

Abstract
The amyloid precursor protein (APP) is a large, ubiquitous integral membrane protein with a small amyloid-β (Aβ) domain. In the human brain, endosomal processing of APP produces neurotoxic Aβ-peptides, which are involved in Alzheimer's disease. Here, we show that the Aβ sequence exerts a physiological function when still present in the unprocessed APP molecule. From the extracellular site, Aβ concentrates APP molecules into plasmalemmal membrane protein clusters. Moreover, Aβ stabilization of clusters is a prerequisite for their targeting to endocytic clathrin structures. Therefore, we conclude that the Aβ domain directly mediates a central step in APP trafficking, driving its own conversion into neurotoxic peptides.
Introduction
The human amyloid precursor protein (APP) gene encodes for a ubiquitously expressed type I integral membrane protein with a large extracellular domain and a short cytoplasmic region (1). Three major isoforms, ranging from 695 to 770 amino acids (aa) in length, have been identified. APP first attracted attention when one of its degradation products, the ∼40-aa large β-amyloid (Aβ) peptide, was found in senile brain plaques isolated from Alzheimer's patients (2). Though the primary function of APP still remains elusive, most research focuses on the function and generation of the Aβ cleavage product.
To date, it is clear how Aβ peptides are generated and also that alternative APP degradation pathways exist. For Aβ production, the enzymes β- and γ-secretase are required to cleave the Aβ sequence at its N- and C-terminal ends, respectively (3). As β-secretase functions in early endosomes (4–6), endocytic uptake of APP molecules is a prerequisite for amyloidogenic processing. Alternatively, the Aβ region is cleaved at its center by plasmalemmal α-secretase, yielding harmless fragments (nonamyloidogenic pathway).
After processing and secretion of Aβ peptides, Aβ oligomers or fibrils play a central role in the development of Alzheimer's disease (7). Moreover, several nonpathogenic activities of Aβ peptides have been discussed (for example, see Baruch-Suchodolsky et al. (8), Igbavboa et al. (9), and Zou et al. (10)). However, so far it is unknown whether the Aβ domain has a function when still present in the unprocessed molecule (for exception, see Tienari et al. (11)).
Interfering with trafficking or processing of APP might reduce the progression of the disease (12,13), for instance, by taking advantage of α- and β-secretase being active at the plasma membrane and in early endosomes, respectively. A block of APP endocytosis would favor α-processing and at the same time avoid β-cleavage, resulting in overall nonamyloidogenic processing (14).
Internalization of APP occurs via a clathrin-mediated pathway (15). Moreover, it is known that endocytosis of APP is regulated by a flotillin-dependent APP clustering step (16). However, flotillin rather acts as a modulator of a more intrinsic clustering mechanism, since clusters become smaller but still remain after flotillin downregulation (16).
Here, we examine the mechanism of APP clustering in the plasma membrane, as understanding APP clustering is important for understanding APP trafficking, which in turn is a prerequisite for the development of therapeutic strategies against Alzheimer's disease.
Materials and Methods
Cloning of APP constructs
The expression vector used for all constructs was pcDNA6.2/C-emGFP/DEST (V355-20, Invitrogen, Carlsbad, CA). N-terminally myc-tagged human APP695 (NCBI reference sequence NM_201414 with VPEQKLISEEDL inserted between positions 19 and 20 (11,17)) was inserted via PCR/Gateway cloning (Invitrogen) into the expression vector, resulting in C-terminally GFP-tagged myc-APP with a KGGRADPAFLYKVVDAVN linker between APP and GFP. Following the same procedure, a C-terminally deleted construct was obtained (APP-ΔC; lacking aa 649–695 of the original APP sequence). For N-terminal deletion, a cDNA sequence was obtained from MWG Eurofins (Ebersberg, Germany) and inserted via PCR/Gateway cloning into the expression vector (APP-ΔN; lacking aa 18–601). The myc-tagged full-length APP was used in Figs. 1 and 2, Fig. S1, Fig. S2, and Fig. S4 C. For Figs. 3–5, Fig. S3, Fig. S5, and Fig. S6, we used APP without myc-tag that was cloned by using the Acc65I and EcoRI restriction sites within the APP sequence. To this end, a double-digested PCR product was inserted into the double-digested pcDNA6.2/C-emGFP/DEST expression vector from the myc-tagged APP, resulting in a construct without the VPEQKLISEEDL insert. From this plasmid, using the restriction sites Acc65I and EcoRI or Acc65I and SgsI, deletion constructs were produced (the deletions Δ22–283, Δ22–435, Δ22–537, Δ22–596, and Δ22–601). In addition, fusion PCR products carrying the mutations N467S and/or T291G, T292G and T576G were amplified and inserted into the expression vector.
