sublethal concentrations of MnCl2 (200 μM), ZnCl2 (100 μM), or buffer (blank) for 5 hr. After treatment, cells were harvested, and total RNA was isolated with RNAzol B (Cinna/Biotecx). Complementary DNA (cDNA) was prepared from approximately 200 ng total RNA with IScript cDNA synthesis kit (Bio-Rad) in a total volume of 40 μl following the manufacturer's instructions. For RT-qPCR experiments, SLC30A10 and YWHAZ expression were compared to the expression of a reference gene, TBP. Specific primers used are reported in Table S1. Two assays were designed for SLC30A10 (termed SLC30A10_Q1 and SLC30A10_Q2). Both RT-qPCR assays were validated by the demonstration of linearity over two orders of magnitude and by observation of a single melt peak by plotting relative fluorescence units (RFU) data with time (T) (−d(RFU)/dT) on the y axis as a function of temperature on the x axis. Reactions (15 μl) contained 200 nM primer, 1X KAPA SYBR FAST qPCR Master Mix, and 0.5 μl cDNA. All reactions were performed in triplicates on a Bio-Rad CFX96 Real-Time System with the following cycle conditions: 3 min initial denaturation at 94°C followed by 30 cycles of 5 s each at 94°C denaturation and 30 s 60°C primer annealing, extension, and RFU data collection. Data were analyzed with CFX Manager Software V1.5 (Bio-Rad). Statistical analysis with TBP/SLC30A10 and TBP/YWHAZ ΔCt values from each sample was done via one-way analysis of variance ([ANOVA] Tukey Honestly Significant Difference [HSD] test).
In Silico Analyses
The transmembrane regions in SLC30A10 and their orientation in the membrane were predicted with the TMHMM Server v. 2.0 and the TMpred program. The closest SLC30A10 homologs were identified by blasting the human sequence with the BLASTp program. Retrieved sequences were saved in FASTA format and aligned with the program ClustalW. GenBank accession numbers for the SLC30A1 and SLC30A10 homologs are reported in the Table S2.
Results
Clinical Studies
The most important clinical features are summarized in Figure 1 and Table 1. In the Italian family, two brothers, born from healthy first cousins, developed adult-onset parkinsonism, hypermanganesemia, polycythemia, and chronic liver disease. The proband (individual SIE-03, IV-1 in Figure 1A), a 60-year-old man, was referred because of a 3 year history of progressive gait disturbances and bradykinesia. Treatment with pramipexole (1.05 mg/day) had failed to improve parkinsonism. His past history revealed he developed thrombocytopenia and polycythemia in his 30s. Our neurological examination showed moderate hypomimia, monotone speech, moderate upper limbs bradykinesia, and mild rigidity (more severe on the right than on the left), global bradykinesia, wide-based gait with freezing and starting hesitation, and moderate postural instability (Movie S1). There was no evidence of tremor, dystonia, or cerebellar or pyramidal disturbances. The Unified Parkinson Disease Rating Scale13 motor score, part III (UPDRS-III) was 27 (maximum 108). Aside from polycythemia (red cell count 6.28 × 1012/mmc, normal range [n.r.] 4.10–5.80) and mild thrombocytopenia (136 × 109/mmc, n.r. 150–400) routine blood tests (including liver functions parameters) were unremarkable. Abdominal ultrasound showed hepatomegaly with liver steatosis and splenomegaly. Alcohol consumption or high-fat diet were not referred. Electrophysiological investigation showed a sensory-motor axonal polyneuropathy. T1-weighted brain magnetic resonance imaging (MRI) showed bilateral, symmetrical hyperintensies at the level of the caudate and lentiform nuclei, thalamus, cortico-spinal tract, medial cerebral peduncle, substantia nigra, posterior pons, and bulbar olives (Figures 1B, 1D, and 1E).
Table 1.
Clinical and Biochemical Findings in Individuals with SLC30A10 Mutations
|
Individual Code |
|||||
|---|---|---|---|---|---|
| SIE-02 | SIE-03 | NIJ005 | NIJ010 | NIJ012 | |
| Gender | male | male | male | male | female |
| Age (last examination, years) | 59 | 60 | 63 | 65 | 46 a |
| Presentation | neurologic | neurologic | neurologic | neurologic | neurologic/hepatic |
| Symptoms onset (years) | 47 | 57 | 2 | 14 | 10 |
| Blood Mn levelsb (unit of measurement); normal range | 104.0 (mcg/l); 3.0–8.0 | 106(mcg/l); 3.0–8.0 | 231.6 (nmol/l); <32.8 | 2626 (nmol/l); 183–352 | n.a. |
| Polycythemia | + | + | + | + | + |
| Dystonia | - | - | + | + | - |
| Parkinsonism | + | + | - | - | - |
| Hepatomegaly | + | + | - | - | + |
| Liver ultrasound | steatosis | hyperplasia | normal | normal | n.a. |
| Liver biopsy | n.a. | n.a. | n.a. | n.a. | cirrhosis |
| Liver serum parameters | normal | normal | normal | normal | n.a. |
| MRI-T1 abnormalities | + | + | + | + | n.a. |
| Unremarkable investigations | EEG, MRS, SPECT, EMG, ENG, HV serology | EEG, MRS, SPECT, HV serology | |||
The following abbreviations are used: n.a., not available; EEG, electroencephalogram; MRS, brain magnetic resonance spectroscopy; EMG, electromyography; ENG, nerve conduction studies; HV serology, negative serology for hepatitis A, B, and C; SPECT, brain ([123I]-FP-CIT) single photon emission computed tomography of the dopamine transporter. A normal striatal uptake of this tracer indicates the integrity of the nigrostriatal dopaminergic pathways.
