

Abstract
Epigenetics refers to the study of mechanisms that alter gene expression without altering the primary DNA sequence. Epigenetic mechanisms are heritable and reversible. Over the last few decades, epigenetics has obtained a large importance in cancer research. Epigenetic alterations are widely described as essential players in cancer progression. They comprise DNA methylation, histone modifications, nucleosome positioning, and small, noncoding RNAs (miRNA, siRNA). They are involved in transcriptional changes and decisive events that will determine cell fate and phenotype. Epigenetics not only offers light into cancer biological processes, but also represents an attractive opportunity of reverting cancer-specific alterations, which may lead, in the future, to a possibility of stopping this disease. Epigenetic changes have been identified as putative cancer biomarkers for early detection, disease monitoring, prognosis, and risk assessment. Other epigenetic alterations are promising therapeutic targets and even therapeutic agents. Emerging discoveries in this area are already contributing to cancer management and monitoring, and a lot more progresses are expected in the future.
Keywords: Cancer, epigenetics, DNA methylation, histones modifications, microRNA
Introduction
In the late 1930s, early 1940s, a biologist, named Conrad Hal Waddington, focused on the embryology field, and discussed the development processes that lie between the genotype and the phenotype, using studies in Drosophila melanogaster. He is given credit to have proposed the term epigenetics as “the causal interactions between genes and their products, which bring the phenotype into being” (Waddington 1942). “Epigenetics” carries the Greek prefix “epi” (that means above, beyond, on top, after, in addition) and another word derived from Greek (genno that means give birth) that refers to the well-known term genetics: representing the inheritance of variation, the science of heredity. Waddington's definition has evolved over time as the phenomenon of epigenetics is implicated in a wide variety of biological processes. The identification of mechanisms above and beyond genetics allowed a broader understanding on how individuals carrying the same genotype exhibit different phenotypes, for example, monozygotic twins (Fraga et al. 2005). There are several current variations on the definition of epigenetics that could be summarized as “the study of heritable changes in gene expression that occur independent of changes in the primary DNA sequence” (Sharma et al. 2010). In this concept of epigenetics, it is important to focus on the key points, such as no change in the original sequence of DNA, heritable, and reversible. A review by Taby and Issa (2010) introduces a comprehensive definition, “This variability in gene expression is heritable through mitosis and potentially meiosis, without any underlying modification in the actual genetic sequence. This alteration in gene expression plays a fundamental role in several aspects of natural development, from embryogenesis, in which a resetting of the ‘epigenetic code’ takes place in the very early moments after conception (Reik et al. 2001), to the determination of cellular fate and its commitment to a particular lineage.” Epigenetics also plays a fundamental role in biological diversity such as phenotypic variation among genetically identical individuals (Morgan et al. 1999), enabling cells to have distinct identities although containing the same genetic information.
The most common phenomena that are classified as epigenetic changes are: DNA methylation (methylation on the cytosine bases of the DNA, on CG dinucleotides, commonly referred to as CpG), histone modifications (posttranslational modifications that alter their interaction with DNA and nuclear proteins, such as acetylation and methylation), and small, noncoding RNAs (miRNA, siRNA). There is a debate whether all histone modifications (and many noncoding RNAs) fall in the classic definition of epigenetics, because it is likely that relatively few of these modifications or RNAs will be self-perpetuating and inherited (Riddihough and Zahn 2010). Nevertheless, they are currently classified as epigenetic modifications, once they fall in the category of biological aspects that alter gene expression without involving DNA sequence alterations.
Cancer is a disease that involves a multi-step process that accumulates alterations, genetic and/or epigenetic (Kinzler and Vogelstein 1996). Knudson's hypothesis (Knudson 1971) postulates that two hits (genetic alterations) are required for the full inactivation of a tumor suppressor gene in the carcinogenesis process, such as intragenic mutations and loss of chromosomal material (loss of heterozygosity (LOH) or homozygous deletion). However, the fact that methylation of CpG islands located in the promoters of genes can cause transcriptional silencing, coupled with the observation that DNA methylation patterns are perturbed in cancer cells, has led to the suggestion that abnormal methylation of the promoters of tumor suppressor genes might be implicated in carcinogenesis (Jones and Laird 1999). Such methylation-mediated silencing of tumor suppressor genes can act as one of the two hits advocated in Knudson's hypothesis. In their review, Jones and Laird (1999) have summarized pathways of tumor suppressor gene inactivation that demonstrate the simultaneous presence of both epigenetic and genetic alterations in various types of cancer.