Figure 1.
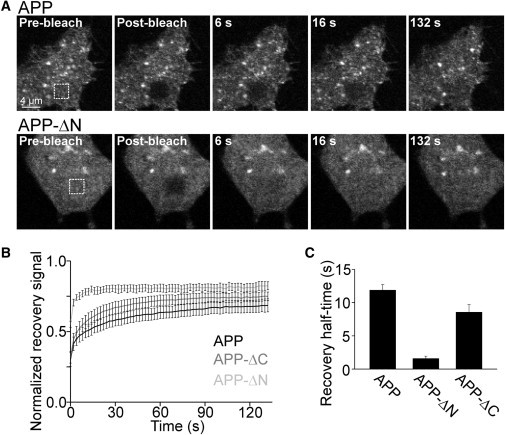
APP mobility is restricted via the N-terminal extracellular domain. (A) Confocal micrographs were recorded from PC12 cells expressing GFP-tagged APP (upper) and variants with an N-terminal deletion (APP-ΔN; lower) or a C-terminal deletion (APP-ΔC; not shown). Fluorescence bleaching was performed in a squared ROI (dotted square) at the basal plasma membrane (compare first and second images). Recovering fluorescence was related to the prebleach value. For one experiment, normalized recovery traces from several cells were averaged for calculation of the half-time of recovery. (B and C) Averaged experiments. Values are given as the mean ± SE (n = 3–5).
Figure 2.

APP disperses after deletion of the extracellular domain. TIRF microscopy images from PC12 cells expressing the same constructs as in Fig. 1, illustrating that APP disperses upon deletion of the extracellular domain (APP-ΔN), whereas it remains clustered without the intracellular C-terminal domain (APP-ΔC). Images are shown at arbitrary scaling.
Figure 3.
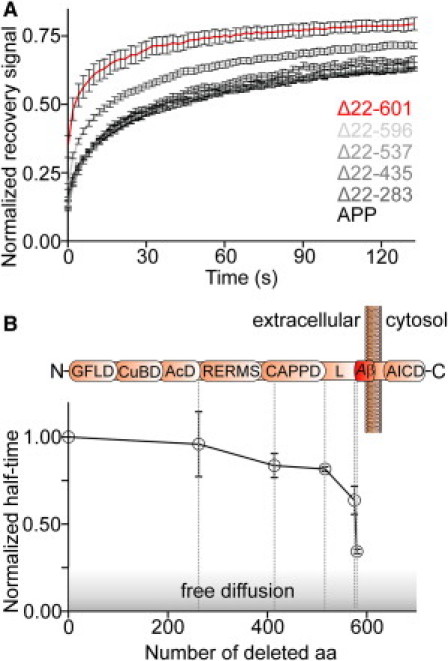
Entire Aβ domain is essential for mobility restriction of APP. (A) FRAP recovery traces (as in Fig. 1B) from APP constructs carrying the indicated deletions within the ectodomain. (B) Half-times were normalized to full-length APP and plotted against the number of deleted aa. The shadowed area indicates the half-time zone for a freely diffusing single-span membrane protein (see also Materials and Methods). The erratic increase in mobility from construct Δ22–596 to Δ22–601 indicates a change from restricted to free diffusion. Please note that the two constructs differ by only five aa from the N-terminus of the amyloid β region. (Upper) Domain structure of APP (not to scale; referring to Reinhard et al. (29)). Dashed lines roughly indicate the position of the last deleted amino acid of the respective construct. GFLD, N-terminal growth-factor-like domain; CuBD, copper-binding domain; AcD, acidic domain; RERMS, five-aa peptide sequence; CAPPD, central APP domain; L, linker; Aβ, amyloid-β peptide sequence; AICD, APP intracellular domain. Values are given as the mean ± SE (n = 3–12).
Figure 4.