Deceased.
The Mn levels were measured by different laboratories and in different times in the different individuals. Thus, different units of measurement and normal ranges are reported.
Plasma ceruloplasmin and urinary copper excretion were normal, whereas plasma copper was slightly increased (1317.79 mcg/l, n.r. 500–1250). Increased blood transferrin (545 mg/dl, n.r. 200–360) and decreased ferritin (16 ng/ml, n.r. 30–400) were also found. Blood and urinary manganese concentrations were markedly increased (106 mcg/l, n.r. 3.0–8.0; 16.0 mcg/l, n.r. 0.5–4.0). Also, muscle manganese content was elevated (0.73 mcg/g; normally undetectable). All possible sources of manganese exposure were excluded. A 5 day course of disodium calcium edetate infusion at a dose of 20 mg/kg twice daily yielded a marked increase of urinary levels of manganese, whereas the blood manganese increase persisted. A clear improvement of the extrapyramidal symptoms was also observed, and the UPDRS-III score decreased from 27 to 13 (Movie S2). This individual was therefore started on monthly 5 day courses of chelation therapy with disodium calcium edetate. After 4 months of treatment, manganese urinary excretion was still markedly elevated (219.0 mcg/l, n.r. 0.5–4.0) and blood manganese levels returned to the normal range (4.2 mcg/l, n.r. 3.0–8.0). Serial neurological evaluation showed progressive improvement in gait, bradykinesia, and posture. The UPDRS-III score at the 5 month follow up was 5/108 (Movie S3). Chelation therapy was well tolerated and did not alter renal function parameters. Longitudinal clinical and biochemical data in this individual are summarized in the Table 2.
Table 2.
Clinical and Biochemical Follow Up in Individual SIE-03 during Monthly 5 Day Courses of Chelation Therapy with Disodium Calcium Edetate, 20 mg/kg twice/day
| 24 Hr Urine Mn (mcg/l) | Blood Mn (mcg/l) | UPDRS-III | Walking Test, 3 m (s) | |
|---|---|---|---|---|
| Baseline | 1.06 | 106.0 | 27 | 18 |
| Month 1 | 30.0 | 133.0 | 13 | 13 |
| Month 2 | 76.0 | 101.0 | 10 | 10 |
| Month 3 | 216.0 | 90.0 | 9 | 10 |
| Month 4 | 219.0 | 4.2 | 8 | 8 |
UPDRS-III is an abbreviation for Unified Parkinson Disease Rating Scale, part III (motor score). Higher scores correspond to more severe parkinsonism; the maximum score is 108.
The younger brother (individual SIE-02, IV-2 in Figure 1A) was a 59-year-old man presenting with a 12 year history of gait difficulties, postural instability with frequent falls, and hypomimia. Polycythemia was diagnosed 2 years earlier. Neurological symptoms did not improve after administration of levodopa/carbidopa (100 + 25 mg, twice a day). On admission, a neurological examination showed mild hypomimia, slurred speech, a wide-based gait with freezing and festination, and postural instability. Formal UPDRS scoring was not applicable because of the marked genu recurvatum that severely invalidated gait and posture. Routine blood tests confirmed polycythemia (red cell count 5.95 × 1012/mmc, normal range 4.10–5.80). An abdominal ultrasound showed mild hepatomegaly with hepatic nodular hyperplasia but no splenomegaly. Alcohol consumption and high-fat diet were not referred. A T1-weighted brain MRI showed bilateral, symmetrical signal hyperintensies with the same distribution as was seen in his brother (Figures 1C and 1F). Markedly increased blood and urinary manganese concentrations (104.0 mcg/l, n.r. 3.0–8.0 and 6.5 mcg/l, n.r. 0.5–4.0, respectively) were found. Manganese content in the muscle was also elevated (0.82 mcg/g, normally absent). Chelation therapy (disodium calcium edetate) was also started with referred clinical improvement without side effects. However, detailed longitudinal data could not be collected because this individual refused follow-up examination.
