

Abstract
Substrate transport in sodium-coupled amino acid symporters involves a large-scale conformational change that shifts the access to the substrate-binding site from one side of the membrane to the other. The structural change is particularly substantial and entails a unique piston-like quaternary rearrangement in glutamate transporters, as evidenced by the difference between the outward-facing and inward-facing structures resolved for the archaeal aspartate transporter GltPh. These structural changes occur over time and length scales that extend beyond the reach of current fully atomic models, but are regularly explored with the use of elastic network models (ENMs). Despite their success with other membrane proteins, ENM-based approaches for exploring the collective dynamics of GltPh have fallen short of providing a plausible mechanism. This deficiency is attributed here to the anisotropic constraints imposed by the membrane, which are not incorporated into conventional ENMs. Here we employ two novel (to our knowledge) ENMs to demonstrate that one can largely capture the experimentally observed structural change using only the few lowest-energy modes of motion that are intrinsically accessible to the transporter, provided that the surrounding lipid molecules are incorporated into the ENM. The presence of the membrane reduces the overall energy of the transition compared with conventional models, showing that the membrane not only guides the selected mechanism but also acts as a facilitator. Finally, we show that the dynamics of GltPh is biased toward transitions of individual subunits of the trimer rather than cooperative transitions of all three subunits simultaneously, suggesting a mechanism of transport that exploits the intrinsic dynamics of individual subunits. Our software is available online at http://www.membranm.csb.pitt.edu.
Introduction
Glutamate transporters (also known as excitatory amino acid transporters (EAATs)) located on neurons and glia remove excessive glutamate from the synapse after neuronal firing, preventing toxicity (1,2) and regulating proper activation of postsynaptic receptors (3). This central role in regulating neurotransmitter activity makes glutamate transporters attractive drug targets for neurological diseases. Transport of glutamate is driven by the symport of three Na+ ions (4). As Na+ ions travel across the membrane from extracellular (EC) to cytoplasmic (CP) regions down their electrochemical gradient, glutamate is concurrently transported against its concentration gradient. Although no human glutamate transporters (i.e., EAATs) have yet been structurally resolved, crystal structures have been determined for the orthologous aspartate transporter GltPh from the archaeon Pyrococcus horikoshii (5–7). Cross-linking experiments (8–10) point to a strong structural similarity between EAATs and GltPh, and the latter is used as a structural model for the former.
GltPh is a homotrimer that is assembled in such a way that its three subunits are arranged symmetrically about a central axis that is normal to the membrane (Fig. 1). Each subunit has two domains: 1), the N-terminal cylinder, or scaffold, consisting of transmembrane (TM) helices TM1–TM6, which form the intersubunit interface; and 2), the C-terminal core domain, consisting of helices TM7 and TM8 and the helical hairpins HP1 and HP2 (5), which are involved in substrate binding and transport (11–13). In the outward-facing (OF) conformation (5,6) shown in Fig. 1 A, the subunits form a solvent-filled basin ∼50 Å deep and 30 Å wide facing the EC region. The subunit structure exhibits a remarkable pseudo-symmetry: Using rigid-body rotations, helices TM1–TM3 of the scaffold can be superimposed on TM4–TM6, and HP1 and TM7 of the core domain can likewise be superimposed onto HP2 and TM8. A homology model of the inward-facing (IF) structure (Fig. 1 B) that we generated from the OF structure by exploiting this symmetry (14) agreed well with the concurrently resolved IF crystal structure (6).
Figure 1.
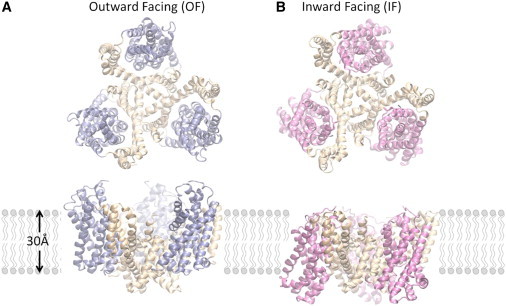
OF and IF structures of GltPh show a large displacement of the core domains. Each subunit of the homotrimer contains a trimerization domain (wheat) and a transport domain (blue and pink). (A) In the OF conformation (PDB code: 2NWL), the transport domains extend into the EC medium, forming an aqueous basin around the trimer interface. (B) In the IF conformation (PDB code: 3KBC), the transport domains are displaced across the membrane to the CP, whereas the trimer interface experiences little conformational change. The top diagrams in panels A and B represent the views from the EC region, and the lower diagrams display the side views within the membrane. Lipid molecules are represented by simple polar heads and hydrophobic tails. Molecular representations were rendered in VMD (47).
In the conventional alternating access model of glutamate transport (15), the transporter binds glutamate on the EC side of the membrane and releases it on the CP side. Chemical and computational studies have uncovered details about several aspects of the transport cycle (Fig. 2). The cycle involves local and global changes in structure. Molecular-dynamics (MD) simulations revealed that the HP2 loop is quite flexible in the OF conformation, and that its opening in the apo form of the subunit exposes several polar residues, prompting Na+ and substrate entry and binding (12). The additional binding of two Na+ accompanied by the dislocation of the ions/substrate stabilizes the closed holo form of the OF conformation (16). These structural changes (HP2 loop opening, substrate/ion binding and relocation) are relatively fast and local events that occur within the timescale that is accessible to full atomic simulations. Next, a global structural change to the IF conformation takes place (Fig. 2, D and E). Although the physical mechanism behind this change has attracted wide interest, it has not yet been elucidated. MD simulations starting from the IF conformation showed that the release of substrate in the IF conformation also involves as series of local rearrangements initiated by the release of a Na+ through the open HP2 loop (17). The opening of the loop and release of sodium ion(s) allows water molecules to enter the binding site and dislodge the substrate, which eventually exits through combined motions of HP1 and HP2. These hairpins' loops presumably close, putting the transporter into an occluded apo state of the IF conformation and allowing it to undergo another global structural change back to the OF conformation, completing the cycle.
Figure 2.
