

Abstract
Non-viral vectors that harness the change in pH in endosomes are increasingly being used to deliver cargoes, including nucleic acids, to mammalian cells. Here we present evidence that the pKa of the β-NH2 in 2,3-diaminopropionic acid (Dap) is sufficiently lowered, when incorporated in peptides, that its protonation state is sensitive to the pH changes that occur during endosomal acidification. The lowered pKa around 6.3 is stabilised by the increased electron withdrawing effect of the peptide bonds, by inter-molecular hydrogen bonding and from contributions arising from the peptide conformation, including mixed polar/apolar environments, Coulombic interactions and inter-molecular hydrogen bonding. Changes of the charged state are therefore expected between pH 5 and 7 and large scale conformational changes are observed in Dap rich peptides, in contrast with analogues containing lysine or ornithine, when the pH is altered through this range. These physical properties confer a robust gene delivery capability on designed cationic amphipathic peptides that incorporate Dap.
Keywords: Circular dichroism, peptides, NMR spectroscopy, pH sensitivity, gene delivery
Introduction
Efficient and non-toxic delivery of therapeutic nucleic acids to mammalian tissues remains a challenge.[1] Non-viral vectors, though supposedly safer, often have relatively poor transfection efficiency and hence efficient non-viral gene delivery vehicles remain highly sought after. Efficient vectors are capable of complexing with nucleic acids and offering protection from nucleases while ensuring that the complexes reach and are taken up by the target cell. Many such vectors enter cells using endosomal pathways but escape of the complexes from endosomes is often observed to be poor and is a considerable limiting factor in their use. Incorporating pH sensitive imidazole groups when designing non-viral vectors has proven to be an effective strategy for increasing the transfection efficiency of compounds that deliver their cargo to mammalian cells via endocytosis.[2] The histidine residues in e.g. the vector peptide LAH4 are uncharged at neutral pH but when the pH of the endosomal lumen drops, the side-chains become protonated.[3, 4] Histidine rich peptides, as with other polyamines with high buffering capacities between pH 5.1 and pH 7.4,[5-12] are capable of acting as a proton sponge while the membrane disturbing behavior and nucleic acid binding ability of the peptide are also dramatically altered.[13, 14] As a result of including the pH sensitive histidines, large numbers of membrane disturbing peptides are released from the nucleic acid-peptide complex during endosomal acidification enhancing the ability of the complex to escape the endosome and reach the cell cytosol and, eventually, nucleus.[14]
Although structurally related to its cationic analogues such as lysine and ornithine, 2,3-diaminopropionic acid (Dap) in its free acid form is characterized by distinctly lower proton ionization constants for its two amino groups. At 6.67, the pKa of the α-NH2 is around 3 pKa units lower than the typical value for α-amino acids (ca. 9.7 to 9.9 at 25°C).[15] The pKa for both the α and β-NH2 groups in the methyl ester of Dap are lowered (4.18 and 7.96 at 37°C respectively) when compared with the free acid form (6.42 and 9.37 at 37°C respectively).[15] Since the lowering of the pKa values in the ester compared with the acid is regarded as being exclusively a result of the greater electron withdrawing ability of the methoxycarbonyl group over the carboxylate[15], we were interested to determine the effect on the pKa of the β-NH2 when the α-NH2 and carbonyl participate in peptide bond formation. Potentiometric experiments revealed the pKa of the β-NH2 in model compounds, simulating the electron withdrawing and hydrogen bonding effects expected in a peptide, to be substantially lowered such that deprotonated Dap side-chains could be expected in certain environments. We hypothesized that a deprotonated side-chain at neutral pH would lead to a peptide with similar properties to those previously described for histidine rich cationic amphipathic peptides.