In the Dutch family, three siblings born from healthy first cousins all suffered from polycythemia from early childhood, requiring regular phlebotomies. In the oldest brother (individual NIJ010, IV-1 in Figure 1G), now 65 years old, neurological problems started at age 14 when he experienced painful limb spasms on walking. Over the years, he developed generalized dystonia and became wheelchair dependent. Except for dystonia, the neurological examination at age 63 showed no other abnormalities (Movie S4). He was recently also diagnosed with multiple myeloma (Kahler's disease) (MIM 254500), a common hematologic malignancy. His liver function and liver and spleen ultrasound were normal. Of note, only a borderline increase of manganese blood levels was reported at recent evaluations (41.7 nmol/l, n.r. < 32.8), and a brain MRI showed no gross abnormalities (Figure 1I). However, markedly increased manganese levels (up to 2626 nmol/l, n.r. 183–352) were documented in the past. Iron levels were normal, whereas total iron binding capacity was mildly elevated (101 μmol/l, n.r. 45–80).
In his younger brother (individual NIJ005, IV-3 in Figure 1G), now 63 years of age, walking difficulties became apparent at the age of 2. Progressive dystonia led to wheelchair dependency at the age of 34. Manganese levels were severely elevated (231.6 nmol/l, n.r. < 32.8). Iron and ferritin levels were normal, whereas total iron binding capacity was also mildly elevated (114 μmol/l, n.r. 45–80). Liver function parameters were normal, as was a liver ultrasound. Spleen ultrasound data were not available. The brain MRI showed hyperintense changes of the pallidus on T1-weighted images (Figure 1H). At the age of 61, neurological examination showed a rather pure, generalized dystonia except for some sequelae of a cervical disc herniation, such as hypesthesia and weakness in the C5–C6 district and an extensor plantar response on the left. Currently, he is in a nursing home because of alcohol-related cognitive and behavioral changes.
Their younger sister (individual NIJ012, IV-2 in Figure 1G) died at the age of 46 because of decompensated liver cirrhosis, portal hypertension, splenomegaly, and hepatic encephalopathy. Information about her neurological conditions is limited. She developed gait difficulties at the age of about 10. At the age of 35, the neurological examination showed gait disturbance and leg sensorimotor disturbances with absent tendon reflexes, without clear documentation of dystonia. A liver biopsy at age 31 showed micro- and macronodular cirrhosis with depositions of metallic- and protein-bound copper. Manganese levels have never been determined, and neuroimaging was never performed.
Genetic Studies
High-density SNP-array genotyping revealed only one locus in the genome, on chromosomal region 1q41-q42, showing extended homozygosity in all the three studied affected individuals (Figure 2 and Figures S1–S2). In individual SIE-02 the homozygous region was flanked by heterozygous SNPs at positions 204188482 and 238837410; in individual SIE-03 the homozygous region was smaller and flanked by heterozygous SNPs at positions 214699571 and 238837450. The analysis of individual NIJ005 narrowed down further the critical region to 12.1 Mb of homozygous sequence, flanked by heterozygous SNPs at positions 215429271 and 227575528 and containing 106 annotated genes.
Figure 2.

Homozygosity Mapping and Detection of SLC30A10 Mutations
(A) Genome-wide SNP arrays scan defines one large homozygous region shared by the affected individuals from the Italian and the Dutch families on chromosomal region 1q41-q42.
(B) Genomic organization of SLC30A10.
(C) Electropherogram showing the SLC30A10 mutations present in the Dutch affected individuals NIJ005 and NIJ010.
(D) Electropherogram showing the SLC30A10 mutation present in the Italian affected individuals SIE-02 and SIE-03.
To identify the disease-causing variant, we initially performed a mutational analysis of the coding regions of all of these 106 genes by exome sequencing in individual NIJ005. However, this approach did not detect any homozygous exonic or splice-site variants, which were not annotated in dbSNP132 or in the 1000 Genomes Project, in the 106 genes located in the critical region (Table S3). Interestingly, one of these genes, termed solute carrier family 30, member 10 (SLC30A10), encodes a poorly characterized protein bearing homology to the SLC30 family of zinc cation transporters (GenBank accession numbers NM_018713.2 and NP_061183.2). Thus, SLC30A10 represented an excellent candidate for this phenotype because the body levels of another divalent cation (manganese) are severely altered in these individuals, which could be because of a defect in the transport of manganese. Interestingly, the inspection of the exome data in individual NIJ005 revealed a very low coverage of the SLC30A10 exon 1 (a very GC-rich region) (Table S4).