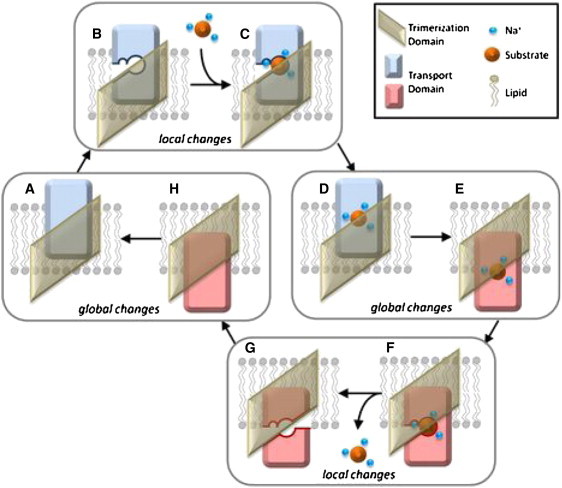
Many of the details of the GltPh transport cycle have been resolved. This cartoon summarizes the process for a single subunit. Starting from the OF unbound state (A), the flexible HP2 loop opens (B), exposing the sodium and substrate-binding sites. Three Na+ ions and the substrate bind (C), starting from the binding of a first Na+ (to the Na3 site), and then bind to a substrate and a sodium ion (to the Na2 site), and finally to the Na1 site. Here, for simplicity, we do not show these intermediate steps. The HP2 loop closes (D) and the transport domain moves through the membrane to the cytoplasm, placing the subunit in the IF closed state (E). The HP2 loop again opens (F), permitting the release of one Na+ ion from the Na1 site and subsequent hydration of the binding pocket. Water molecules aid in substrate dissociation by promoting the opening of HP1 and flooding the binding site, eventually leading to exit of substrate and the remaining Na+ ions (G). When the HP loops close (H), the transport domain moves back across the membrane to the EC face, completing the cycle. The global conformational changes D-E and G-A are the focus of this study.
Still unresolved are the mechanisms of the global structural change between OF and IF conformations, which have eluded MD studies owing to the prohibitively large size of the molecule and magnitude of the structural transition. Coarse-grained models, such as elastic network models (ENMs), appear to be useful for circumventing this issue. In particular, the anisotropic network model (ANM) (18,19) can usually be used to investigate the global conformational changes of bimolecular systems at a manageable level of complexity, at the cost of losing precision on a local (atomic) scale (20). With the publication of the IF structure (7), however, it became clear that neither these motions nor a combination of other low-energy ANM modes could adequately account for the nearly 18 Å displacement of the core domain through the membrane.
We hypothesize that the omission of the influence of membrane on the protein dynamics is a source of discrepancy between ANM predictions and experimental data, i.e., the inability of ANM to account for the structural change between the OF and IF substates of GltPh is proposed to be rooted in the lack of inclusion of membrane effects in the canonical ANM. Studies in which ANM was applied to a number of membrane proteins suggested that the membrane plays a minor role in the global dynamics of potassium channels (21) and G-protein-coupled receptor (22). However, other work indicated that it has a measurable influence on the dynamics of the nuclear pore complex (23) and porins (24), suggesting that the effect of the lipid bilayer may become more or less pronounced depending on the type of (protein) structure-encoded motions involved, with some motions experiencing stronger resistance than others. The motions that are intrinsically accessible to GltPh trimeric structure may entail such strong resistance, and the suitable incorporation of the constraints exerted by the membrane on the intrinsic dynamics of the transporter may help elucidate the collective motions that enable the alternating access of the transport core to the EC and CP regions during its neurotransmitter transport cycle, consistent with the two known endpoints.
The test this hypothesis, we explored the global conformational transition of GltPh using two novel (to our knowledge) methods for incorporating membrane effects into the ENM. The first method, termed implicit membrane ANM (imANM), uses a modification of the force constants to preferentially restrain radial motions, thus mimicking the constraining effects of the surrounding lipid molecules. The second method, which we call explicit membrane ANM (exANM), models the membrane as a collection of point masses, much like the amino acids in the network. We find that in both cases, the predicted dynamics of the modified ENMs accounts for the observed OF-IF transition better than the canonical ANM. Starting from one (known) conformer (OF or IF), the endpoint (IF or OF) is predicted to be along the lowest-energy, or softest, modes of motions that are intrinsically accessible to GltPh in the presence of the lipid environment.
The spatial anisotropy resulting from the membrane is thus observed to play an important role in selectively modulating the motions of GltPh. Of most importance, the predicted soft modes explain for the first time (to our knowledge) the physical mechanism of transition between these two endpoints (OF and IF), which differ by a root mean-square deviation (RMSD) of 9.7 Å. A previous computational prediction of the IF state starting from the OF state, performed by cutting/pasting symmetrically related structural elements, had left us with the unresolved puzzle of how the molecule undergoes such reconfigurations during its allosteric cycle. The results presented here show that there is indeed a single mode/path, favored by the trimeric architecture, that enables the cooperative transition of GltPh from the OF to the IF state, provided that the implicit/explicit effect of the membrane is incorporated into the model.
Materials and Methods
Structural preparation and alignment
Protein structures and membrane information were taken from the Orientations of Proteins in Membranes (OPM) database (25). When required, residues were removed so that all structures would have identical sequence, yielding two structures of 1194 residues each.
ANM
In the ANM, each residue constitutes a node of the network, and nearby nodes are connected by uniform Hookean springs. The total potential is a sum of pairwise interactions:
| (1) |
where γ is the spring constant between nodes, Rij is the instantaneous distance between nodes i and j, Rij0 is their equilibrium distance, rc is a cutoff distance of 11 Å, and H(x) is the Heaviside step function (equal to 1 if x > 0, and 0 otherwise). If Δr is the 3N-dimensional vector of mass-weighted displacements of the N nodes in the system from their equilibrium positions, then the dynamics of the protein obeys the set of 3N equations of motion written in compact notation as
| (2) |
Here H is the 3N×3N Hessian matrix of second derivatives of the potential with respect to the components of r. From Eq. 1, the 3×3 super-element of H corresponding to the interaction between residues i and j is
| (3) |
where the zero superscripts indicate equilibrium values. The diagonal super-elements satisfy
| (4) |
Diagonalization of H yields six eigenvalues that are identically zero, corresponding to the rigid-body rotations and translations of the molecule, which carry no internal energetic cost. The nonzero eigenvectors, v(k) (1 ≤ k ≤ 3N-6), of H define the 3N directions of collective fluctuations (modes of motion) near equilibrium state for the system of N nodes, and the corresponding eigenvalues, λk, are the squares of the oscillatory frequencies in units that are determined by the spring constant, γ. The kth eigenvalue represents the curvature of the potential energy surface along the kth mode direction (18): modes with large eigenvalues have high energetic curvature and indicate rigid, high-frequency motions, whereas those with smaller eigenvalues have lower curvature and correspond to low-frequency soft motions. The lowest-frequency oscillations thus require the least energy for a given deformation. As a result, motions along those directions are most easily accessible to the system, and we are usually interested in these softest modes, which are known to be uniquely defined by the structure and have been postulated to facilitate protein function.