We have tested this hypothesis by comparing the gene delivery capabilities and biophysical properties of a series of peptides with the primary sequence KKLAXALXLLALLWLXLAXALKKA-NH2 where X is either lysine (LAK), ornithine (LAO), histidine (LAH) or Dap (LADap) as well as comparing the pKa of the β-NH3 group of Dap with that of a series of model compounds allowing the contributions of differing electronic environments to the stabilization of a lowered pKa to be determined. In transfection assays where we tested the ability of peptides to deliver luciferase reporter plasmid to SV40 transformed human lung fibroblasts (MRC5-V2), both the histidine and Dap containing peptides were found to function effectively as DNA delivery peptides while the two analogues containing either ornithine or lysine were around 250 times less efficient. The transfection efficiency of LADap was comparable with L-PEI, an example of a commercially available delivery system. The transfection efficiency of LADap and LAH4-L1, the most efficient vector peptide tested in our studies so far, was strongly affected by the presence of bafilomycin A1, an indicator of endocytosis driven gene transfer which prevents acidification of the endosome.[16, 17] The ability of the Dap containing peptide to deliver DNA at the same levels as the histidine rich analogue, the sensitivity of this process to inhibitors of endosomal acidification and the observed changes in secondary structure and oligomerisation strongly support the hypothesis that incorporation of Dap in a peptide confers pH sensitivity in the range between pH 5 and 7.
Results and Discussion
Model compounds
In order to explore our hypothesis regarding the effect of the altered electronic environment in the peptide, as opposed to the free acid form of Dap, we obtained or prepared three model compounds designed to show the effect of first one and then two amide bonds on the pKa of the β-NH2. The structures of the compounds (Scheme 1) are given. Compound 1 is the methyl ester of Dap and is used to relate this work to the previous study.[15] Compound 2 differs from compound 1 at the α-NH2 position where the amine is acetylated forming the first amide bond. In compound 3 the methyl ester is replaced with a primary amide group. Although this does not replicate the secondary amide that is present in the peptide, addition of electron donating groups such as a methyl group to create a secondary amide might have a considerable effect on the intramolecular hydrogen bonding between the amine and amide groups. Such effects are observed in ethylenediamine units in PEG block catiomers.[18] The electron withdrawing effect of the two amide groups is acknowledged when using the pKa prediction tool MarvinView 5.1.4 which indicates that at 8.5, 8.3 and 8.1 the pKa of the β-NH2 in compounds 1, 2 and 3 respectively are almost 2 units lower than that of lysine. The prediction tool however, does not take into account possible intramolecular hydrogen bonding that may stabilize an even lower pKa. Furthermore, the exceptionally low pKa of both the α and β-NH2 in Dap and its methyl ester may, in part, be due to stabilization by hydrogen bonding between the vicinal groups. Therefore, the pKa of the β-NH2 was determined at 25°C using potentiometric methods for all three compounds. This allows the combined effect of both electron withdrawal and intramolecular hydrogen bonding from the carbonyl and amide, rather than amine, groups that would be present in the corresponding peptide to be determined. The determined values for the pKa for the β-NH2 in each of compounds 1, 2 and 3 were 8.29, 7.84 and 7.82 respectively at 25°c which can be extrapolated to give estimates of 7.94, 7.49 and 7.49 respectively at 37°C. These values not only offer experimental confirmation that the pKa of the β-NH2 is depressed by the electron withdrawing effects of the amide groups, but also demonstrate that the pKa is depressed by a further 0.3 units through hydrogen bonding from nearby groups that would be present in the LADap peptide.
Scheme 1.
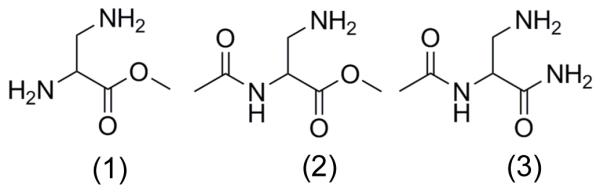
Model compounds used to determine the effect of electron withdrawing amide bonds on the pKa of the β-amino group. Methyl 2,3-diaminopropanoate (1), methyl 2-acetamido-3-aminopropanoate (2) and 2-acetamido-3-aminopropanamide (3)
Biophysical characterisation
Circular Dichroism (CD) spectroscopic investigations of the four amphipathic peptide analogues in solution (Fig. 1) lend support to the notion that the Dap side-chain is sensitive to pH changes between pH 5 and 7. Both histidine and Dap rich peptides adopt α-helical conformations at neutral pH (Figure 1A/B) yielding spectra with characteristic negative bands at around 210 and 220 nm and, importantly, an intense positive band around 190 nm. In contrast, the lysine and ornithine rich peptides adopt an unordered conformation (Figure 1C/D) with only a weak positive band at around 190 nm, a weak negative band around 220 nm and the negative band between 200 and 210 nm shifted to shorter wavelength. Using the CDPro suite of spectral analysis programs the α-helical content was estimated for LAH and LADap, at each pH where spectra were obtained, and plotted as a function of pH (Fig. 1E). In order for intramolecular hydrogen bonding to contribute to a depressed pKa for the Dap side chain, hydrogen bonding donors and/or acceptors should be available in the peptide backbone and not be involved in interactions stabilizing secondary structure. Some evidence for this is observed in the CD spectra. Interestingly, at the most basic pH the calculated α-helical content of the Dap rich peptide was somewhat lower than that of the histidine rich peptide even though a plateau in conformational change is reached between pH 6.78 and pH 7.23. This suggests that helix stabilizing intramolecular hydrogen bonding may not be optimal in the Dap rich peptide despite its propensity for forming such a conformation. Hence the amide group from the N-1 residue as well as the carbonyl for the N+1 reside, relative to Dap in the peptide sequence, may have some ability to participate in intramolecular hydrogen bonding in a similar fashion to the model compounds, albeit at a lower level.