In this review, we described and exemplified some of the main epigenetic changes that have been associated with cancer.
DNA methylation alterations in cancer
DNA methylation alterations are probably the most widely studied epigenetic alterations in cancer. Those were the first epigenetic alterations to be linked to cancer. Riggs and Jones in 1983 (Riggs and Jones 1983), after studying the participation of DNA methylation in mammalian chromosome X inactivation (Riggs 1975), established a role for this mechanism in gene regulation in cancer. In the same year, Feinberg and Vogelstein (1983) described the absence of DNA methylation (hypomethylation) as a characteristic that was able to distinguish tumor cells from their normal counterparts.
DNA methylation, that is, the addition of a methyl group to the 5-carbon position of a cytosine (C), occurs almost exclusively in Cs located 5′ to a guanine (G), and this pair of nucleotides is known as CpG dinucleotide. Traditionally, regions of >200 bases that have clusters of CpG dinucleotides (at least 50% of G + C content, and a ratio of 0.6 or more between observed and expected [o/e] CpG dinucleotide frequency) are called CpG island, by definition. Lately, more stringent criteria are being created for the definition of a CpG island, with the intent of excluding some repeats regions; for example, a minimum size of 500 bp (base pairs) with at least 55% of G + C content and CpG o/e ratio increased to 0.65 (Takai and Jones 2002; Wang and Leung 2004). CpG islands are observed in about 60% of human genome promoter regions, although the occurrence of the CpG dinucleotides themselves is predicted to be around 1% in mammalian genomes. The establishment of the distribution of the 5-methylcytosine content in a cell type occurs during the embryonic development. DNA methylation patterns in normal tissues are in part dependent on the relative levels and activities of DNA methylation-related enzymes such as DNA methyltransferases (DNMT) and DNA demethylases, whose expressions are regulated at both the transcriptional and posttranscriptional level (Jost and Bruhat 1997; Turker and Bestor 1997; Kim et al. 2010).
CpG island methylation is associated with the suppression of gene expression. Tree variations of DNA methylation, that is, hypomethylation, loss of imprinting (LOI), and hypermethylation, have been clearly linked to cancer.
DNA hypomethylation refers to the loss of DNA methylation in a region where it usually occurs and it is generally assumed to be a genome-wide phenomenon; thus it is globally assessed. DNA hypomethylation has been described in several tumor types (Feinberg and Vogelstein 1983; Bedford and van Helden 1987; Ehrlich et al. 2002; Ehrlich 2002; Smith et al. 2007; Netto et al. 2008; Kim et al. 2010; Wolff et al. 2010). DNA methylation occurring along the gene body is positively correlated with expression (Hellman and Chess 2007). It has been proposed that it might be related to elongation efficiency and prevention of spurious initiations of transcription (Zilberman et al. 2007). It has also been more recently described as the mechanism by which genes that show oncogenic properties, such as Cancer Testis Antigens, that exhibit a methylated promoter region in normal cells, can become reactivated in cancer cells by the loss of this methylation, resulting in hypomethylation (Simpson et al. 2005; Glazer et al. 2009; Smith et al. 2009; Straussman et al. 2009).
LOI refers to the loss of parental allele-specific monoallelic expression of genes. Imprinting refers to parental allele-specific expression of genes. In this situation, hypermethylation at one of the two parental alleles leads to monoallelic expression of the gene (Kacem and Feil 2009). LOI accounts for the loss of this differential expression of parental alleles, and has been often observed in tumors with embryonic origin (Reik and Surani 1989; Ferguson-Smith et al. 1990; Rainier et al. 1993; Reik et al. 2001; Hajkova et al. 2002; Oosterhuis and Looijenga 2005).
DNA hypermethylation refers to the gain of methylation in a locus originally unmethylated. It tends to occur at specific regulatory sites in the promoter region and may show a tumor-specific pattern (Bird 1992; Baylin et al. 1998; Costello et al. 2000; Esteller et al. 2001). It has been observed in cancer that the presence of methylation in the promoter region of genes with tumor-suppressive characteristics can result in a complete repression of the transcription process (Merlo et al. 1995). However, it is not a direct relationship; there are gene/region-specific densities and frequencies or “hotspots” where the methylation has to occur for it to block transcription. Promoter methylation of tumor suppressor genes is recognized as a strong and important method of the inactivation of tumor suppressor genes (Momparler 2003; Hoque et al. 2008; Sharma et al. 2010).