imANM
We introduce a simple modification to the ANM to implicitly incorporate the perturbation in protein dynamics induced by the presence of the membrane. The idea is to take account of the resistance to reconfiguration exerted by the lipid bilayer when the movement of the protein entails a deformation against the membrane. Toward that end, we first rewrite Eq. 3 as
| (5) |
where the uniform spring constant γ has been replaced by the direction-dependent spring constants γx, γy, and γz, allowing us to independently alter the force constants along each Cartesian coordinate. We mimic the radial constraining effect of the membrane by adopting the relation
| (6) |
where s > 0 is a scaling factor for radial motions (along the x- and y-directions, normal to the membrane surface being along the (axial) z-direction). Radial motions of the protein are suppressed when s > 1 and enhanced when s < 1. Although this constraint may be readily assumed to depend on the position (elevation) along the membrane, we adopted as a first approximation a uniform scaling factor s = 16 for all residues (see Fig. S1 in the Supporting Material). More elaborate models, based on the work of Cantor (26,27), which involve the force constants varying with penetration depth, do not significantly alter the results.
The RTB method (28,29) was used to minimize unrealistic distortions in the protein structure. This technique consists of grouping residues into rigid blocks and expressing the normal modes in terms of rotations and translations of the blocks. Our aim was to avoid the distortion of transmembrane hydrogen-bonded secondary structures, and to that end we defined each helix identified by the DSSP (30) as a rigid block. This procedure eliminates the unphysical stretching of TM helices. We note that kinks in helices, such as those that appear in TM7 and TM8, are recognized by the DSSP as a disruption of secondary structure and therefore remain flexible in our model. To avoid an unrealistic deformation of backbone virtual bonds, we increased the spring constant between adjacent residues along the backbone by 100-fold, consistent with previous work (31–33) that demonstrated that the adoption of stiffer springs for bonded interactions improves the agreement between predicted and experimentally observed mean-square fluctuations.
exANM
A more elaborate and computationally more complex method of incorporating the effects of the lipid bilayer involves explicit inclusion of the lipid molecules as additional nodes in the ENM. The assumption here is that several lipid atoms occupying a small volume in the membrane can be represented by a single coarse-grained site, or centroid. Protein-lipid interactions are then represented by harmonic interactions between residues and membrane centroids. This model is not meant to reproduce the complicated physics of the lipid bilayer, but to represent the membrane through a discretized mean-field approximation so as to incorporate the anisotropic protein/membrane interactions near equilibrium in the ENM (see Fig. S2). The centroids are represented by spheres of radius 3.1 Å each, and arranged in a face-centered cubic lattice with a depth of 27 Å (obtained from the OPM database) and a radial extent of 80 Å. The size of the centroids was selected to give approximately the same node density as is present in the protein. The membrane thickness and axial position were taken from the OPM database (25), and an 80 Å radius was used for the membrane surface because it extends significantly beyond the edge of the protein (see Fig. S2). We calculated the motions of the protein under the influence of the membrane by integrating over the degrees of freedom provided by the membrane (32,34). Force constants were assigned separately for the membrane and the protein, and a membrane force constant twice as stiff as that of the protein was empirically found to yield good results.
The source code is available online at http://www.membranm.csb.pitt.edu.
Results and Discussion
The intrinsic motions of GltPh distinguish between trimerization and transport domains
We explored the dynamics of GltPh using the ANM and two membrane-specific variants, imANM and exANM, as described in Materials and Methods. For the OF conformation, the first three ANM modes are pincer-type openings and closings of the trimer characterized by radial motions of the core domains with respect to the scaffold (see Fig. 3 A, Movie S1 and Movie S2). The first two modes are degenerate and asymmetrical, resulting in two core domains moving in one radial direction as the third moves oppositely. The third mode is a nondegenerate symmetrical motion that moves all three core domains identically. When combined, these motions describe the independent radial opening/closing motions of the core domains with respect to the trimer interface, in accord with experimental evidence suggesting that the three core domains function independently of each other (35–40). The first two ANM modes of the IF conformation are asymmetric twisting of the core domains with respect to the trimer interface (Fig. 3 A and Movie S3), and the third ANM mode of the IF conformation is a nondegenerate symmetrical twisting of the cores with respect to the trimer interface (Movie S4). These modes are analogous to the first modes of the OF conformation, but with the outward opening/closing motion replaced by a twisting motion that increases the exposure to the CP region.
Figure 3.
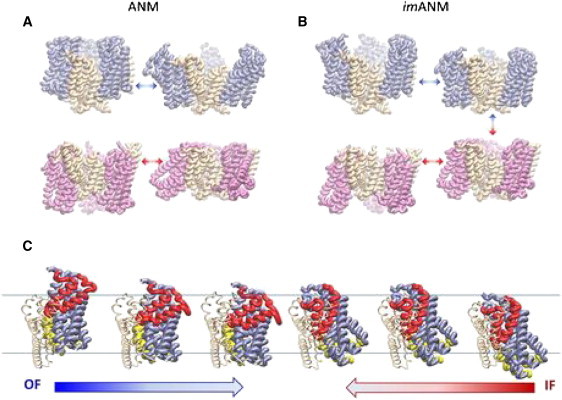
First three modes of imANM suggest a mechanism for conformational change. (A) The slowest nondegenerate ANM mode for the OF conformation of GltPh (top) is a pincer-type opening/closing of the trimer through motions of the transport domains. The same mode in the IF conformation (bottom) is a coordinated twisting of the transport domains. (B) In imANM, the first nondegenerate mode is a piston-type axial motion of the transport domains for both the OF and IF conformations. It allows both the OF and IF conformations to move toward a transition intermediate, indicated by the arrows at far right. In all cases, the first twofold degenerate mode is the same as the mode shown, but with one of the subunits phase-shifted by 180°. (C) The transition for a single subunit. Starting from the OF crystal structure at far left, the motion along the first nondegenerate mode brings the transport domain into the membrane, as shown by the blue arrow. Starting from the IF crystal structure at right, the slowest nondegenerate mode moves the transport domain up toward the EC region. The change that is not captured by this mode is the difference between the two centermost structures. HP1 and HP2 are colored yellow and red, respectively.