Figure 1.

pH dependent conformational changes in the four cationic amphipathic helical peptides. Dap containing LADap (A) and histidine containing LAH (B) undergo a helical to unordered transformation as the pH is lowered from pH 7. In contrast, lysine containing LAK (C) and ornithine containing LAO (D) retain an unordered conformation throughout the pH range tested. CDPro analysis of the pH dependent change in conformations for both LAH and LADap (E) indicates that the conformational change begins at a much higher pH for LADap than LAH.
pH titration experiments, performed through small additions of perchloric acid, caused both histidine and Dap rich peptides to unfold into the unordered state, seen for the ornithine and lysine rich peptides, with LADap unfolding at a noticeably higher pH (Fig. 1E). The apparent pKa derived from these measurements reveals an acidic pKa of 5.34 for the conformational change driven by the protonation of histidine. The pKa derived for the same conformational change driven by protonation of Dap in the peptide is a similarly acidic 6.33 when compared with the expected value of 7.49 at 37°, obtained by extrapolation from the pKa observed for the model compound at 25°C. The histidine side chain pKa in proteins is known to vary considerably according to its environment.[19] Titratable histidines in proteins have been identified with a side-chain pKa as high as 9.2 and as low as 4.6.[19] Importantly, Coulombic interactions between neighboring side-chains in space, as predicted for amphipathic peptides[20], would also be expected to depress the pKa. Hence an acidic value for the side-chain pKa of histidine in LAH is neither unexpected nor is it likely to be as a result of either intra- or intermolecular hydrogen bonding. It follows that, since the LADap peptide is analogous to the LAH peptide, the Dap side chains can be expected to be affected in a similar way. The obtained value for the apparent pKa is therefore reasonable when compared with the study of the model compounds and when considered alongside that obtained for histidine and the analogue. Furthermore, as for the LAH peptide, the majority of the discrepancy between the pKa observed in the model compound and the peptide could be accounted for by Coulombic interactions between the four Dap side chains.
Although the CD analysis is not an exclusive probe of the pKa, inclusion of the most appropriate controls, analogous peptides where only the Dap residues are replaced with amino acids of known pH sensitivity, in the experimental setup indicates that the observed pH sensitive change in conformation for LADap is exclusively due to the incorporated Dap residues. Since the Dap, lysine, ornithine and histidine residues themselves differ only in respect of the side chain then it follows that the pH dependent behavior of the peptide must itself be due to the Dap side chain. In order to identify the origin of the observed changes in secondary structure, fluorescence emission spectra of the intrinsic tryptophan probe were obtained to probe the environment of Trp14 in each of the pH sensitive peptides. These experiments (Fig. 2) indicated that the α-helical conformation, observed for both LAH and LADap at basic pH, represents a self associated state. Tryptophan emission spectra with maxima at shorter wavelengths are indicative of more hydrophobic environments whereas those at longer wavelengths suggest a more polar environment. Furthermore, the blue shifted maxima are usually of greater intensity than those observed in more polar environments.[21] At neutral pH the emission maximum for LAH/LADap was blue shifted (343/345 nm) and of much greater intensity when compared with that observed at acidic pH (351/353 nm) indicating a more hydrophobic environment for the tryptophan probe as expected for an aggregated versus monomeric peptide (Fig 2A/B).