There are several proposed mechanisms by which methylation can inhibit gene expression, such as by direct blockage of transcription factors, by preventing them from binding to their target sites, and by recruitment of methyl-binding domain (MBDs) proteins (Klose and Bird 2006; Hoque et al. 2008). These MBDs are present in transcription co-repressor complexes that can interact with other gene repressing mechanisms involving other members of the epigenetic machinery such as histone deacetylases (HDAC) and histone methyltransferases (HMT), resulting in chromatin reconfiguration and gene silencing (Nan et al. 1998; Lopez-Serra and Esteller 2008; Taby and Issa 2010). In contrast, unmethylated CpG islands generate a chromatin structure favorable for gene expression by recruiting Cfp1, which associates with HMT Setd1, creating domains rich in the histone methylation mark H3K4 trimethylation (Portela and Esteller 2010; Tomson et al. 2010).
DNMTs are enzymes that catalyze the transfer of the methyl group from S-adenosyl-L-methionine (SAM) to the C in the CpG nucleotide (Vucic et al. 2008). This enzyme family is divided by function. DNTM1 is responsible for the maintenance of methylation, whereas DNMT3A and DNMT3B play a role in de novo methylation, being responsible for creating the methylation pattern necessary during embryonic development. These two members of the DNMT family are found highly expressed in embryonic stem cells and down-regulated in differentiated cells (Esteller 2007). The maintenance DNMT, DNMT1, has a 30- to 40-fold preference for hemimethylated DNA, and also has de novo DNMT activity. DNMT1 is the most abundant DNMT in the cell and is transcribed mostly during the S phase of the cell cycle. It is most often needed to methylate hemimethylated sites that are generated during semi-conservative DNA replication (Portela and Esteller 2010).
Aberrant DNA methylation is widely accepted as a common mechanism used by tumor cells to silence tumor suppressor genes (Jones and Baylin 2007). A tremendous amount of data has been published in different tumor types. The following paragraphs will mainly highlight a few recent reviews that gathered this data by tumor type, and exemplify a tiny amount of several potential DNA methylation biomarker candidates with meaningful clinical correlation.
In colon cancer, several genes have been reported to be hypermethylated in tumor tissue versus normal (and in all stages from adenoma to carcinoma), as well as in bodily fluids, and stool (reviewed by Kim et al. 2010). Genes involved in different cellular functions have been described as epigenetically altered in colorectal cancer: cell cycle (p14 and p16), signal transduction (RASSF1A), DNA repair (MGMT, hMLH1) are just a few examples. The genes that displayed the highest sensitivity in colon cancer detection are SFRP2 and ALX4. In this tumor type, a CpG island methylator phenotype (CIMP) has been described, and the tumors can be divided into CIMP-, the ones that show very few or no genes methylated, and CIMP+, the tumors that show an accumulation of methylation (reviewed by Kim et al. 2010). Currently, there is enough evidence that colorectal cancers with high degrees of methylation (CIMP+) represent a clinically and etiologically distinct group that seem to have a distinct epidemiology, histology, and molecular features (Issa 2004).
Ovarian carcinoma is a disease that is mostly detected in late stages, and there is an urgent need for early detection biomarkers. Hypermethylation of APC, p16, and DAPK has been detected in serum or plasma from early stage ovarian carcinoma patients, becoming potential biomarkers for early detection in this tumor type. Another important characteristic in this tumor type is therapeutic response prediction. The DNA repair gene MGMT has been associated with positive response to platinum-based therapy although another DNA repair gene hMLH1 has been associated with resistance to this same chemotherapy in ovarian carcinoma (reviewed by Maldonado and Hoque 2010). The authors also described other epigenetic mechanisms described in this tumor type.