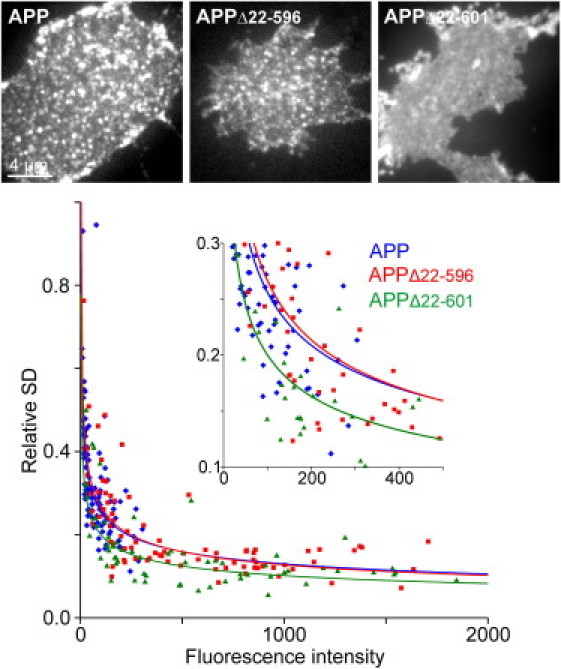
Truncation of Aβ is accompanied by cluster dispersion. Fixed membrane sheets generated from cells expressing the constructs as indicated. From individual membrane sheets the SD of the mean intensity was determined in a ROI and related to the mean background-corrected fluorescence, yielding the relative SD, which is a quantitative measure for the degree of clustering (18). For each construct, data from three to four independent experiments were pooled. From individual membrane sheets, the relative SD was plotted against the mean intensity and a function f(x) = a×xb was fitted (APP, a = 0.9961 and b = −0.295; APPΔ22–596, a = 1.169 and b = −0.321; APPΔ22–601, a = 0.7785 and b = −0.295). Please note that maximal and minimal values obtained at low and high expression levels are a consequence of mathematical processing and do not necessarily indicate that clusters disappear at high expression levels (the same effect is expected if clusters just become more numerous). Full-length APP (blue line) and APPΔ22–596 (red line) have the same degree of clustering, whereas the predominantly uniform distribution of APPΔ22–601 (green line) is indicated by a shift of the fitted line toward lower values. For each condition, 62–105 individual membrane sheets were analyzed. Images are shown at arbitrary scaling.
Figure 5.
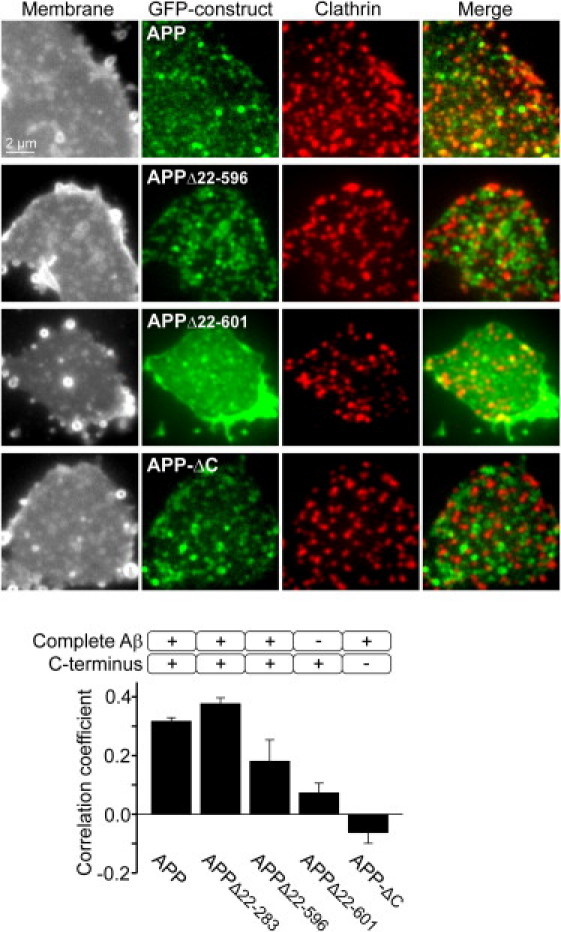
Targeting of APP into clathrin structures requires the entire Aβ- and the intracellular domain. Membrane sheets from PC12 cells expressing GFP-tagged APP, APPΔ22–283 (not shown), APPΔ22–596, APPΔ22–601, or APP-ΔC were fixed and immunostained for clathrin. Shown from left to right are TMA-DPH staining visualizing the integrity of the plasma membrane; the GFP channel showing the distribution of the respective APP construct; clathrin staining indicating endocytic clathrin structures and an overlay of GFP/clathrin signals. From central areas, the correlation between the GFP and the clathrin staining patterns was quantified by calculation of the Pearson correlation coefficient. The coefficient yields 1 in the case of two identical pictures (realistic values are much lower due to technical reasons), 0 when signals are unrelated, and −1 in the case of a picture and its negative. On the level of individual spots, the value of 0.32 obtained for full-length APP corresponds to 54% of APP clusters that are positive for clathrin (Fig. S6). Values are given as the mean ± SE (n = 3–4). Images are shown at arbitrary scaling.
Using the NCBI reference sequence NM_201414 as reference, the APP coding sequence of all constructs was verified by sequencing. Comparative fluorescence recovery after photobleaching (FRAP) analysis showed no difference between the two full-length APP constructs (with and without myc-tag; data not shown).