Figure 2.
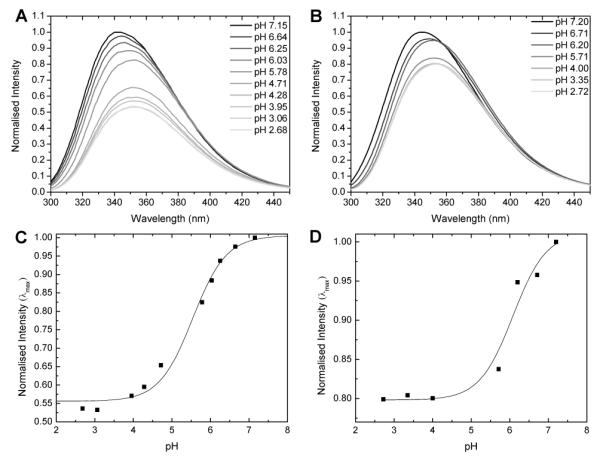
Intrinsic tryptophan fluorescence of LAH (A) and LADap (B) in 5 mm Tris buffer at various pH. LAH/LADap have blue shifted emission maxima at 343/345 nm around neutral pH and red shifted maxima (351/353 nm) at acidic pH. The changes in maximum intensity for LAH (C) and LADap (D) are plotted against pH to determine apparent pK’s of 5.5 ± 0.1 for LAH and 6.1 ± 0.2 for LADap.
Since the peptide is in a self associated state when the Dap side chain is deprotonated, intermolecular hydrogen bonding may also be implicated in stabilizing the lowered pKa of the β-NH2 although, again the data for LAH, where hydrogen bonding is a less likely source of stabilization, suggests Coulombic interactions make the major contribution. Emission spectra of the tryptophan in LAH were of a much lower intensity at acidic pH than those of LADap. This is likely a result of the enhanced ability of histidine residues to quench tryptophan fluorescence when protonated.[22] Plotting the change in intensity of the emission maxima as a function of pH allows determination of a pK for the transition (Fig. 2C/D). The values of 5.5 ± 0.1 and 6.1 ± 0.2 for LAH and LADap, respectively, are in agreement with those obtained from the CD investigation above.
Finally, 1H NMR was used to probe protonation events in the histidine or Dap side chains of the LAH/LADap peptides (see supplementary material). For LAH, histidine resonances could be assigned and traced from pH 3 through to pH 7.5. Plotting chemical shift, for resonances assigned to the ε1 and δ2 protons, as a function of pH revealed a pKa of 5.2 ± 0.1 for the histidine side chains in LAH. For LADap, assignments could only be made for the β protons below pH 6 as above this pH, the resonances became broadened even with the application of magic angle spinning line narrowing techniques. The initial changes in chemical shift are enough to allow only an approximate pKa of 6.2 for the Dap β-NH2 to be determined. However this information, coupled with the line broadening observed lends further support to the hypothesis that LADap is sensitive to pH changes in this range. The observed side-chain protonation in LAH and LADap supports a model whereby the nominal net positive charge in both peptides is increased from +5 to +9 as the pH is lowered, increasing repulsion between monomers and driving disassociation and unfolding. Therefore, although NMR measurements are commonly used for the measurement of pKa, the self association observed for the peptides renders NMR and, similarly, potentiometric methods impracticable. The CD methodology applied here is well suited to describe peptide oligomerisation and folding[23] and is the most appropriate technique for studying the conformational changes that are closely linked to the changes in charge state of the cationic side-chains.