In genitourinary tumors, specifically in prostate cancer, one of the most promising cancer-related hypermethylated genes is GSTP1, which has been demonstrated to be hypermethylated in about 90% of prostate tumors, in contrast to bladder or renal cancer, where only around 10% of the tumors showed this alteration. It is important to compare tumors from the same system, when thinking about a detection test, because not only the test aims at detecting cancer, but also would even have more utility if it could distinguish its organ of origin. In prostate cancer bodily fluids samples, the specificity of this marker is 100% both for urine and serum, but the sensitivity is variable between 27% and 40% in urine and above 40% in serum. When adding other cancer-related hypermethylated genes to GSTP1 for the detection of prostate cancer, it is possible to increase the sensitivity in urine up to 87%, without decreasing the excellent specificity, by adding INK4a, ARF, MGMT (reviewed by Cairns 2007; Hoque 2009). In bladder cancer, our group selected six genes (CCNA1, MINT1, CCND2, PGP9.5, CRBP, and AIM1) that were differentially methylated (higher than 30%) in tumor when compared with normal bladder tissues (<5% frequency). Moreover, by creating a methylation index (number of genes methylated divided by genes analyzed), we observed a significantly higher index level in invasive tumors when compared with noninvasive bladder cancer samples (Brait et al. 2008).
The best known example of epigenetic alteration/DNA promoter hypermethylation and therapeutic response is MGMT promoter methylation that has been associated with clinical response of gliomas to alkylating agents; these patients have longer survival (both overall and progression-free) (Esteller et al. 2000). Alkylating agents kill cells by forming cross-links between adjacent strands of DNA, thus inhibiting DNA replication. Tumors present inherent or acquired resistance to these agents, and it seems to rely on the DNA repair mechanism of MGMT. The main mechanism of MGMT inactivation is DNA promoter methylation. These observations highlight the importance of MGMT methylation as a specific predictive biomarker for the responsiveness to chemotherapy with alkylating agents. This epigenetic alteration has been associated with response to alkylating agents, as a combination of the latter and radiotherapy (reviewed by Natsume et al. 2010). There are tests offered to glioma/glioblastoma patients in a clinical setting to assess MGMT methylation status to determine if they will benefit from the addition of an alkylating agent (usually temozolomide) to the standard therapy (radiotherapy) (Costa 2010a).
The foregoing examples are a few of the potentially clinically useful DNA methylation-based potential bio-markers that have been described in cancer.
Histone modifications and cancer
Within the cell's nucleus, the DNA is packaged together with proteins, making a complex, known as chromatin. The main proteins participating in the chromatin are histones. Histones determine the structure of chromatin and play a central role in gene regulation. There are five types of histones in the chromatin: H1/H5, H2A, H2B, H3, and H4 (Bhasin et al. 2006). The basic unit of the chromatin is the nucleosome and it is composed of an octamer (eight histone complex) of the four core histones (H3, H4, H2A, H2B) around which 147 bp of DNA are wrapped (Kouzarides 2007). Nucleosomes are linked to each other by 10 to 90 bp of DNA, which is occupied by a single copy of a “linker” histone, H1, or seldom H5 depending on the cell type (Georgel and Hansen 2001). Due to this packaging, the access to genetic information contained in the nucleus DNA is restricted by having this protective structure. Histones contain a globular C-terminal domain and an unstructured N-terminal tail (Luger et al. 1997).
Histone modifications work by either changing the accessibility of chromatin or by recruiting and/or occluding non-histone effector proteins, which decode the message encoded by the modification patterns. The mechanism of inheritance of this histone code, however, is still not fully understood. Histone modifications can lead to either activation or repression depending upon which residues are modified and the type of modifications present (Sharma et al. 2010). Overall, histone modifications impact on chromatin conformation and consequently influence gene transcription, DNA repair, DNA replication, and cell cycle checkpoints (Sawan et al. 2008).
One of the most studied histone modifications is the deacetylation, which occurs in H3 and H4. In the repressed state of the chromatin, histones are deacetylated and contribute to a tight configuration of the DNA packaging. Once the histones become acetylated (addition of an acetyl group to the N-terminal tail), their lysine residues have their positive charge neutralized, and the connection between the histone and the negatively charged DNA becomes less rigid, allowing accessibility to the transcription factor and other players on the transcription processes (Struhl 1998). HDAC are the enzymes that mediate the loss of acetylation, they remove the acetyl radical present in the lysines of the histones, HAT (histone acetyltransferases) are the enzymes that do the opposite: they add acetyl radicals to those positions. Alterations in HDACs have been described in cancer as genetic alterations like mutations in HDACs (Ropero et al. 2006), and changes in the levels of expression (Zhu et al. 2004; Lucio-Eterovic et al. 2008). Alterations in HAT expression levels have also been correlated with cancer (Isharwal et al. 2008). HDAC interact with DNMT and can also be regulated by miRNA; thus, the whole epigenetic machinery acts in conjunction to assure the chromatin conformation and level of accessibility.