The softest modes divide each of the three subunits into two substructures that move almost rigidly with respect to each other: the transport domain and the trimerization domain (Fig. 4). The transport domain is composed of the core domain elements TM7, TM8, HP1, and HP2, and the TM helices TM3 and TM6 (colored blue and pink in Fig. 1, A and B). It undergoes concerted radial opening/closing motions with respect to the trimerization domain, whereas the trimerization domain (consisting of TM1, TM2, TM4, and TM5; colored wheat in Fig. 1) remains rigidly embedded in the membrane.
Figure 4.
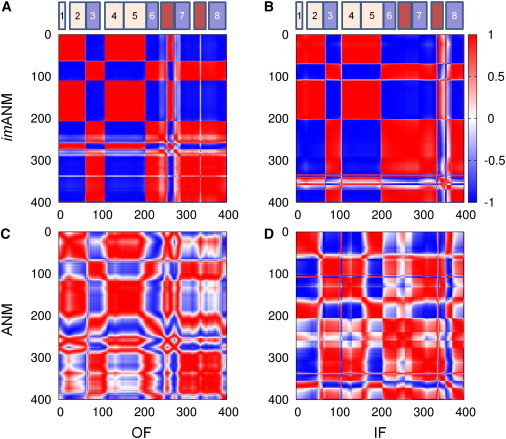
Separation of each subunit into transport and trimerization domains is visualized by using the cosines of the angles between residue motions (Eq. S4). Each matrix element indicates the cosine of the angle between motions of two residues, as calculated from the second mode of imANM (A and B) or ANM (C and D). Red indicates comoving (fully correlated) residues, and blue indicates oppositely moving (fully anticorrelated) residues. (A) The second imANM mode moves TM1, TM2, TM4, and TM5 in one direction, and TM3, TM6, TM7, HP2, and TM8 in the opposite direction. The coupling of TM3 and TM6 to the transport domain is contrary to the original domain definition provided by Boudker and co-workers (6). For reference, schematics of the locations of the eight TM helices are also shown, with the transport domain in blue, the trimerization domain in wheat, and the HP loops in dark red. (B) A similar sharp domain separation is seen for imANM mode 2 of the IF conformation. (C and D) The ANM results produce a less clear demarcation.
It is worth noting that the ANM-predicted parsing of the protein into these two domains is different from the core and scaffold domains originally proposed by Boudker and co-workers (6) based on the examination of one static (OF) structure. The main difference between the two definitions is the coupling of TM3 and TM6 to the transport domain rather than to the scaffold. This finding is consistent with a definition of domains that Reyes et al. (7) inferred by comparing the crystallographically resolved OF and IF structures. Specifically, they noted that TM3 and TM6 were reoriented en bloc together with the core domain during the transition between OF and IF conformations. This gave rise to the definition of a transport domain that contains these helices in addition to the originally defined core domain. The fact that the ANM results are consistent with the latter domain parsing suggests that this particular dynamics-based domain separation is intrinsically favored by the architecture of the individual subunits.
The softest modes predicted in the presence of the membrane account for half of the transition between OF and IF conformations
In imANM, the radial motions of the transport domains are severely diminished (if not completely suppressed), whereas axial motions become more pronounced. Instead of the twisting or pincer-type opening and closing motions predicted by the ANM, the transport domains under the imANM potential move in piston-like risings and fallings with respect to the trimer interface (Fig. 3 B, Movie S5, Movie S6, Movie S7, and Movie S8). Analogously to the ANM, the first two modes define a doubly degenerate asymmetric motion of the transport domains with respect to the trimer interface, and the third mode is a nondegenerate symmetric motion of all three transport domains in unison. The piston-type motion observed in imANM holds for both OF and IF conformations, suggesting a merging pathway for the conformation change starting from either endpoint. Notably, the overlap (see Supporting Material) between the first two modes that are accessible to the OF conformation and those that are accessible to the IF conformation is 0.821 in imANM (and 0.647 in ANM), and the overlap between the third mode that is accessible to the OF and IF conformations is 0.855 in imANM (and 0.587 in ANM). These similarities between the soft modes obtained with imANM for both endpoints (OF and IF) suggests that the imANM potential defines an energetically favorable path that fits the observed global conformational change. Fig. 3 C illustrates snapshots along this pathway.
The contrast between the slowest modes of ANM and imANM is most distinctive when we compare the modes with the observed deformation between the OF and IF conformations (Fig. 5). Because they are asymmetric, the first two modes do not contribute to the symmetric deformation between the conformations, but the third, which is the first nondegenerate imANM mode, contributes disproportionately to the deformation between these conformations. When calculated from the OF and IF conformations, respectively, this mode has an overlap of 0.521 and 0.549, alone, with the deformation vector. The equivalent cumulative overlaps obtained using the ANM potential are 0.161 and 0.064, respectively. Interestingly, combining the third mode of the IF conformation with that of the OF conformation does not significantly improve the overlap. When both structures are deformed simultaneously along the third imANM mode, the resultant motion has a maximum overlap of 0.565 with the observed deformation (compared with 0.178 for ANM). In terms of RMSD, the distance between structures drops from its initial value of 9.712 Å to 8.014 Å when both conformations are deformed along the third imANM mode (an RMSD of 9.577 Å is attained using the third ANM mode for both conformations).
Figure 5.