Gene delivery properties
Having demonstrated that, in solution, the conformational state of LADap closely mimicked that of LAH, albeit with pH dependent conformational changes occurring at an elevated pH, we were interested to test whether a Dap containing peptide would behave similarly when complexed with nucleic acids and hence would possess similar gene delivery properties to those observed for histidine rich peptides.[4, 13, 24] The ability of LADap to mediate delivery of the reporter gene luciferase to MRC5-V2 cells was assessed by means of a luciferase based transfection assay (Fig. 3). The transfection efficiency mediated by LADap was similar to but not significantly greater than that observed with the histidine rich analogue LAH (Fig. 3A). Similar analogues containing either lysine or ornithine in place of Dap, for which no pH dependent conformational changes had been observed, were much less efficient at mediating luciferase expression (Fig. 3A), indeed expression mediated by LADap was 246 times greater than that mediated by LAK (p < 0.05). We then compared the transfection efficiency of LADap with that of LAH, LAH4-L1, the most efficient vector peptide from our previous studies[13], and linear polyethylenimine (L-PEI), another cationic non-viral vector that is available commercially (Fig. 3B). In this experiment the transfections were performed either in the presence or absence of bafilomycin A1. As an inhibitor of endocytosis which prevents acidification of the endosome, this treatment would allow the role of the pH dependent activity of each vector to be assessed. The substantial and significant (p < 0.05) reductions in LADap mediated transfection efficiency in the presence of bafilomycin A1 confirmed the pH dependent behavior of LADap during the transfection process.
Figure 3.

Transfection of MRC5-V2 cells with luciferase reporter using a series of cationic amphipathic peptides (A). The transfection efficiency of the peptide containing Dap (LADap) is not significantly different than that of the analogous peptide containing histidine (LAH) and is much greater than that of the two peptides containing either lysine (LAK) or ornithine (LAO). * indicates differences are significant (p < 0.05) with respect to LADap. Transfection is sensitive to inhibition with bafilomycin A1 (B) indicating that the gene delivery process is dependent on endosomal acidification and that the vector is sensitive to the change in pH in the endosome. PEI mediated transfection is reduced by around 99% and peptide mediated transfection by between 76 and 87%. * indicates treatment reduced transfection efficiency (p < 0.05). Transfections experiments were performed in serum free media.
Conclusion
In conclusion, we present clear evidence that when incorporated into peptides, the β-NH2 group of Dap is unlikely to be protonated at neutral pH. Furthermore the pKa determined here for Dap in a designed amphipathic peptide lends itself to applications requiring functional groups sensitive to pH changes, as seen in mammalian cell endosomes, such as non-viral nucleic acid delivery.
Experimental Section
Materials for synthesis of model compounds
Methyl 2-amino-3-((tert-butoxycarbonyl)amino)propanoate hydrochloride and peptide synthesis reagents were from Bachem, (Weil am Rhein, Germany). 2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-((tert-butoxycarbonyl)amino)propanoic acid was purchased from Novabiochem/Merck (Nottingham, UK). All other reagents were analytical grade or better.
Synthesis of methyl 2,3-diaminopropanoate (1)
Methyl 2-amino-3-((tert-butoxycarbonyl)amino)propanoate hydrochloride(0.26 g, 1 mmol) was treated with a 1:1 mixture of TFA/DCM (2 ml) for 3 hours at room temperature. The solvent was removed in vacuo, the residue dissolved in 10% acetic acid solution and freeze dried for 24 hours. The residue was then re-dissolved in water and freeze dried for 24 hours, three times, to afford a crystalline solid (yield 85%). 1H NMR (D2O, 400 MHz): δ 4.42 (m, 1H), 3.78 (s, 3H), 3.5 (m, 2H).
Synthesis of methyl 2-acetamido-3-aminopropanoate(2)
Methyl 2-amino-3-((tert-butoxycarbonyl)amino)propanoate hydrochloride (0.26 g, 1 mmol) was dissolved in DMF (2 ml) and triethylamine (0.25 g, 2.5 mmol) was added to the stirred solution and a white solid formed immediately. After 10 minutes acetic anhydride (0.26 g, 2.5 mmol) was added to the reaction mixture and stirred for 4 hours at room temperature. The trimethylammonium chloride was removed by filtration. The solvent was removed in vacuo, and the residue was treated with 5% NaHCO3 solution. The aqueous phase was extracted with diethylether, and the ethereal phase washed with NaHCO3 solution and and dried over Na2SO4. The solvent was evaporated and the residue dried under high vacuum overnight at room temperature. Deprotection of the Boc group was then achieved as described for compound (1) (Yield 38 %). 1H NMR (CDCl3, 400 MHz): δ 4.69(m, 1H), 3.77 (s, 3H), 3.3 (m, 2H), 2.02 (s, 3H).