Another well-studied histone modification comprises histone methylation. Different from the lysine acetylation mentioned above, lysine methylation leads to transcriptional activation or repression, depending on which residue is modified and the density of methylation (Sharma et al. 2010). Methylation takes place on the arginine residues as well. Histone methylation is now considered as a reversible process. There are two distinct effects: the ones that cause histone methylation and the ones that erase it. In a recent review on histone modification in the context of cancer, Chi et al. (2010) thoroughly exemplifies the different scenarios, that is, specific histone methylation and its consequence, describing the roles of HMT as well as histone demethylases (HDM), which add and remove the methyl group, respectively. Here, we appreciated some of the examples given. H3K4 methylation is established by the SET1 and mixed lineage leukemia (MLL) family of HMTs, and removed by the lysine-specific histone demethylase 1 (LSD1) and jumonji AT-rich interactive domain 1 (JARID1) family of HDMs. For histone H3, methylation has been observed at multiple lysine sites, including H3K4, K9, K27, K36, and K79, and the addition of up to three methyl groups at each lysine produces a total of four methylation states: unmethylated, monomethylated, dimethylated, or trimethylated. These histone methylation states exhibit a distinct distribution pattern in the mammalian genome. H3K4 trimethylation (H3K4me3) is strongly associated with transcriptional competence and activation, with the highest levels observed near transcriptional start sites (TSS) of highly expressed genes, whereas H3K27 trimethylation (H3K27me3) is frequently associated with gene silencing, especially the repression of unwanted differentiation programs during lineage specification. The distribution patterns of H3K4me3, H3K27me3, and their associated histone marks underlie the diversity of cellular states for pluripotency and lineage differentiation. Another example is the HMT Polycomb group protein EZH2 that catalyzes H3K27me3 (Cao et al. 2002). EZH2 expression has been suggested as a tumor biomarker in several tumor types and functional studies indicate its capacity of promoting growth in vivo and in vitro (Kleer et al. 2003; Yu et al. 2007; Martinez-Garcia and Licht 2010). In contrast, monovalent active or repressive histone marks are often found on these genes in differentiated cell lineages. The bivalent chromatin state has been suggested as a mechanism for retaining chromatin and cellular plasticity at early stages of development (Chi et al. 2010).
Other posttranslational modifications that occur in histones include: ubiquitination, phosphorylation, and sumoylation. Alterations in the abundance of “linker” histones have been recently associated to chromatin configuration (Clapier and Cairns 2009). These distinct modifications that play repressive and/or activating roles in histones are part of the regulatory mechanisms that determine cell fate, by controlling and participating in the regulation of gene expression (Sharma et al. 2010).
Role of chromatin remodeling
The definition of nucleosome, the basic unit of the chromatin, was mentioned earlier in this review in the context of histone modifications. In this section, the role of chromatin remodeling and nucleosome repositioning and how it affects gene expression are discussed in more detail.
Chromatin remodeling has gained attention due to evidences of its involvement in histone modifications as well as DNA methylation (Jones and Baylin 2002; Esteller and Almouzni 2005; Martin and Zhang 2005; Baylin and Ohm 2006; Ting et al. 2006; Jones and Baylin 2007).
The combination of nucleosome positions and their chemical and compositional modifications is key to genome regulation. Recent technologies such as microarrays and massive parallel DNA sequence allowed the identification with high precision, of the nucleosomes throughout the genome. There is emerging evidence that nucleosomes regulate transcriptional initiation, and therefore understanding how nucleosomes are positioned has implications for how cells respond to external stimuli or how misregulation of nucleosome positioning leads to developmental defects and cancer (Jiang and Pugh 2009b). It has been shown that nucleosomes adopt canonical positions around promoter regions and more random positions in the interior of genes (Mavrich et al. 2008). The understanding of the mechanisms that surround this preferential positioning is still to be determined.
The 5′ and 3′ ends of the genes present nucleosome-free regions, which might be explained by the need of exposition of the binding sites for the transcription machinery. Thus, the presence or absence of a nucleosomes in such regulatory regions will determine the possibility of gene activation (Yuan et al. 2005; Schones et al. 2008; Jiang and Pugh 2009a,b).