Cell culture
PC12 cells were maintained, propagated, and plated onto poly-L-lysine coated coverslips essentially as previously described (18). HepG2 cells were propagated in EMEM (BE12-992F, Lonza, Basel, Switzerland) supplemented with 10% fetal calf serum and 1% Pen/Strep at 37°C and 5% CO2. For transfection, we used a Microporator MP100 (Digital Bio, Seoul, South Korea) or a Neon-Transfection System (Invitrogen) applying 100-μl gold tips, adding 10–25 μg plasmid/transfection tip and adjusting settings to 1410 V and 30 ms for PC12 cells or 1200 V and 50 ms for HepG2 cells. For Fig. S4, cells were treated before the FRAP experiment with deglycosylating enzymes α-neuraminidase (0.5 U/ml) and peptide-N-glycosidase (0.1 U/ml) (N2876 and G5166, respectively, Sigma, St. Louis, MO) and β-galactosidase (0.16 U/ml; P07305, New England Biolabs, Ipswich, MA) in serum-free medium for 1 h at 37°C in the cell incubator. Then cells were mounted into a microscopy chamber in Ringer solution (130 mM NaCl, 4 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 48 mM glucose, and 10 mM HEPES, pH 7.4). Cells were used ∼8 h (Figs. 3–5, Fig. S3, Fig. S4, Fig. S5, and Fig. S6), 24 h (Figs. 1 and 2 and Fig. S2), and 8–24 h (Fig. S1) after transfection.
FRAP analysis
FRAP measurements in Ringer solution at room temperature (RT) and analyses were performed using an Olympus Fluo View 100 laser scanning microscope essentially as described previously (18). In brief, the pixel size was adjusted to 207 nm and the image size to 100 × 100 pixel, and bleaching was performed in a 15 × 15 pixel region of interest (ROI) using the 488-nm and 405-nm laser lines simultaneously. Depending on the experiment, bleaching duration varied from 1–2 s. In total, a sequence of 70 images was recorded, including three prebleach and 67 postbleach images. For analysis, apart from the bleaching ROI, values were obtained from a reference ROI in an unbleached area and a background value was determined. Recovery traces were background-subtracted and normalized to the average of the prebleach values. Vertical drift during the measurement was indicated by a loss of fluorescence in the reference ROI. When the value of the reference ROI decreased by >15%, the measurement was excluded.
The half-time of recovery was obtained by fitting a hyperbola (y(t) = offset + maxrec × t/(t + t1/2)) using Origin8Pro (OriginLab, Northampton, MA). For one experiment and condition, 3–11 cells were analyzed and half-times and normalized recovery traces were averaged.
The diffusion coefficient of a freely diffusing single-span membrane protein has been reported to be 0.23 μm2/s (19), which corresponds to the value 0.13 of the normalized half-time (Fig. 3).
TIRF microscopy
Total internal reflection fluorescence (TIRF) microscopy was performed essentially as already described (18). Cells were imaged at RT in Ringer solution (n = 3–4 independent experiments).
Wheat germ agglutinin staining
For Fig. S4, cells were pretreated with deglycosylating enzymes as above. Then membrane sheets were generated (18) and fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, and 8.1 mM Na2HPO4, pH7.4) for 1 h. PFA was quenched with 50 mM NH4Cl in PBS and membrane sheets were incubated with 5 μg/ml Alexa594-labeled wheat germ agglutinin (WGA) (W11262, Invitrogen) in PBS. Membrane sheets were washed with PBS and imaged in PBS containing 1-(4-tri-methyl-ammonium-phenyl)-6-phenyl-1,3,5-hexatriene p-toluenesulfonate (TMA-DPH) (T204, Molecular Probes, Eugene, OR). Imaging and image analysis (using the program ImageJ) were performed as described (18). For each independent experiment and condition, 24–39 membrane sheets were analyzed and values were averaged.
Immunostaining of membrane sheets
Immunostaining was performed according to a protocol essentially as previously described (18). For clathrin staining, a goat antibody directed against the clathrin heavy chain (sc-6579, Santa Cruz Biotechnology, Santa Cruz, CA) and, as secondary antibody, a donkey-anti-goat antibody coupled to Alexa594 (A11058, Invitrogen) were used. Membrane sheets were imaged in the presence of TMA-DPH to illustrate the integrity of the membrane. Membrane sheets with the expressed constructs were identified in the green channel and then imaged in the blue, green, and red channels to show the lipid staining (TMA-DPH fluorescence), GFP-fluorescence, and clathrin staining, respectively.