Synthesis of 2-acetamido-3-aminopropanamide(3)
Fmoc-XAL-PEG-PS resin (0.16 mmol/g, 2.0g) was treated with 20% (v/v) piperidine in DMF for 10 minutes and then washed with DMF. Successful deprotection was confirmed by Kaiser (ninhydrin) and picrylsulphonic acid tests[25]. The acylation step was performed using three equivalents of 2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-((tert-butoxycarbonyl)amino)propanoic acid, preactivated with 2-(6-Chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU) and collidine in DMF in a molar ratio of 1:0.95:2 respectively overnight over a continuous nitrogen flow. Next the FMOC group was deprotected as described and the free amino group was treated with acetic anhydride (3 equivalents) and DIPEA in DMF for 3 hours. The resin was washed with DMF and methanol and dried in vacuo for three hours at room temperature. Finally the resin was treated with a mixture of 88% trifluoroacetic acid (TFA), 5% H2O, 5% phenol and 2% triisopropylsilane for three hours at room temperature. Thereafter, the resin was filtered and the filtrate was concentrated under reduced pressure. The residue was extracted with diethyl ether and water. The aqueous phase was washed with ether three times and finally freeze dried. The product was purified using a Water Sep-Pak Vac 35cc C18 cartridge and monitored by HPLC. The product eluted in 0.1 % TFA. (Yield 70%). 1H NMR (CD3OD, 400 MHz): δ 4.71 (m, 1H), 3.41 (m, 2H), 2.06 (s, 3H).
Potentiometric titration
Potentiometric titration was performed for the three model compounds in 12.1 ml KCl (0.1M) solution. 400 ml HPLC grade water was boiled for 15 minutes and then cooled under nitrogen gas to eliminate CO2. 200 ml of this water was then used to prepare a 0.1M KCL solution. The reservoir of the titrator was filled with 0.1M KCl and an argon gas bubbler was used to prevent potassium hydroxide solution absorbing CO2 from the air. Prior to titration of the model compounds, a blank titration is performed. Briefly, 100 ml 0.2092M volumetric standard HCl (Aldrich) was added to 10 ml 0.1 M KCl solution. Argon gas was passed though the vessel to displace any CO2 present. When the thermostat registered 25 °C, the program titror was activated to automatically collect pH values and KOH volume data. Generally, the potential range for electrode calibration is 250 to −250mV. This process takes approximately 15 minutes; the electrode was rinsed by deionized water and was then ready for the experiment. The electrode E0 and S are then optimized by GLEE[26] until the average error is less than 0.05 mV. The precise concentration of the KOH is also obtained in this process. Simulations were carried out using HYSS[27] to check the weight of the sample needed to create a detectable buffer effect on the titration curve in comparison with the blank titration curve. The model compounds were dissolved in 0.05 ml DMSO, added to 0.1M KCl solution (12.1 ml), sealed in the vessel and titrated as described above. The obtained titration curve was imported into HYPERQUAD[28] and the pKa values were optimized according to the defined dissociation equilibrium model.
Circular dichroism and fluorescence experiments
Peptides were purchased as “desalted” grade (EZBiolab, Westfield IN, USA) and further purified by reverse phase HPLC using an acidified water/methanol gradient. Peptides were dissolved in 5 mM Tris pH 7.29 at a final concentration of between 30 and 50 μM. The samples were titrated down by adding 0.3% or 1% (v/v) HClO4 solution in micro-litre amounts. CD spectra were acquired on a Chirascan™ Spectrometer (Applied Photophysics, Leatherhead, UK) with samples maintained at 310 K. Spectra were recorded from 260 to 180 nm using a 0.5 mm path length and were processed using Chirascan software where a spectrum of the peptide free solution was subtracted and Savitzky-Gorlay smoothing with a convolution width of 5 points applied. Fluorescence emission spectra were acquired on a Varian Cary Eclipse Spectrophotometer (Palo Alto CA, USA) using an excitation wavelength of 280 nm and excitation and emission slit widths of 5 nm. Samples were maintained at 310 K in a 4 × 10 mm cuvette (Hellma, Müllheim, Germany).