We therefore undertook Sanger sequencing of the SLC30A10 coding region. Other genes within the critical region were not analyzed by Sanger sequencing. Different homozygous mutations were detected in the affected members of the Italian and Dutch families and predicted to lead to frameshift and premature protein truncation (Figure 2). In the Dutch individuals NIJ005 and NIJ010 (IV-3 and IV-1 in Figure 1G), a homozygous c.507delG mutation is present in exon 1, predicted to cause frameshift for 21 amino acids, followed by a premature stop at codon 191, thereby removing a large portion (295 amino acids) of SLC30A10 (p.Pro170Leufs∗22). In addition, these individuals also carry a homozygous substitution in exon 1, c.500T>C, encoding the p.Phe167Ser alteration resulting from the missense mutation in SLC30A10. In the Italian affected individuals SIE-02 and SIE-03 (IV-2 and IV-1 in Figure 1A), a homozygous c.1235delA mutation is present in exon 4 and predicted to cause a frameshift for 25 amino acids, followed by a premature stop at codon 437 that removes the last 49 amino acids of SLC30A10 (p.Gln412Argfs∗26). Unfortunately, DNA from the third affected member in the Dutch family (NIJ012, IV-2 in Figure 1G) and the unaffected parents in both families was not available. Furthermore, RNA, which could be used to test whether the mutant transcripts undergo nonsense-mediated mRNA decay, was not available from any members of the two families.
These mutations were not present in dbSNP132, the 1000 Genomes Project, or in the Exome Variant Server database. We also tested 996 chromosomes from Italian control individuals for the mutation found in the Italian affected individuals, and 668 control chromosomes (294 Dutch and 374 Italian) for the mutations present in the Dutch affected individuals. None of these mutations was detected. Furthermore, only one additional heterozygous variant was detected, in one Italian control individual (c.1273G>A, predicted to lead to the p.Val425Ile alteration resulting from the missense mutation, which is not expected to be pathogenic), highlighting a high conservation of SLC30A10 coding sequence.
Immunohistochemistry Studies
SLC30A10 spans 14,388 nucleotides on chromosomal region 1q41 (GenBank NM_018713.2). The principal transcript contains four exons for a total of 2,854 nucleotides, and it encodes an ∼52 kD protein of 485 amino acids, SLC30A10. In humans, the highest levels of expression of SLC30A10 mRNA are documented in the liver and the central nervous system (CNS), but data concerning the SLC30A10 protein are lacking. In order to characterize the localization of SLC30A10, we performed immunohistochemistry in normal human liver and the central nervous system and in the liver biopsy from one of the Dutch affected individuals by using a commercially available polyclonal antibody (Figure 3). In the normal liver, a punctuate pattern of immunoreactivity was detected in the cytoplasm of the hepatocytes (Figure 3A) and in the epithelium of bile ducts (Figure 3C); here, the plasma membrane facing the lumen of the bile ducts was also strongly immunoreactive. The above-mentioned staining pattern is compatible with localization of SLC30A10 in different compartments of the secretory pathway (including the Golgi system, endosomes, and the plasma membrane). Of note, immunoreactivity was almost absent in the liver tissue from the affected individual (Figures 3B and 3D), confirming the severe, deleterious effect of the mutations. Also of note, in the nervous system, the strongest immunoreactivity with similar cytoplasmic punctuate pattern was detected in the neurons in the globus pallidus (Figures 3E and 3F). However, other neurons, such as the motoneurons of the spinal cord were also immunoreactive (Figure 3G).
Figure 3.
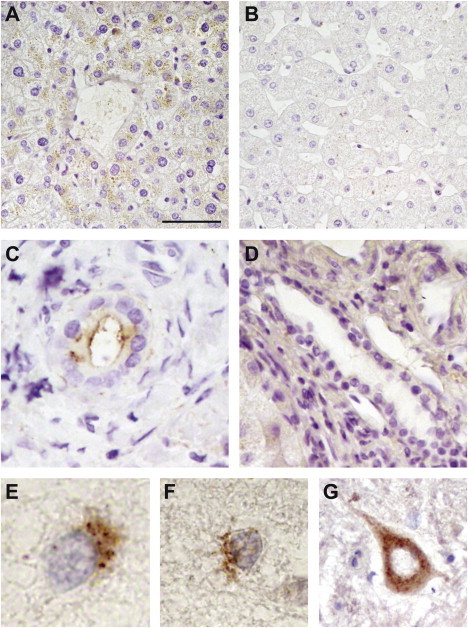
Localization of SLC30A10 in the Liver and Nervous System
Immunohistochemistry was performed with an anti-SLC30A10 antibody (Santa Cruz); the images shown are from normal liver tissue (A and C); a liver biopsy from the NIJ012 affected individual (B and D); neurons from globus pallidus (E and F) and spinal cord (G) of a normal donor. In the liver, immunoreactivity is present in the normal hepatocytes (A) and normal bile ducts (C), but absent in the tissue from the affected individual (B and D). The scale bars represent 16.5 μm for all panels.