To correct for lateral shifts occurring during channel switching, and to ensure that the focus was properly adjusted, we added 100 nm TetraSpeck beads (T7279, Invitrogen/Molecular Probes) to the coverslip that were detectable in all channels. The correlation coefficient was determined using ImageJ, applying the ImageJ plugin Colocalization Indices (20). For each independent experiment and condition, 4–14 membrane sheets were analyzed and their values were averaged. For full-length APP, we also analyzed the overlap of APP and clathrin on the level of individual spots (Fig. S6) and corrected for accidental colocalization as previously described (21).
Detection of constructs at the cell surface
For detection of GFP-labeled APP, APPΔ22–596, and APPΔ22–601 at the cell surface, we followed a protocol essentially as previously described (16). In brief, PC12 cells expressing the respective constructs were cooled down in the cold room on ice, and all the steps described here were performed with ice-cold solutions. Cells were washed with Hank's BSS (HBSS; cat# H15-008, PAA Laboratories, Cölbe, Germany) and incubated for 30 min with a mouse monoclonal primary antibody (1:100 in HBSS; SIG-39300, Covance, Princeton, NJ) reactive for aa 1–16 of the Aβ peptide. Then cells were washed and incubated for 40 min with secondary antibody donkey-anti-mouse coupled to Alexa594 (1:200 in HBSS; A21203, Invitrogen). Cells were washed and fixed overnight in 4% PFA in PBS. Quenching of PFA with NH4Cl and embedding in Mowiol (6 g glycerol, 2.4 g Mowiol (4-88, Hoechst, Frankfurt, Germany), 6 ml ddH2O, and 12 ml of 200 mM Tris, pH 7.2) saturated with Dabco (0718.1, Carl Roth, Karlsruhe, Germany) was performed at RT. For imaging, we used the Olympus Fluo View 100 laser scanning microscope, applying the laser line 488 for GFP and the laser line 543 for Alexa594. In addition, cells were imaged in the differential interference contrast (DIC) mode. For analysis, we used ImageJ. In brief, using the DIC image as reference, a linescan was placed at the rim of the cell. In the green (GFP) and red (immunostaining) channels, the average intensities per pixel were measured and background-corrected. Pooling data from several independent experiments, immunostaining intensity was plotted against GFP fluorescence for each cell.
Results and Discussion
Clustered membrane proteins are restricted in diffusion (see, e.g., Zilly et al. (18) and He and Marguet (22)). Therefore, to probe the clustered state of APP molecules, we analyzed its diffusional mobility by FRAP. For fluorescent labeling, GFP was fused to the C terminus of the 695-aa large neuronal APP isoform and the construct was expressed in neuronal and liver model cell lines. For FRAP analysis, we focused on the basal plasma membrane, bleached fluorescence in a squared ROI, and monitored the recovery of fluorescence over time. The recovery rate was much slower than expected for free diffusion, although it was somewhat dependent on the time after expression, becoming accelerated between 8 and 24 h after transfection (Fig. S1). To test which protein domains restrict mobility in neuronal cells, we analyzed diffusion of a deletion construct lacking the complete extracellular domain (APP-ΔN) and found a severalfold higher mobility when compared to full-length APP (Fig. 1). We then asked whether this increased mobility is indeed due to a change from clustered to uniform molecule organization. To this end, we applied TIRF microscopy, which selectively illuminates the plasma membrane region, avoiding the imaging of deeper-lying intracellular compartments. As shown in Fig. 2, the majority of APP fluorescence was concentrated into punctate structures, whereas upon removal of the ectodomain (APP-ΔN) the distribution became uniform with additional spotty signals occasionally seen. This is in line with our assumption that fast and slow diffusion in FRAP experiments correspond to low and high degrees of protein clustering, respectively. Next, we deleted the intracellular domain (construct APP-ΔC, with the full extracelluar domain intact). The mobility was slightly increased (Fig. 1, B and C), and the distribution of APP-ΔC still had a clustered appearance in TIRF micrographs, although it was less clear than for APP (Fig. 2). Analysis of the deletion constructs in liver cells (Fig. S2) revealed identical results, implying that neuronal factors are not involved in the basic APP clustering mechanism.
So far, the data suggest that the extracellular domain of APP is essential for concentrating the protein into clusters in the membrane. In the absence of the intracellular domain, clusters still form but become more dynamic, in line with the presence of modulators stabilizing APP clusters from the intracellular side.