NMR 1.5 mg of peptide was solubilized in 600 μl D2O/H2O (10%/90%) containing 5 mM Tris buffer, pH 7.4. Titration was as above, however data points above pH 7.4 were recorded by adding several microlitres of a 4% (w/v) NaOH solution. Spectra were recorded on a 500 MHz Bruker Avance spectrometer equipped with a cryoprobe, using a standard 1D WATERGATE pulse sequence (p3919gp), at 310 K. 16 scans were accumulated, with a recycle delay of 1 second. Additional spectra were recorded under high resolution magic angle spinning (HR-MAS) conditions on a 400 MHz Bruker Avance spectrometer equipped with a 4 mm HR-MAS probe. 512 scans were acquired at a MAS frequency of 5 kHz.
Cell culture and DNA transfection
Dulbecco’s modified Eagle medium (DMEM; Gibco-BRL) was supplemented with 2 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin and 10% of fetal calf serum (FCS; HyClone). 100,000 SV40 transformed human fetal lung fibroblasts (MRC-5 V2) per well were plated in 24-well plates (Costar) one day before transfection. SMD2-LucΔITR (7.6 kb) is an expression plasmid encoding the firefly luciferase gene under the control of the human cytomegalovirus (CMV) immediate-early promoter. 4 μg of plasmid DNA and the desired amount of peptide were each diluted in 100 μl of 150 mM NaCl and gently mixed. DNA complexes were prepared by varying the peptide/DNA w/w ratios in order to find out which ratio allows for maximal reporter gene expression. Optimal ratios were 8.75 : 1 for both LAH and LADap, and 5 : 1 and 7.5 : 1 for LAO and LAK respectively. 9 μl of L-PEI (22 kDa) was used. The particle size for peptide/DNA complexes produced in this way were measured using a Zetasizer nano ZS (Malvern). DNA (3 μg) and the desired amount of peptide were prepared in 150 μl of 150 mM NaCl and left for 20 min at 35°C. Particle sizes were between 1370 nm and 2000 nm but the samples were polydisperse and no significant differences could be determined between complexes prepared with the four peptides. Similar experiments were performed for peptides alone. Smaller particles between 148 and 409 nm were observed but, again, the samples were polydisperse and no significant differences could be observed between the four peptides. Transfection reagent and DNA solution were mixed in a final volume of 0.2 ml and incubated for 15 min at room temperature. Before adding the complexes to the cells, the mixtures were diluted with medium to a final volume of 1 ml. The transfection was performed in serum-free conditions in duplicates (0.5 ml/well). After about 3 h, the transfection medium was replaced with fresh medium containing 10% serum. Luciferase activity was assayed about 30 h after transfection as described previously.[24] Cellular toxicity from treatment, either with DNA/vector complex or bafilomycin A1, was assessed by comparing the protein content of untreated or treated cells. No significant effects of either treatment were observed. Statistical analysis was by ANOVA with Bonferonni post-hoc t-test, one tailed.
Supplementary Material
Acknowledgements
This work was supported by the Medical Research Council (NIRG to AJM), the Wellcome Trust (VIP Award to AJM and Capital Award for the KCL Centre for Biomolecular Spectroscopy), the University of London Central Research Fund (AR/CRF/B) and Applied Photophysics. VA is the holder of a Maplethorpe Postdoctoral Fellowship of the University of London.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- [1].Li SD, Huang L. Gene Ther. 2006;13:1313–1319. doi: 10.1038/sj.gt.3302838. [DOI] [PubMed] [Google Scholar]
- [2].Midoux P, Pichon C, Yaounac J-J, Jaffrès P-A. Br. J. Pharmacol. 2009;157:166–178. doi: 10.1111/j.1476-5381.2009.00288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bechinger B. J. Mol. Biol. 1996;263:768–775. doi: 10.1006/jmbi.1996.0614. [DOI] [PubMed] [Google Scholar]