In Silico and In Vitro Studies
SLC30A10 belongs to the cation diffusion facilitator (CDF) superfamily of metal transporters. CDF proteins transport divalent metal cations, such as iron, zinc, copper, nichel, cobalt, manganese, and cadmium.14 Most CDF transporters share a common topology, which is also predicted for SLC30A10 by our in silico analyses, and consist of six transmembrane domains (TMD), cytosolic N and C termini, and a conserved C-terminal domain termed the cation efflux domain (Pfam 01545) (Figure 4A). Most CDF proteins function as antiporters, exchanging divalent cations with H+ or K+ ions, and catalyzing the efflux of metals from the cytosol to the extracellular space or to subcellular organelles.15 Within the CDF superfamily, zinc-specific transporters are more abundant and better characterized than manganese-specific ones. SLC30A10 displays the highest homology to SLC30A1 (Znt-1), a plasma membrane zinc transporter.16 However, we noted that key structural features shared by known zinc transporters14,17 are not present in SLC30A10 and its homologs (Figure 4B). Indeed, the SLC30A1 group of zinc transporters share two conserved HxxxD motifs (where x = any amino acid) located in the TMD II and V and a histidine-rich region, either in the cytosolic loop between TMD IV and V or at the N or C termini. In the human SLC30A10 and its homologs in other species, the conserved histidine in the HxxxD motif in TMD II is consistently replaced by an asparagine. Moreover, the histidine-rich region is absent, replaced by a sequence rich in arginines, lysines, and serines. Another CDF member, the plant ShCDF8 protein from Stylosanthes hamata known to be involved in manganese tolerance, also lacks the histidine-rich region.18 Conversely, some key features present in other CDF members known to be involved in manganese transport are consistently present in the SLC30A10 cluster (Figure 4B).18 For example, a very conserved cysteine residue at the cytosolic side of TMD IV is characteristic of manganese-specific transporters, and it is also consistently present in the SLC30A10 cluster (and not in the SLC30A1 cluster). These arguments support strongly the contention that SLC30A10 and its homologs differ functionally from the zinc transporters and are specific for a different cation substrate, most likely manganese.
Figure 4.
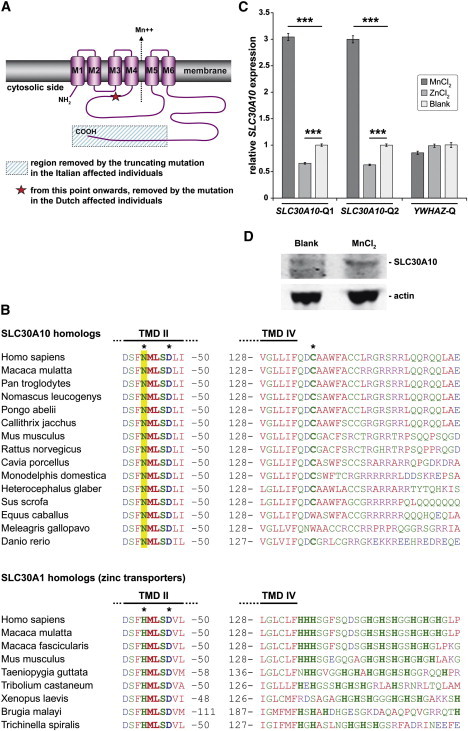
In Silico Analysis of SLC30A10 and In Vitro Expression Studies
(A) Schematic topology of SLC30A10. The predicted effects of the disease-causing mutations are depicted.
(B) Alignment of the SLC30A10 and the SLC30A1 homologs.
(C) The expression of SLC30A10 is induced in hepatocellular carcinoma cells (HepG2) after exposure to sublethal concentrations of manganese. The cDNA levels of the target gene (SLC30A10) and those of an unrelated gene (YWHAZ) are compared to those of a reference gene (TBP). Two assays are performed for SLC30A10 (Q1 and Q2) yielding similar results. The data shown represent means ± standard error of the mean of experiments performed in triplicates. The statistical analysis that used TBP/SLC30A10 and TBP/YWHAZ ΔCt values from each sample was done with one-way ANOVA (Tukey HSD test). ∗∗∗p < 0.001
(D) Immunoblotting analysis of HepG2 cells shows increased abundance of a band of the predicted size of SLC30A10 after exposure of the cells to sublethal concentrations of MnCl2. Actin is used as a loading control.
The expression of several CDF transporters is under transcriptional control by the concentration of their cation substrate. For example, the expression of SLC30A1 (MIM 609521) is strongly induced by extracellular zinc.19,20 In order to explore the mechanism regulating SLC30A10, we exposed HepG2 hepatocellular carcinoma cells to sublethal levels of manganese or zinc. After 5 hr of manganese exposure, a marked 3-fold increase in the SLC30A10 mRNA expression was detected (Figure 4C). Immunoblotting analysis (Figure 4D) confirmed an increased abundance of a band corresponding to the size of SLC30A10 (∼52 kD). Exposure to zinc led to a small decrease (∼40%) in SLC30A10 expression. These data document robust expression of SLC30A10 and the presence of the encoded protein in human cells of hepatic origin, and, more importantly, they show that the SLC30A10 is under tight control by the extracellular manganese levels. Taken together, our results provide, therefore, functional evidence that SLC30A10 is a bona fide manganese transporter in humans.
Discussion
The discovery of two different homozygous, clearly deleterious mutations in a highly conserved protein that cosegregate with disease in two independent families and are absent in large numbers of control chromosomes establishes that the loss of the function of SLC30A10 is the cause of this disease.