The evidence of a sequence of interactions between different epigenetic mechanisms was provided by Morey et al. (2008) in acute promyelocytic leukemia (APL). Their work demonstrated a direct role of the nucleosome remodeling and deacetylase (NuRD) complex in the establishment and transmission of epigenetic repressive marks as a precise sequence of events, ultimately resulting in aberrant repression of target genes. They showed that the translocation PML-RARa (present in human APL) represses gene transcription through several distinct mechanisms, including histone deacetylation, DNA methylation, histone modification, chromatin compaction, and heterochromatinization. Their data reveal that NuRD complex plays an essential role in both the establishment and maintenance of aberrant epigenetic silencing imposed by PML-RARa. This work confirmed and further elucidated earlier findings that depicted a mechanistic interaction between epigenetic and genetic factors, participating in early carcinogenesis steps, also showing evidence that the PML-RARa oncogenic fusion protein recruits DNMT, consequently inducing DNA hypermethylation (Di Croce et al. 2002).
Another important protein complex that participates in nucleosome positioning is the Swi/Snf complex. These proteins target specific promoter regions and play a role in either activating or repressing transcription via three biochemical processes: nucleosome remodeling, nucleosome sliding, and octamer transfer. As mentioned earlier, it is still not clear how such positioning and complex formation is triggered, or how the histone–DNA interaction is established. Recent data indicate that it is probably a combination of proximal genetic sequences (“cis effect”) or by other mechanisms operated by ATP-dependent nucleosome remodeling complexes in a sequence-independent manner (“trans effect”) (Längst and Becker 2004; Segal et al. 2006; Weissman and Knudsen 2009; Taby and Issa 2010).
MicroRNAs in cancer
MicroRNAs (miRNAs) are small noncoding RNAs (ncRNA) (around 22 nucleotides in size) that are found in both plants and animals. They regulate gene expression at the posttranscriptional level (Bartel 2004, 2009). There are two main ways miRNAs interact with their targets, depending on the degree of complementarity between them. When the complementarity is perfect between the miRNA and the target mRNA sequence, the RNA interference (RNAi) pathway is triggered, and the target mRNA is degraded. In contrast, if the miRNA binds to the 3′ untranslated region (UTR) of their mRNA targets through partial complementarity, it blocks translation of the target mRNA. The induction of RNAi and degradation of the target RNA was originally observed with exogenously introduced double-stranded (ds) RNA. At that time, the effector 22-nt RNA responsible for the degradation of the target RNA was termed siRNA. However, the initial distinction made between miRNA and siRNA becomes less relevant because they are both processed by the same machinery and use similar or identical protein complex (RNA-induced silencing complex or RISC) for their function. Consistent with translational control, miRNAs that use this mechanism reduce the protein levels of their target genes, but the mRNA levels of these genes are barely affected (reviewed by Esquela-Kerscher and Slack 2006). Some groups have also shown potential evidence for a third mechanism by which miRNAs could regulate gene transcription by targeting promoter-associated ncRNAs (Omoto and Fujii 2005; Gonzalez et al. 2008). Figure 1 shows the two modes of action of small ncRNA-mediated gene silencing discussed above: miRNA-mediated translation inhibition pathway and siRNA-mediated mRNA degradative pathway.
Figure 1.