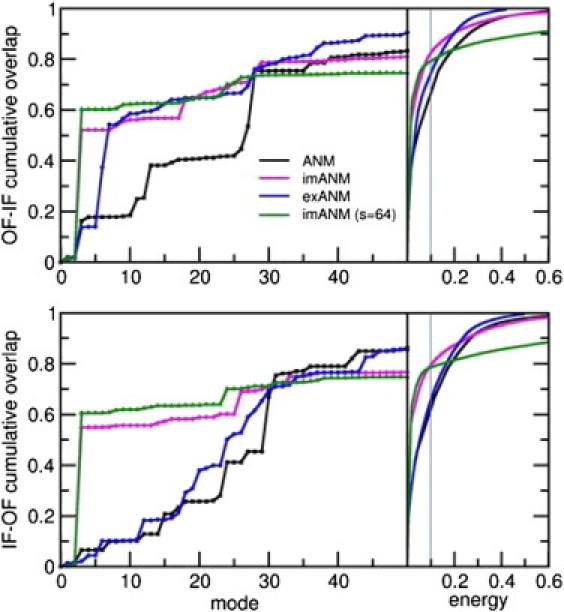
Inclusion of the membrane improves the efficiency of the modes in reproducing the structural deformation. In the left panels, the cumulative overlap of the deformation vector is plotted against the slowest 50 modes using different ENMs. The ANM (black) shows a steady increase in overlap with the number of modes considered. In contrast, the first nondegenerate mode of the imANM (magenta) accounts for more than half of the deformation in either direction. The exANM (blue) shows behavior similar to that of the imANM for the OF-IF transition, and ANM-like behavior for the IF-OF transition. A larger overlap can be obtained by increasing the imANM membrane scaling factor, but this results in loss of overlap in higher modes (green). In the right panels, the cumulative overlap is plotted against the normalized energy of deformation between the two conformations. The membrane-augmented models (magenta and blue) allow for greater displacements at lower energies compared with ANM (black). The energetic cost of using the imANM eventually overtakes that of ANM.
The behavior of the explicit membrane model is somewhere between that of ANM and imANM (see Fig. 5). Starting from the OF conformation, the first three emANM modes closely resemble those of the ANM, but modes 6 and 7 have a combined overlap of 0.524 with the deformation. The overlap curve mirrors that of imANM beyond the seventh mode. Starting from the IF conformation, the emANM results for the IF-OF transition are nearly identical to those of ANM through the first 50 modes. The difference in behavior of the two membrane models is likely due to the difference in propensities for the OF and IF conformations to exhibit radial motions. As shown in Fig. S3, the slowest modes of the OF conformation have a larger radial component (detailed in the Supporting Material) than those of the IF conformation. By design, the imANM inhibits radial motions of both conformations, whereas the emANM only suppresses those radial motions that occur in the absence of membrane. The implicit membrane and explicit membrane models produce similar behavior in the OF conformation because both models suppress the intrinsic radial motions of the OF molecule. The more subdued radial motions of the IF conformation are mostly unaffected by the explicit membrane, but are preemptively suppressed by the implicit membrane.
The presence of the membrane lowers the energetic cost of the GltPh conformational change
In contrast to the improved performance of emANM in the higher modes, imANM results are tepid beyond the third mode. Indeed, higher imANM modes contribute very little to the OF-IF deformation, whereas the contributions of higher ANM modes accumulate steadily. Around mode 30 the cumulative overlap of the ANM modes with the deformation overtakes that of imANM (Fig. 5). Physically, the imANM modes degrade into highly localized motions much earlier than the ANM modes do, resulting in insignificant contributions toward the deformation. We observed that this trend depends on the value of the scaling constant that determines the membrane constraints (see Materials and Methods). As the scaling factor s increases, the contribution of the first nondegenerate mode to the deformation increases, but the contributions of the higher modes decrease.
The energetic costs of the conformational deformation using various models are shown in the right panels in Fig. 5. By rescaling the eigenvalues of the Hessian matrix so that the trace of the covariance matrix is unity, we can directly compare the energy associated with motions across different models. For example, we see that in ANM, ∼65% of the OF → IF change can be accounted for by using only 10% of the total energy required to complete the transition, or by moving in a subspace spanned by <30 modes; the remaining energy is used in specific local motions. Notably, the same fractional energy allows one to achieve ∼82% of the global change using imANM, because the modification introduced by the imANM potential decreases the initial energy gradient for the transition. Eventually, the energetic cost of deformation in imANM exceeds that of ANM. This happens after 97% of the conformational change is complete, which takes ∼275 modes from either model. Interestingly, the full transition requires less energy using exANM than it does using ANM. However, the accuracy of coarse-grained ENMs is known to diminish at high modes (41), so we limit our analysis to relatively low energies. For comparison, we display in Fig. 5 the results obtained using imANM with a scaling factor of s = 64. This increases the overlap between the third mode and the deformation (see Fig. S1), but leads to a sharp increase in energy at the higher modes. We therefore adopt a scaling factor of 16 because it produces a considerable overlap with the deformation with a rather small energy requirement.
The lowest-energy transitions involve the motion of a single subunit at a time
The degenerate modes could conceivably contribute to the motion of a single subunit, such as would be observed if the three subunits functioned independently. To explore the effects of the degenerate modes on the motions of individual subunits, we generated two hypothetical intermediate structures described as outward-dominant (OF) and inward-dominant (ID). In the OD structure, two subunits are in the OF conformation and the third is in the IF conformation, whereas in the ID structure, two subunits are in the IF conformation and the third is in the OF conformation. The overlap of modes with the deformations to and from these modeled intermediate structures is presented in Fig. 6. It can be seen that in several cases a single slow mode contributes disproportionately to the deformation. The imANM curves are nearly the same regardless of whether the transition endpoints are crystal structures or modeled intermediates. This suggests that, in terms of transitions permitted by the slowest modes, the imANM potential does not favor the crystal structures over the modeled intermediates. The ANM and exANM curves often show better overlap with deformations involving the modeled intermediate conformations compared with those involving the crystal conformations. For example, the OF conformation shows better overlap with the transition to the OD intermediate than to the IF conformation, and the IF conformation shows better overlap with the ID intermediate than with the OF conformation. These observations hint that the OD and ID conformations may be energetically favored transition intermediates between the OF and IF conformations.