Detailed quantitative RT-PCR profiling in rodents documented that the highest expression of the SLC30A10 ortholog was in the liver, followed by the intestine, and then by the CNS.21 In humans, there is evidence of expression of SLC30A10 in the liver, the CNS, and other organs in both fetal and adult life (Unigene expressed sequence tag profile at National Center for Biotechnology Information).22 Furthermore, microarray data on the regional expression of SLC30A10 in the human brain are available online in the ALLEN Human Brain Atlas. In the Atlas, SLC30A10 expression levels are highest in the globus pallidus, subthalamic nucleus, and deep cerebellar nuclei and lower in the putamen and other diencephalic and cortical areas, whereas low expression levels are documented in the hippocampus and cerebellar cortex. Our immunohistochemistry studies in normal human liver and CNS provide data on the localization of SLC30A10. Furthermore, we document the dramatic loss of the protein immunoreactivity in the liver biopsy from one of the affected individuals in the family with SLC30A10 mutations. These data are in substantial agreement with the mRNA expression profile data, and taken together, they fit well with the profile of organs involved in the metabolism of manganese (particularly the liver) and with the phenotype associated with loss-of-function mutations in the gene.
Manganese is an essential cation for the function of several enzymes, including some crucially important for the metabolism of neurotransmitters and other neuronal metabolic pathways, such as glutamine synthetase, the mitochondrial superoxide dismutase 2 (SOD2), arginase, and pyruvate decarboxylase.2 Excessive levels of manganese induce cell toxicity by a multitude of effects, including competition with other cations for binding to key proteins, effects at the level of DNA replication and transcription, oxidative stress, interference with the mitochondrial oxidative phosphorylation, inhibition of the complex I, ATP depletion, and apoptotic cell death.1 There is also evidence of direct effects of manganese excess on different neurotransmitter systems, including GABAergic, glutamatergic, and dopaminergic (also implicated in basal ganglia dysfunctions, such as dystonia, and parkinsonism).1
Manganese levels in the body must therefore be tightly regulated, and several transporters have been proposed to play a role.1,2 Yet, the exact mechanisms of manganese transport have not been conclusively determined. In particular, a protein responsible for manganese efflux from the cytosol has not been identified yet. Our findings nominate SLC30A10 as a very important manganese transporter in man and other species.
The high expression of SLC30A10 and its encoded protein in the human liver is in keeping with a predicted important role for this organ in the manganese homeostasis.21 In individuals with homozygous loss-of-function SLC30A10 mutations, the manganese excretion is severely defective, leading to metal accumulation in the liver, the bloodstream, the brain, and other peripheral tissues. The polycythemia is likely secondary to hypermanganesemia because manganese is known to act on the erythropoietin system in a manner similar to hypoxia,23 and polycythemia was reported to resolve after chelation therapy.10 The hypermanganesemia is by itself a cause of brain toxicity, as shown in the persons with professional manganese intoxications3 or those with chronic liver disease.4 However, in individuals with SLC30A10 mutations, we speculate that the consequences of hypermanganesemia for the brain might be particularly severe because SLC30A10 is also highly expressed in the brain, particularly within the basal ganglia, probably to prevent the neurotoxic effects of manganese excess there.
Thus, the metabolic signature in individuals with SLC30A10 mutations is the extreme hypermanganesemia with secondary polycythemia. Furthermore, the disease leads to a variety of neurological or hepatic disturbances that vary in severity and onset age, even within the members of the same family. The neurological presentation ranges from juvenile-onset dystonia (in the Dutch family reported here and a family reported previously)10 to late-onset akinetic-rigid parkinsonism (in the Italian family reported here). Additional neurological pictures might include juvenile-onset spastic paraparesis, as reported previously in another individual.11 Interestingly, the mutations present in our individuals with juvenile-onset dystonia are predicted to have more deleterious effects on SLC30A10 than the mutations found in our individuals with late-onset parkinsonism (Figure 4A), suggesting a possible source of genotype-phenotype correlation. However, marked intrafamilial variability is also present. Two of the three Dutch sibs had severe, generalized dystonia, whereas the third had no obvious extrapyramidal involvement. Also, liver cirrhosis eventually led to early death in one of the Dutch sibs (neurologically the least affected), but liver function was completely normal in the other two. The Italian affected individuals had only asymptomatic hepatic involvement detected by ultrasound. A similar wide variability in types of neurological and hepatic presentation and clinical severity is characteristic of Wilson's disease (MIM 277900), a disorder of copper transport.24 Whether the SLC30A10 mutations, the hypermanganesemia, or both, played a role in the pathogenesis of the multiple myeloma developed in individual NIJ010 (IV-1 in Figure 1G) remains unknown, but it cannot be excluded.