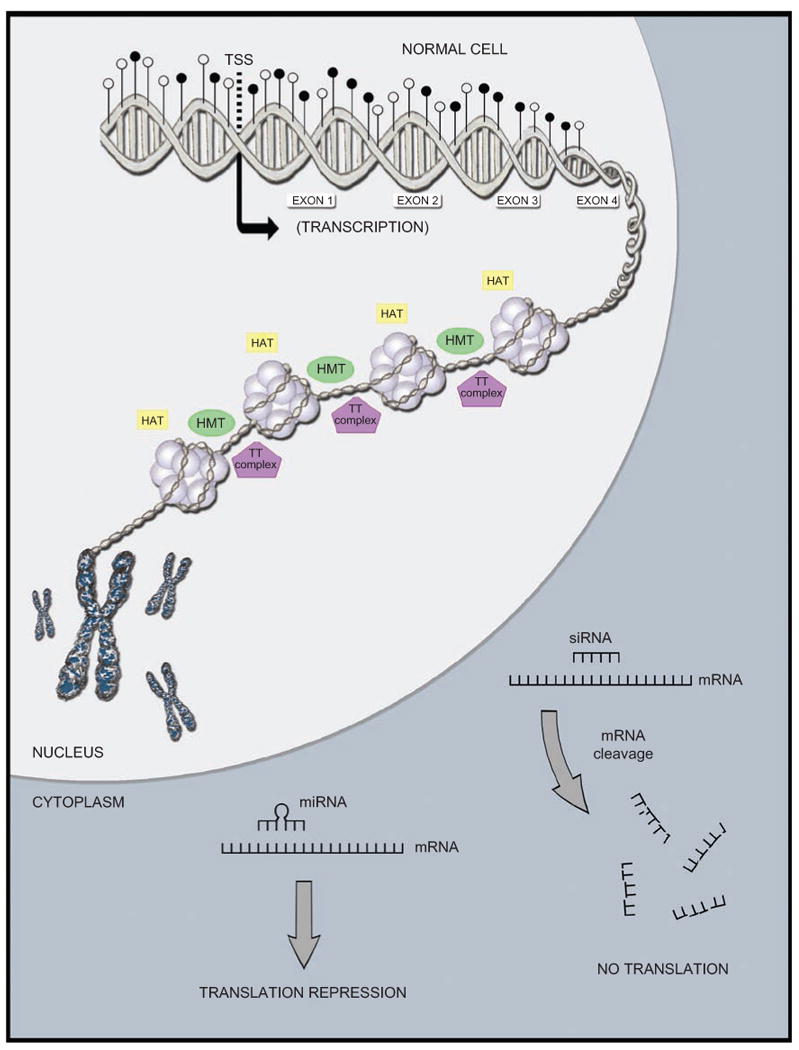
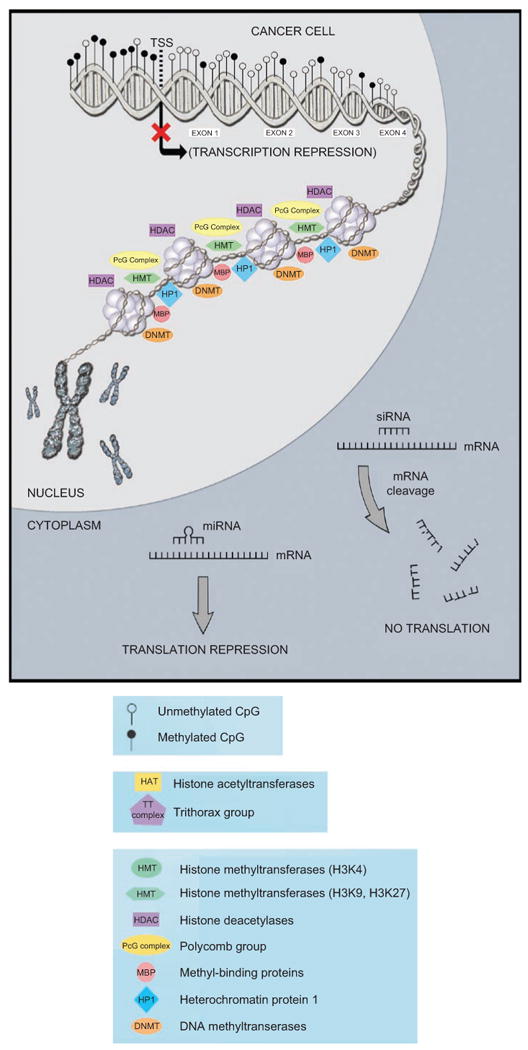
Representative examples of epigenetic processes occurring in a cell (promoter DNA methylation, global methylation, histone modifications, nucleosome positioning, miRNA in action). (A) Normal cell. In the nucleus, one tumor suppressor gene promoter shown as unmethylated, gene body shown as heavily methylated, euchromatin (less compacted chromatin), leading to accessibility (we observed histone-modifying enzymes that play a role in this state), a transcriptionally active form that will result in gene expression. In the cytoplasm, we observed two mechanisms by which ncRNAs/miRNAs repress expression. (B) Cancer cell. We observed some changes in determined processes, when compared with the normal cell. In the nucleus, one tumor suppressor gene promoter shown methylated, gene body shown as hypomethylated, heterochromatin (highly compacted chromatin), leading to restricted accessibility (we observed histone-modifying enzymes that play a role in this state), a transcriptionally inactive form that will result in loss of gene expression. In the cytoplasm, we observed two mechanisms by which ncRNAs/miRNAs repress expression (occurring the same way in both cells, since these small molecules have been shown to have tumor-suppressive as well as oncogenic activities). TSS = transcriptional start site.