The extracellular domain could mediate APP clustering by several mechanisms. For instance, APP contains glycosylation sites in the ectodomain (23,24) that could interact with or be part of the extracellular glycocalyx. Other possibilities are that the bulkiness of the ectodomain could restrict mobility, or a special protein domain could be responsible for clustering. To test the first possibility, we performed FRAP experiments on cells with APP harboring point mutations at the glycosylation sites (Fig. S3) or cells with a degraded glycocalyx (Fig. S4). However, APP diffused normally, ruling out a role of the glycocalyx. We then performed FRAP analysis with progressively shorter constructs (Fig. 3). Over a wide sequence range, the deletions caused only small mobility enhancements approximately linear to the number of removed aa. This observation might be due to friction forces, which can be expected to be reduced with the smaller geometric sizes of the extracellular domains. In any case, because deletion of almost the entire ectodomain (construct APPΔ22–537) raised the mobility only modestly (Fig. 3), we can conclude that the globular size of the ectodomain does not produce a clustering pattern due to restriction by extracellular diffusion barriers (for a suggested mechanism based on intracellular diffusion barriers, see Kusumi et al. (25)). Notably, the mobility changed upon further deletion, with a remarkable stepwise increase caused by removing the first five aa of the Aβ region. These findings strongly suggest that APP clustering is mediated primarily through a specific interaction involving the N-terminal region of the Aβ domain, although some stabilizing effect of the more upstream-lying aa is notable. To verify that the protein disperses upon Aβ-shortening, we compared the lateral distribution of APP to the distributions of the two shortest variants, APPΔ22–596 and APPΔ22–601, applying fixed plasma membrane sheets (Fig. 4). Plasma membrane sheets were generated by brief ultrasound treatment of glass-adhered cells, leaving behind the basal plasma membrane. This preparation allows for high signal/noise ratio analysis of fluorescence signals. Moreover, unlike TIRF microscopy, it can be used to exclude the possibility that uniform fluorescence results from cytosolic GFP or that punctate signals arise from undocked vesicles close to the membrane (see also Fig. S5 for a surface staining showing the presence of the constructs within the plasma membrane). As shown in Fig. 4, both APP and APPΔ22–596 show a strongly clustered pattern, whereas APPΔ22–601 is predominantly uniformly distributed. This corroborates the conclusions drawn from the FRAP experiments, that the first aa of the Aβ-region dramatically affect the lateral organization of APP by immobilizing APP into clusters.
We then asked if clustering is an event associated with endocytosis and compared the APP clustering pattern to the distribution of clathrin. A high degree of overlap was measured only for constructs possessing both the Aβ and the intracellular domain, though the still clustered, but already more dynamic, APPΔ22–596 less efficiently correlated with clathrin. Compared to full-length APP, the construct with the partially deleted Aβ domain was almost fivefold less targeted to endocytic structures. This indicates that a certain degree of cluster stability is required for efficient entry into the endocytic pathway. We also tested the construct lacking the intracellular domain (APP-ΔC). As expected, APP-ΔC did not incorporate into clathrin structures, instead even showing some trend toward being excluded from the clathrin staining pattern (Fig. 5).
In summary, the data show that a few aa within the Aβ region are sufficient for the formation of APP clusters. In contrast, the intracellular domain of APP is incapable of forming clusters on its own, though it gives additional stabilization to Aβ-formed clusters from the intracellular site.
As shown by our clathrin staining experiments, the intracellular portion of APP directs the molecule into endocytic structures. At present, it is unclear whether clathrin is recruited to the site of clustering or whether clustered APP becomes trapped into clathrin assemblies, but we can safely conclude that the process is only effective upon APP immobilization by Aβ. Probably, clathrin structures interfere with the dynamics of APP clusters, explaining the slight increase in APP mobility upon removal of the intracellular domain. In any case, the data show that tight clustering is a prerequisite for directing APP efficiently to the endocytic pathway. Therefore, we conclude that APP clustering and immobilization is a key step in APP trafficking from the plasma membrane via clathrin-coated vesicles to early endosomes (Fig. 6).
Figure 6.
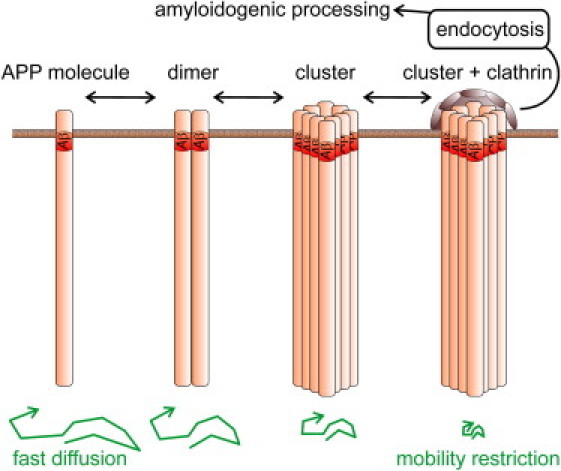
APP cluster formation within the plasma membrane. Unprocessed APP molecules reach the cell surface through the constitutive secretory pathway, where they cluster via the N-terminal aa of Aβ. APP dimers may form via alternative mechanisms involving regions of the extracellular domain (30–32) or consecutive GxxxG motifs more downstream of the Aβ N-terminus (28). With increasing cluster size, APP molecules become immobile and locally provide more binding sites for the endocytic machinery, favoring full clathrin assembly or sorting to clathrin structures, internalization, and amyloidogenic processing. Interference with the clustering reaction from the extracellular site would increase the time APP resides in the plasma membrane and thereby the chance to be cleaved by α-secretase, resulting in nonamyloidogenic processing.