Of note, chelation therapy was able to normalize the manganese blood levels in the Italian individual and led to dramatic clinical improvement of this otherwise potentially lethal disease. Similar beneficial effects were previously reported in another individual.10 An early diagnosis is therefore crucial, and genetic testing of SLC30A10 is warranted in individuals with the neurological or hepatic presentation, or in those with polycythemia, in combination with hypermanganesemia. Variability in SLC30A10 might also modulate susceptibility in subjects with professional exposure to manganese, and further studies are therefore warranted in this direction.
In conclusion, our identification of SLC30A10 mutations as the cause of this pleomorphic syndrome delineates it as a primary disease of manganese metabolism and identifies SLC30A10 as a critical regulator of the manganese transport. Our findings have important implications for understanding the physiology of this essential metal and its pathophysiology in disparate organs such as liver, blood, and the nervous system.
Acknowledgments
The authors thank all the individuals involved for their contributions and Tom de Vries-Lentsch, Erasmus MC, Rotterdam, for artwork. This study was supported by the Internationaal Parkinson Fonds-Netherlands and the Netherlands Organization for Scientific Research VIDI grant (to V.B.).
Supplemental Data
One affected individual from the Italian family with SLC30A10 mutations (SIE-03, IV-1 in Figure 1A) before the beginning of the chelation therapy. Severe global bradykinesia, gait disturbances with starting hesitation, and postural disturbance are present.
The affected individual from the Italian family with SLC30A10 mutations (SIE-03, IV-1 in Figure 1A) after 5 days of chelation therapy. Bradykinesia, gait and postural disturbances show initial, clear improvements.
The affected individual from the Italian family with SLC30A10 mutations (SIE-03, IV-1 in Figure 1A) after 5 months under a chelation therapy regimen. Marked improvement of bradykinesia, gait, and postural disturbances are shown.
One affected individual from the Dutch family with SLC30A10 mutations (NIJ010, IV-1 in Figure 1G) with severe generalized dystonia.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
ALLEN Human Brain Atlas, http://www.brain-map.org
Exome Variant Server database, http://evs.gs.washington.edu/EVS/
Genome Browser (NCBI), http://www.ncbi.nlm.nih.gov/sites/entrez?db=Genome
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
TMHMM Server, http://www.cbs.dtu.dk/services/TMHMM/
TMpred program, http://www.ch.embnet.org/software/TMPRED_form.html
References
- 1.Roth J.A. Homeostatic and toxic mechanisms regulating manganese uptake, retention, and elimination. Biol. Res. 2006;39:45–57. doi: 10.4067/s0716-97602006000100006. [DOI] [PubMed] [Google Scholar]
- 2.Bowman A.B., Kwakye G.F., Hernández E.H., Aschner M. Role of manganese in neurodegenerative diseases. J. Trace Elem. Med. Biol. 2011;25:191–203. doi: 10.1016/j.jtemb.2011.08.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang C.C., Chu N.S., Lu C.S., Wang J.D., Tsai J.L., Tzeng J.L., Wolters E.C., Calne D.B. Chronic manganese intoxication. Arch. Neurol. 1989;46:1104–1106. doi: 10.1001/archneur.1989.00520460090018. [DOI] [PubMed] [Google Scholar]
- 4.Butterworth R.F. Metal toxicity, liver disease and neurodegeneration. Neurotox. Res. 2010;18:100–105. doi: 10.1007/s12640-010-9185-z. [DOI] [PubMed] [Google Scholar]
- 5.Chalela J.A., Bonillha L., Neyens R., Hays A. Manganese encephalopathy: An under-recognized condition in the intensive care unit. Neurocrit. Care. 2011;14:456–458. doi: 10.1007/s12028-010-9476-5. [DOI] [PubMed] [Google Scholar]
- 6.Sanotsky Y., Lesyk R., Fedoryshyn L., Komnatska I., Matviyenko Y., Fahn S. Manganic encephalopathy due to “ephedrone” abuse. Mov. Disord. 2007;22:1337–1343. doi: 10.1002/mds.21378. [DOI] [PubMed] [Google Scholar]
- 7.Stepens A., Logina I., Liguts V., Aldins P., Eksteina I., Platkājis A., Mārtinsone I., Tērauds E., Rozentāle B., Donaghy M. A Parkinsonian syndrome in methcathinone users and the role of manganese. N. Engl. J. Med. 2008;358:1009–1017. doi: 10.1056/NEJMoa072488. [DOI] [PubMed] [Google Scholar]
- 8.Calne D.B., Chu N.S., Huang C.C., Lu C.S., Olanow W. Manganism and idiopathic parkinsonism: Similarities and differences. Neurology. 1994;44:1583–1586. doi: 10.1212/wnl.44.9.1583. [DOI] [PubMed] [Google Scholar]
- 9.Huang C.C., Chu N.S., Lu C.S., Chen R.S., Schulzer M., Calne D.B. The natural history of neurological manganism over 18 years. Parkinsonism Relat. Disord. 2007;13:143–145. doi: 10.1016/j.parkreldis.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Tuschl K., Mills P.B., Parsons H., Malone M., Fowler D., Bitner-Glindzicz M., Clayton P.T. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia-A new metabolic disorder. J. Inherit. Metab. Dis. 2008;31:151–163. doi: 10.1007/s10545-008-0813-1. [DOI] [PubMed] [Google Scholar]