Figure 6.
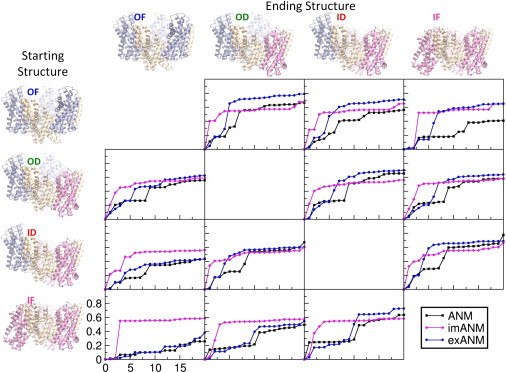
Cumulative overlap curves suggest possible transitions to intermediate conformations. Each plot shows the overlap of the first 20 modes of ANM (black), imANM (magenta), and exANM (blue), with the deformation between the starting structure shown at left and the ending structure shown at top.
This idea gains support from the analogous energy curves shown in Fig. S4. The plot shows log energy versus normalized deformation for all transitions using the three ENMs. In each panel, the lowermost curve indicates the least energetic transition. Starting from the OF conformation, the easiest transition is to the OD conformation (green curves), from which the easiest transition is back to the OF conformation (blue curves). This result, which is independent of the ENM used, implies that a conformational change involving a single subunit is favored to the simultaneous motion of two or three subunits. Similarly, the favored motion from the IF conformation is to the ID intermediate (red curves), which favors motion back to the IF conformation (magenta curves). Again we see that the energetically favored motion is of a single subunit, rather than multiple subunits in unison. Our results further indicate that the energetically easiest transitions are those between the OD and ID intermediates, suggesting that the molecule exists primarily in either the OF or IF state (or both) and that the OD and ID states are transitory.
Although our models provide relative barrier heights for conformational transitions, they cannot predict free-energy differences between conformations. We note that the wild-type GltPh forms crystals in the OF conformations (5), but a double cysteine mutant is required to form crystals in the IF conformation (7), suggesting that the OF conformer is more stable. This information together with the predicted relative transition energies leads us to propose that the resting state of GltPh is the OF conformation, with all subunits facing the EC side of the membrane. Upon substrate binding, a single subunit changes its conformation, allowing its transport domain to translocate toward the cytoplasm. When transport is complete, the empty subunit moves back to the EC side of the membrane. This mechanism allows two subunits to bind substrate on the EC side while the third subunit is transporting.
We note that conformational transitions depend specifically on the binding states of the subunits, and not entirely on the global conformation of the transporter. Exactly how substrate and ion binding influences the molecule's energy function is still unknown. One possibility is that it shifts the curves of Fig. S4 to promote transitions to otherwise unfavorable states; however, the highly localized structural changes induced by substrate or inhibitor binding are manifested primarily in the side chains and are not expected to significantly alter the backbone conformation. The resolution of the coarse-grained ENMs used in this study is too low to investigate these effects, but a more detailed study of the reaction kinetics using all-atom MD may shed light on the role of substrate binding in transporter function. Pinpointing the effects of substrate or inhibitors on the global conformational change has proved to be a challenge. To date, our laboratory has performed MD simulations totaling >360 ns for the GltPh trimer (12,13,17), yet not one instance of the global conformational change has been observed. Pending the outcome of microsecond-scale simulations, we are driven toward a multiscale approach to the problem that exploits the complementarity of the coarse-grained and all-atom models.
Is the method applicable to other membrane proteins?
The two models and corresponding analyses are applicable to any membrane protein with known structure, and the software is accessible online. We attribute the sensitivity of GltPh under the modified potentials to the molecule's intrinsic propensity toward radial motion (see Fig. S3). This result suggests that the net effect of the membrane on structural protein dynamics is to stifle radial motion rather than to inhibit all motions. The influence of the membrane is thus rather pronounced in GltPh, raising the question of whether other membrane proteins exhibit similar perturbations in their global dynamics. The application of the ANM to several membrane proteins has yielded global modes that are consistent with known (experimentally observed) structural changes (20). It is worth noting, however, that most of these membrane proteins tend to undergo global torsion/twisting motions around their longitudinal (z) axis (perpendicular to the membrane) in their softest nondegenerate ANM mode (42). This type of collective motion exerts shear forces against the membrane and can be accommodated by local redistribution of the membrane. In contrast, the normal (radial) forces exerted against the membrane by GltPh necessitate significant displacements to create an extra volume within the membrane environment. Examples of membrane proteins that favor global torsion/twist motions successfully described by the canonical ANM include rhodopsin (22), potassium channels (21), mechanosensitive channel MscL (43), nicotinic acetylcholine receptor (44), and gramicidin (45). The application of exANM to such cases would lead to minimal, if any, alteration in the predicted mode distribution, consistent with their negligibly small radial movements (which we verified for rhodopsin). Calculations performed for other transporters (e.g., sodium/hydantoin transporter Mhp1 and ATP binding cassette MalFGK) that have been structurally resolved in two distinctive functional states (IF and OF) showed that their selected softest modes did not exhibit a significant change due to membrane effects. Both of these transporters were verified to have significantly lower radial components in their global modes compared with GltPh (see Fig. S5). In summary, our analysis supports the view that membrane constraints become significant, and should be included in ANM analyses of global dynamics, when the softest modes that are intrinsically favored by the membrane protein architecture have important radial components, as typified by GltPh. Both of the proposed models (exANM and imANM) can aid in the selection or identification of the particular modes that are most likely to be selected, after suitable perturbation, in the membrane environment.
Conclusion
We explored the global conformational change of GltPh and found that the constraining effects of the membrane help guide the piston-like motion of the subunits during transport. When the membrane is taken into consideration, the softest ENM modes show significant overlap with the global conformational change, alternating access between the EC and CP regions, which is required for transporter function. We furthermore predict, based on the available structural data, that the GltPh subunits act independently, with each subunit transitioning to the IF conformation when substrate and sodium are bound. This type of sequential (as opposed to all-or-none) transition mechanism of the individual subunits is consistent with recent experimental observations (46).