- [4].Kichler A, Mason AJ, Bechinger B. Biochim. Biophys. Acta. 2006;1758:301–307. doi: 10.1016/j.bbamem.2006.02.005. [DOI] [PubMed] [Google Scholar]
- [5].Dinçer S, Türk M, Pişkin E. Gene Ther. 2005;12:S139–S145. doi: 10.1038/sj.gt.3302628. [DOI] [PubMed] [Google Scholar]
- [6].Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr J-P. Proc. Natl. Acad. Sci. USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Behr J-P. CHIMIA. 1997;51:34–36. [Google Scholar]
- [8].Sonawane ND, Szoka FC, Verkman VS. J. Biol.Chem. 2003;278:44826–44831. doi: 10.1074/jbc.M308643200. [DOI] [PubMed] [Google Scholar]
- [9].Zhong Z, Song Y, Engbersen JFJ, Lok MC, Hennink WE, Feijen J. J. Control. Release. 2005;109:317–329. doi: 10.1016/j.jconrel.2005.06.022. [DOI] [PubMed] [Google Scholar]
- [10].Lin C, Zhong Z, Lok MC, Jiang X, Hennink WE, Feijen J, Engbersen JFJ. J. Control. Release. 2006;116:130–137. doi: 10.1016/j.jconrel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- [11].Lin C, Blaauboer C-J, Timoneda MM, Lok MC, van Steenbergen M, Hennink WE, Zhong Z, Feijen J, Engbersen JFJ. J. Control. Release. 2008;126:166–174. doi: 10.1016/j.jconrel.2007.11.012. [DOI] [PubMed] [Google Scholar]
- [12].Zelphati O, Szoka FC. Proc. Natl. Acad. Sci. USA. 1996;93:11493–11498. doi: 10.1073/pnas.93.21.11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mason AJ, Martinez A, Glaubitz C, Danos O, Kichler A, Bechinger B. FASEB J. 2006;20:320–322. doi: 10.1096/fj.05-4293fje. [DOI] [PubMed] [Google Scholar]
- [14].Prongidi-Fix L, Sugawara M, Bertani P, Raya J, Leborgne C, Kichler A, Bechinger B. Biochemistry. 2007;46:11253–11262. doi: 10.1021/bi700766j. [DOI] [PubMed] [Google Scholar]
- [15].Hay RW, Morris PJ. J. Chem. Soc. 1971:3562–3569. [Google Scholar]
- [16].Simões S, Slepushkin V, Pires P, Gaspar R, Pedroso de Lima MC, Düzgünes N. Gene Therapy. 1999;6:1798–1807. doi: 10.1038/sj.gt.3301015. [DOI] [PubMed] [Google Scholar]
- [17].Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. J. Biol. Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- [18].Kanayama N, Fukushima S, Nishiyama N, Itaka K, Jang W-D, Miyata K, Yamasaki Y, Chung U-i., Kataoka K. ChemMedChem. 2006;1:439–444. doi: 10.1002/cmdc.200600008. [DOI] [PubMed] [Google Scholar]
- [19].Edgcomb SP, Murphy KP. Proteins. 2002;49:1–6. doi: 10.1002/prot.10177. [DOI] [PubMed] [Google Scholar]
- [20].Johnsson K, Alleman RK, Widmer H, Benner SA. Nature. 1993;365:530–532. doi: 10.1038/365530a0. [DOI] [PubMed] [Google Scholar]
- [21].Lakowicz JR. Principles of Fluorescence Spectroscopy. Plenum Press; New York: 1983. [Google Scholar]
- [22].Chen Y, Barkley MD. MD. Biochemistry. 1998;37:9976–9982. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- [23].Wilcox W, Eisenberg D. Protein Science. 1992;1:641–653. doi: 10.1002/pro.5560010510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mason AJ, Leborgne C, Moulay G, Martinez A, Danos O, Bechinger B, Kichler A. J. Control. Release. 2007;118:95–104. doi: 10.1016/j.jconrel.2006.12.004. [DOI] [PubMed] [Google Scholar]
- [25].Kaiser E, Colescott RL, Bossinger CD, Cook PI. Analytical Biochemistry. 1970;34:595. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- [26].Gans P, Sullivan B. Talanta. 2000;51:33–37. doi: 10.1016/s0039-9140(99)00245-3. [DOI] [PubMed] [Google Scholar]
- [27].Alderighi L, Gans P, Ienco A, Peters D, Sabatini A, Vacca A. Coordination Chemistry Reviews. 1999;184:311–318. [Google Scholar]
- [28].Gans P, Sabatini A, Vacca A. Talanta. 1996;43:1739–1753. doi: 10.1016/0039-9140(96)01958-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.