- 11.Gospe S.M., Jr., Caruso R.D., Clegg M.S., Keen C.L., Pimstone N.R., Ducore J.M., Gettner S.S., Kreutzer R.A. Paraparesis, hypermanganesaemia, and polycythaemia: A novel presentation of cirrhosis. Arch. Dis. Child. 2000;83:439–442. doi: 10.1136/adc.83.5.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brna P., Gordon K., Dooley J.M., Price V. Manganese toxicity in a child with iron deficiency and polycythemia. J. Child Neurol. 2011;26:891–894. doi: 10.1177/0883073810393962. [DOI] [PubMed] [Google Scholar]
- 13.Fahn S., Elton R.L., Members of the UPDRS Development Committee . Macmillan; New York: 1987. Unified Parkinson's Disease Rating Scale. Recent Developments in Parkinson's Disease. pp. 153–163. [Google Scholar]
- 14.Montanini B., Blaudez D., Jeandroz S., Sanders D., Chalot M. Phylogenetic and functional analysis of the Cation Diffusion Facilitator (CDF) family: Improved signature and prediction of substrate specificity. BMC Genomics. 2007;8:107. doi: 10.1186/1471-2164-8-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohana E., Hoch E., Keasar C., Kambe T., Yifrach O., Hershfinkel M., Sekler I. Identification of the Zn2+ binding site and mode of operation of a mammalian Zn2+ transporter. J. Biol. Chem. 2009;284:17677–17686. doi: 10.1074/jbc.M109.007203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palmiter R.D., Findley S.D. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 1995;14:639–649. doi: 10.1002/j.1460-2075.1995.tb07042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukunaka A., Suzuki T., Kurokawa Y., Yamazaki T., Fujiwara N., Ishihara K., Migaki H., Okumura K., Masuda S., Yamaguchi-Iwai Y. Demonstration and characterization of the heterodimerization of ZnT5 and ZnT6 in the early secretory pathway. J. Biol. Chem. 2009;284:30798–30806. doi: 10.1074/jbc.M109.026435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delhaize E., Kataoka T., Hebb D.M., White R.G., Ryan P.R. Genes encoding proteins of the cation diffusion facilitator family that confer manganese tolerance. Plant Cell. 2003;15:1131–1142. doi: 10.1105/tpc.009134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devergnas S., Chimienti F., Naud N., Pennequin A., Coquerel Y., Chantegrel J., Favier A., Seve M. Differential regulation of zinc efflux transporters ZnT-1, ZnT-5 and ZnT-7 gene expression by zinc levels: A real-time RT-PCR study. Biochem. Pharmacol. 2004;68:699–709. doi: 10.1016/j.bcp.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 20.Urani C., Melchioretto P., Gribaldo L. Regulation of metallothioneins and ZnT-1 transporter expression in human hepatoma cells HepG2 exposed to zinc and cadmium. Toxicol. In Vitro. 2010;24:370–374. doi: 10.1016/j.tiv.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Sreedharan S., Stephansson O., Schiöth H.B., Fredriksson R. Long evolutionary conservation and considerable tissue specificity of several atypical solute carrier transporters. Gene. 2011;478:11–18. doi: 10.1016/j.gene.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 22.Seve M., Chimienti F., Devergnas S., Favier A. In silico identification and expression of SLC30 family genes: An expressed sequence tag data mining strategy for the characterization of zinc transporters' tissue expression. BMC Genomics. 2004;5:32. doi: 10.1186/1471-2164-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ebert B.L., Bunn H.F. Regulation of the erythropoietin gene. Blood. 1999;94:1864–1877. [PubMed] [Google Scholar]
- 24.Pfeiffer R.F. Wilson's disease. Handb. Clin. Neurol. 2011;100:681–709. doi: 10.1016/B978-0-444-52014-2.00049-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
One affected individual from the Italian family with SLC30A10 mutations (SIE-03, IV-1 in Figure 1A) before the beginning of the chelation therapy. Severe global bradykinesia, gait disturbances with starting hesitation, and postural disturbance are present.
The affected individual from the Italian family with SLC30A10 mutations (SIE-03, IV-1 in Figure 1A) after 5 days of chelation therapy. Bradykinesia, gait and postural disturbances show initial, clear improvements.
The affected individual from the Italian family with SLC30A10 mutations (SIE-03, IV-1 in Figure 1A) after 5 months under a chelation therapy regimen. Marked improvement of bradykinesia, gait, and postural disturbances are shown.
One affected individual from the Dutch family with SLC30A10 mutations (NIJ010, IV-1 in Figure 1G) with severe generalized dystonia.