The results presented here also demonstrate that the large-scale structural change that occurs between the OF and IF conformations of glutamate transporters can be understood with the use of simple physics-based methods. The overall architecture of the transporter favors a collective change that enables the well-known alternating access mechanisms. Our study provides evidence that physically plausible modifications to an ENM potential energy function can result in global conformational changes that agree closely with those observed experimentally. Notably, a single, softest mode of motion selected by GltPh in the presence of membrane constraints effectively drives more than half of the functional reconfiguration at a minimal energetic cost.
Acknowledgments
This research was funded by National Institutes of Health grants 5R01 GM086238-03 and 1U54GM087519-01A1.
Supporting Material
This movie shows the slowest mode calculated from applying ANM to the outward-facing conformation of the GltPh trimer. This mode is twofold degenerate.
This movie shows the second mode calculated from applying ANM to the outward-facing conformation of the GltPh trimer. This is a non-degenerate mode.
This movie shows the slowest mode calculated from applying ANM to the inward-facing conformation of the GltPh trimer. This mode is twofold degenerate.
This movie shows the second mode calculated from applying ANM to the inward-facing conformation of the GltPh trimer. This is a non-degenerate mode.
This movie shows the slowest mode calculated from applying imANM to the outward-facing conformation of the GltPh trimer. This mode is twofold degenerate.
This movie shows the second mode calculated from applying imANM to the outward-facing conformation of the GltPh trimer. This is a non-degenerate mode.
This movie shows the slowest mode calculated from applying imANM to the inward-facing conformation of the GltPh trimer. This mode is twofold degenerate.
This movie shows the second mode calculated from applying imANM to the inward-facing conformation of the GltPh trimer. This is a non-degenerate mode.
References
- 1.Tanaka K., Watase K., Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 2.Zerangue N., Kavanaugh M.P. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- 3.Amara S.G., Fontana A.C. Excitatory amino acid transporters: keeping up with glutamate. Neurochem. Int. 2002;41:313–318. doi: 10.1016/s0197-0186(02)00018-9. [DOI] [PubMed] [Google Scholar]
- 4.Groeneveld M., Slotboom D.-J. Na(+):aspartate coupling stoichiometry in the glutamate transporter homologue Glt(Ph) Biochemistry. 2010;49:3511–3513. doi: 10.1021/bi100430s. [DOI] [PubMed] [Google Scholar]
- 5.Yernool D., Boudker O., Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- 6.Boudker O., Ryan R.M., Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445:387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 7.Reyes N., Ginter C., Boudker O. Transport mechanism of a bacterial homologue of glutamate transporters. Nature. 2009;462:880–885. doi: 10.1038/nature08616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brocke L., Bendahan A., Kanner B.I. Proximity of two oppositely oriented reentrant loops in the glutamate transporter GLT-1 identified by paired cysteine mutagenesis. J. Biol. Chem. 2002;277:3985–3992. doi: 10.1074/jbc.M107735200. [DOI] [PubMed] [Google Scholar]
- 9.Leighton B.H., Seal R.P., Amara S.G. Structural rearrangements at the translocation pore of the human glutamate transporter, EAAT1. J. Biol. Chem. 2006;281:29788–29796. doi: 10.1074/jbc.M604991200. [DOI] [PubMed] [Google Scholar]
- 10.Qu S., Kanner B.I. Substrates and non-transportable analogues induce structural rearrangements at the extracellular entrance of the glial glutamate transporter GLT-1/EAAT2. J. Biol. Chem. 2008;283:26391–26400. doi: 10.1074/jbc.M802401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slotboom D.J., Konings W.N., Lolkema J.S. Structural features of the glutamate transporter family. Microbiol. Mol. Biol. Rev. 1999;63:293–307. doi: 10.1128/mmbr.63.2.293-307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shrivastava I.H., Jiang J., Bahar I. Time-resolved mechanism of extracellular gate opening and substrate binding in a glutamate transporter. J. Biol. Chem. 2008;283:28680–28690. doi: 10.1074/jbc.M800889200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu Y., Shrivastava I.H., Bahar I. Molecular simulations elucidate the substrate translocation pathway in a glutamate transporter. Proc. Natl. Acad. Sci. USA. 2009;106:2589–2594. doi: 10.1073/pnas.0812299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crisman T.J., Qu S., Forrest L.R. Inward-facing conformation of glutamate transporters as revealed by their inverted-topology structural repeats. Proc. Natl. Acad. Sci. USA. 2009;106:20752–20757. doi: 10.1073/pnas.0908570106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell P. A general theory of membrane transport from studies of bacteria. Nature. 1957;180:134–136. doi: 10.1038/180134a0. [DOI] [PubMed] [Google Scholar]
- 16.Huang Z., Tajkhorshid E. Identification of the third Na+ site and the sequence of extracellular binding events in the glutamate transporter. Biophys. J. 2010;99:1416–1425. doi: 10.1016/j.bpj.2010.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeChancie J., Shrivastava I.H., Bahar I. The mechanism of substrate release by the aspartate transporter GltPh: insights from simulations. Mol. Biosyst. 2011;7:832–842. doi: 10.1039/c0mb00175a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doruker P., Atilgan A.R., Bahar I. Dynamics of proteins predicted by molecular dynamics simulations and analytical approaches: application to α-amylase inhibitor. Proteins. 2000;40:512–524. [PubMed] [Google Scholar]
- 19.Atilgan A.R., Durell S.R., Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bahar I., Lezon T.R., Shrivastava I.H. Normal mode analysis of biomolecular structures: functional mechanisms of membrane proteins. Chem. Rev. 2010;110:1463–1497. doi: 10.1021/cr900095e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shrivastava I.H., Bahar I. Common mechanism of pore opening shared by five different potassium channels. Biophys. J. 2006;90:3929–3940. doi: 10.1529/biophysj.105.080093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isin B., Schulten K., Bahar I. Mechanism of signal propagation upon retinal isomerization: insights from molecular dynamics simulations of rhodopsin restrained by normal modes. Biophys. J. 2008;95:789–803. doi: 10.1529/biophysj.107.120691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lezon T.R., Sali A., Bahar I. Global motions of the nuclear pore complex: insights from elastic network models. PLoS Comput. Biol. 2009;5:e1000496. doi: 10.1371/journal.pcbi.1000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Besya A.B., Mobasheri H., Ejtehadi M.R. Membrane interactions control residue fluctuations of outer membrane porins. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2010;81:051911. doi: 10.1103/PhysRevE.81.051911. [DOI] [PubMed] [Google Scholar]