Inhibition of β-secretase function has already been suggested (13,26) as a means to prevent Alzheimer's disease, but it would require a pharmaceutical with intracellular activity and would carry the risk of possible side effects due to inhibition of an enzyme that also has physiological relevance (27). In contrast, prevention of APP clustering into endocytic structures requires the pharmaceutical to act only from the extracellular site (Fig. 6). Moreover, in analogy to a mechanism suggested to produce shorter Aβ peptides when interfering with GxxxG-motif-mediated APP dimerization (28), dispersed APP may allow γ-secretase to bind the APP molecule farther upstream, leading to the production of shorter and less harmful Aβ peptides.
In a nutshell, our data show that for APP clustering, the Aβ-domain is essential. Moreover, clustering is a prerequisite for targeting APP to endocytic structures. These findings indicate what we consider a novel central step in APP trafficking, which is intriguing for two reasons: first, the Aβ domain initiates its own intracellular production, and second, the finding is medically relevant, as it suggests a new route for battling Alzheimer's disease.
Acknowledgments
The authors acknowledge a plasmid gift encoding for myc-tagged human APP695 from Dr. Stefan Kins (Zentrum für Molekulare Biologie der Universität Heidelberg, Heidelberg, Germany) and thank Dr. Michael Famulok (University of Bonn, Bonn, Germany) for providing HepG2 cells and Dr. Thomas Quast for support with the confocal microscope. We also thank Dr. Ines Raschke, Dr. Jan-Gero Schloetel, and David Walrafen for helpful comments on the manuscript.
This project was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB645).
Supporting Material
References
- 1.Li Q.X., Fuller S.J., Masters C.L. The amyloid precursor protein of Alzheimer disease in human brain and blood. J. Leukoc. Biol. 1999;66:567–574. doi: 10.1002/jlb.66.4.567. [DOI] [PubMed] [Google Scholar]
- 2.Walsh D.M., Selkoe D.J. A β oligomers—a decade of discovery. J. Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 3.O'Brien R.J., Wong P.C. Amyloid precursor protein processing and Alzheimer's disease. Annu. Rev. Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He X., Cooley K., Tang J. Apolipoprotein receptor 2 and X11 α/β mediate apolipoprotein E-induced endocytosis of amyloid-β precursor protein and β-secretase, leading to amyloid-β production. J. Neurosci. 2007;27:4052–4060. doi: 10.1523/JNEUROSCI.3993-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinoshita A., Fukumoto H., Hyman B.T. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J. Cell Sci. 2003;116:3339–3346. doi: 10.1242/jcs.00643. [DOI] [PubMed] [Google Scholar]
- 6.Rajendran L., Honsho M., Simons K. Alzheimer's disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakono M., Zako T. Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS J. 2010;277:1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x. [DOI] [PubMed] [Google Scholar]
- 8.Baruch-Suchodolsky R., Fischer B. Aβ40, either soluble or aggregated, is a remarkably potent antioxidant in cell-free oxidative systems. Biochemistry. 2009;48:4354–4370. doi: 10.1021/bi802361k. [DOI] [PubMed] [Google Scholar]
- 9.Igbavboa U., Sun G.Y., Wood W.G. Amyloid β-protein stimulates trafficking of cholesterol and caveolin-1 from the plasma membrane to the Golgi complex in mouse primary astrocytes. Neuroscience. 2009;162:328–338. doi: 10.1016/j.neuroscience.2009.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou K., Gong J.S., Michikawa M. A novel function of monomeric amyloid β-protein serving as an antioxidant molecule against metal-induced oxidative damage. J. Neurosci. 2002;22:4833–4841. doi: 10.1523/JNEUROSCI.22-12-04833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tienari P.J., De Strooper B., Dotti C.G. The β-amyloid domain is essential for axonal sorting of amyloid precursor protein. EMBO J. 1996;15:5218–5229. [PMC free article] [PubMed] [Google Scholar]
- 12.Cirrito J.R., Kang J.E., Holtzman D.M. Endocytosis is required for synaptic activity-dependent release of amyloid-β in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vassar R., Kovacs D.M., Wong P.C. The β-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J. Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carey R.M., Balcz B.A., Slack B.E. Inhibition of dynamin-dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid β protein. BMC Cell Biol. 2005;6:30. doi: 10.1186/1471-2121-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nordstedt C., Caporaso G.L., Greengard P. Identification of the Alzheimer β/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J. Biol. Chem. 1993;268:608–612. [PubMed] [Google Scholar]