- 25.Lomize M.A., Lomize A.L., Mosberg H.I. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 26.Cantor R.S. Lipid composition and the lateral pressure profile in bilayers. Biophys. J. 1999;76:2625–2639. doi: 10.1016/S0006-3495(99)77415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cantor R.S. The influence of membrane lateral pressures on simple geometric models of protein conformational equilibria. Chem. Phys. Lipids. 1999;101:45–56. doi: 10.1016/s0009-3084(99)00054-7. [DOI] [PubMed] [Google Scholar]
- 28.Tama F., Gadea F.X., Sanejouand Y.H. Building-block approach for determining low-frequency normal modes of macromolecules. Proteins. 2000;41:1–7. doi: 10.1002/1097-0134(20001001)41:1<1::aid-prot10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 29.Li G., Cui Q. A coarse-grained normal mode approach for macromolecules: an efficient implementation and application to Ca(2+)-ATPase. Biophys. J. 2002;83:2457–2474. doi: 10.1016/S0006-3495(02)75257-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 31.Hinsen K., Petrescu A.-J., Kneller G.R. Harmonicity in slow protein dynamics. Chem. Phys. 2000;261:25–37. [Google Scholar]
- 32.Ming D., Wall M.E. Allostery in a coarse-grained model of protein dynamics. Phys. Rev. Lett. 2005;95:198103. doi: 10.1103/PhysRevLett.95.198103. [DOI] [PubMed] [Google Scholar]
- 33.Kondrashov D.A., Cui Q., Phillips G.N., Jr. Optimization and evaluation of a coarse-grained model of protein motion using x-ray crystal data. Biophys. J. 2006;91:2760–2767. doi: 10.1529/biophysj.106.085894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng W.J., Brooks B.R. Probing the local dynamics of nucleotide-binding pocket coupled to the global dynamics: myosin versus kinesin. Biophys. J. 2005;89:167–178. doi: 10.1529/biophysj.105.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larsson H.P., Tzingounis A.V., Kavanaugh M.P. Fluorometric measurements of conformational changes in glutamate transporters. Proc. Natl. Acad. Sci. USA. 2004;101:3951–3956. doi: 10.1073/pnas.0306737101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grewer C., Balani P., Rauen T. Individual subunits of the glutamate transporter EAAC1 homotrimer function independently of each other. Biochemistry. 2005;44:11913–11923. doi: 10.1021/bi050987n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koch H.P., Larsson H.P. Small-scale molecular motions accomplish glutamate uptake in human glutamate transporters. J. Neurosci. 2005;25:1730–1736. doi: 10.1523/JNEUROSCI.4138-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Groeneveld M., Slotboom D.J. Rigidity of the subunit interfaces of the trimeric glutamate transporter GltT during translocation. J. Mol. Biol. 2007;372:565–570. doi: 10.1016/j.jmb.2007.06.067. [DOI] [PubMed] [Google Scholar]
- 39.Koch H.P., Brown R.L., Larsson H.P. The glutamate-activated anion conductance in excitatory amino acid transporters is gated independently by the individual subunits. J. Neurosci. 2007;27:2943–2947. doi: 10.1523/JNEUROSCI.0118-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leary G.P., Stone E.F., Kavanaugh M.P. The glutamate and chloride permeation pathways are colocalized in individual neuronal glutamate transporter subunits. J. Neurosci. 2007;27:2938–2942. doi: 10.1523/JNEUROSCI.4851-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Potestio R., Caccioli F., Vivo P. Random matrix approach to collective behavior and bulk universality in protein dynamics. Phys. Rev. Lett. 2009;103:268101. doi: 10.1103/PhysRevLett.103.268101. [DOI] [PubMed] [Google Scholar]
- 42.Bahar I. On the functional significance of soft modes predicted by coarse-grained models for membrane proteins. J. Gen. Physiol. 2010;135:563–573. doi: 10.1085/jgp.200910368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valadié H., Lacapcre J.J., Etchebest C. Dynamical properties of the MscL of Escherichia coli: a normal mode analysis. J. Mol. Biol. 2003;332:657–674. doi: 10.1016/s0022-2836(03)00851-9. [DOI] [PubMed] [Google Scholar]
- 44.Taly A., Delarue M., Changeux J.P. Normal mode analysis suggests a quaternary twist model for the nicotinic receptor gating mechanism. Biophys. J. 2005;88:3954–3965. doi: 10.1529/biophysj.104.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miloshevsky G.V., Jordan P.C. The open state gating mechanism of gramicidin a requires relative opposed monomer rotation and simultaneous lateral displacement. Structure. 2006;14:1241–1249. doi: 10.1016/j.str.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 46.Jiang J., Shrivastava I.H., Amara S.G. Large collective motions regulate the functional properties of glutamate transporter trimers. Proc. Natl. Acad. Sci. USA. 2011;108:15141–15146. doi: 10.1073/pnas.1112216108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This movie shows the slowest mode calculated from applying ANM to the outward-facing conformation of the GltPh trimer. This mode is twofold degenerate.
This movie shows the second mode calculated from applying ANM to the outward-facing conformation of the GltPh trimer. This is a non-degenerate mode.
This movie shows the slowest mode calculated from applying ANM to the inward-facing conformation of the GltPh trimer. This mode is twofold degenerate.
This movie shows the second mode calculated from applying ANM to the inward-facing conformation of the GltPh trimer. This is a non-degenerate mode.
This movie shows the slowest mode calculated from applying imANM to the outward-facing conformation of the GltPh trimer. This mode is twofold degenerate.
This movie shows the second mode calculated from applying imANM to the outward-facing conformation of the GltPh trimer. This is a non-degenerate mode.
This movie shows the slowest mode calculated from applying imANM to the inward-facing conformation of the GltPh trimer. This mode is twofold degenerate.
This movie shows the second mode calculated from applying imANM to the inward-facing conformation of the GltPh trimer. This is a non-degenerate mode.