- 16.Schneider A., Rajendran L., Simons M. Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J. Neurosci. 2008;28:2874–2882. doi: 10.1523/JNEUROSCI.5345-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soba P., Eggert S., Beyreuther K. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005;24:3624–3634. doi: 10.1038/sj.emboj.7600824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zilly F.E., Halemani N.D., Lang T. Ca2+ induces clustering of membrane proteins in the plasma membrane via electrostatic interactions. EMBO J. 2011;30:1209–1220. doi: 10.1038/emboj.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kenworthy A.K., Nichols B.J., Lippincott-Schwartz J. Dynamics of putative raft-associated proteins at the cell surface. J. Cell Biol. 2004;165:735–746. doi: 10.1083/jcb.200312170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamura K., Watakabe A., Kaneko T. Transiently increased colocalization of vesicular glutamate transporters 1 and 2 at single axon terminals during postnatal development of mouse neocortex: a quantitative analysis with correlation coefficient. Eur. J. Neurosci. 2007;26:3054–3067. doi: 10.1111/j.1460-9568.2007.05868.x. [DOI] [PubMed] [Google Scholar]
- 21.Lang T., Margittai M., Jahn R. SNAREs in native plasma membranes are active and readily form core complexes with endogenous and exogenous SNAREs. J. Cell Biol. 2002;158:751–760. doi: 10.1083/jcb.200203088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He H.T., Marguet D. Detecting nanodomains in living cell membrane by fluorescence correlation spectroscopy. Annu. Rev. Phys. Chem. 2011;62:417–436. doi: 10.1146/annurev-physchem-032210-103402. [DOI] [PubMed] [Google Scholar]
- 23.Påhlsson P., Shakin-Eshleman S.H., Spitalnik S.L. N-linked glycosylation of β-amyloid precursor protein. Biochem. Biophys. Res. Commun. 1992;189:1667–1673. doi: 10.1016/0006-291x(92)90269-q. [DOI] [PubMed] [Google Scholar]
- 24.Perdivara I., Petrovich R., Przybylski M. Elucidation of O-glycosylation structures of the β-amyloid precursor protein by liquid chromatography-mass spectrometry using electron transfer dissociation and collision induced dissociation. J. Proteome Res. 2009;8:631–642. doi: 10.1021/pr800758g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kusumi A., Nakada C., Fujiwara T. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu. Rev. Biophys. Biomol. Struct. 2005;34:351–378. doi: 10.1146/annurev.biophys.34.040204.144637. [DOI] [PubMed] [Google Scholar]
- 26.Rajendran L., Schneider A., Simons K. Efficient inhibition of the Alzheimer's disease β-secretase by membrane targeting. Science. 2008;320:520–523. doi: 10.1126/science.1156609. [DOI] [PubMed] [Google Scholar]
- 27.Prox J., Rittger A., Saftig P. Physiological functions of the amyloid precursor protein secretases ADAM10, BACE1, and Presenilin. Exp Brain Res. 2011 doi: 10.1007/s00221-011-2952-0. Epub ahead of print November 27, 2011. [DOI] [PubMed] [Google Scholar]
- 28.Munter L.-M., Voigt P., Multhaup G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Aβ42. EMBO J. 2007;26:1702–1712. doi: 10.1038/sj.emboj.7601616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reinhard C., Hébert S.S., De Strooper B. The amyloid-beta precursor protein: integrating structure with biological function. EMBO J. 2005;24:3996–4006. doi: 10.1038/sj.emboj.7600860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beher D., Hesse L., Multhaup G. Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the binding sites on APP and collagen type I. J. Biol. Chem. 1996;271:1613–1620. doi: 10.1074/jbc.271.3.1613. [DOI] [PubMed] [Google Scholar]
- 31.Scheuermann S., Hambsch B., Multhaup G. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer's disease. J. Biol. Chem. 2001;276:33923–33929. doi: 10.1074/jbc.M105410200. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y., Ha Y. The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol. Cell. 2004;15:343–353. doi: 10.1016/j.molcel.2004.06.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.