Bioinformatics studies revealed that there is a region of eight nucleotides (“seed region”), at the 5′ end that is decisive to miRNA function (Bartel 2004; Grimson et al. 2007). So far, around 1000 miRNA have been described encoded by the human genome (http://mirnamap.mbc.nctu.edu.tw/) (Costa 2010b). Costa (2010b) discusses a model how this class of molecules can interact with other epigenetic mechanisms (DNA methylation, histone acetylation), illustrating the whole idea of the epigenetic machinery working in conjunction to control gene expression.
In cancer, there is evidence of miRNAs displaying tumor suppressor as well as oncogenic effects; miRNAs have been observed up- and down-regulated in many types of tumors and interacting with genes of the most diverse cellular pathways (Zhang et al. 2007; Baek et al. 2008). miRNAs can play important roles in carcinogenesis by a plethora of mechanisms, such as by modulating angiogenesis, apoptosis, as well as the expression of genes involved in cell migration/invasion, and so on. For example, miR-221 and miR-222 are highly expressed in various cancerous conditions, such as human thyroid papillary carcinomas (Visone et al. 2007), glioblastoma (le Sage et al. 2007), and they target and down-regulate the p27 (Kip1). High levels of miR-221and miR-222 appear in these cancers and correlate with low levels of p27 (Kip1). p27 (Kip1) alterations have been frequently detected in human neoplasms, and a reduced or absent p27 (Kip1) expression has been shown in the most aggressive ones (Visone et al. 2007). The miR-17–92 cluster was the frst oncogenic miRNAs (oncomirs) identified in human (Hayashita et al. 2005), and it contains a number of oncogenic miRNAs, such as miR-17, miR-18, miR-19a, miR-19b-1, miR-20a, and miR-92-1. The major known function of the miR-17–92 cluster is related to transcriptional factors c-Myc. Recently, the oncogenic contribution of each individual miRNA in the cluster has been reported. It appears that the individual miRNAs in the cluster exert their oncogenic function by targeting different aspects of oncogenesis trigger. For example, miR-17/20a targets E2F1 (a cell cycle and apoptosis regulator), miR-92 targets BIM (a pro-apoptotic gene that counteracts the anti-apoptotic activity of genes such as Bcl-2), and miR-19 targets PTEN (a negative regulator of the oncogenic pro-survival PI3K/AKT signaling pathway) (reviewed by van Haaften and Agami 2010).
The let-7 family of miRNAs has also been well-studied. Alterations in its members have been correlated to several types of cancer (Takamizawa et al. 2004; Pillai et al. 2005; Chang et al. 2008; Boyerinas et al. 2010). In lung cancer, the down-regulation of a few members of the let-7 family of miRNAs was observed to significantly correlate with a poorer prognosis after surgery (Takamizawa et al. 2004). In another study, one of the members of this family: let-7a-3 has been shown to be regulated by DNA promoter methylation: they observed that hypomethylation in an associated CpG island leads to augmented expression in vitro and was heavily methylated in normal tissue when compared with tumor tissue, suggesting an oncogenic role for this miRNA in this tumor type (Brueckner et al. 2007). These data taken together reveal potential uses of microRNA as cancer biomarkers for diagnosis and prognosis prediction (Wang et al. 2009).
miR-21 is another example of a largely studied miRNA in cancer. It is overexpressed in diverse cancers including brain tumors (Wurdinger and Costa 2007), in head and neck carcinomas (Chang et al. 2008), in prostate, pancreatic, and lung cancer (Volinia et al. 2006). miR-21 has been associated with tumor cell invasiveness and resistance to apoptosis. Many important tumor suppressor genes are known to be regulated by this miRNA. These evidence makes miR-21 an attractive therapeutic target. By inhibiting it, there is enough evidence to the belief that a lot of tumor-suppressive pathways could be reactivated (Krichevsky and Gabriely 2009).
Two of the well-known tumor suppressor miRNAs include miR-15a and miR-16-1. They target the expression of the anti-apoptotic protein Bcl-2, thereby promoting apoptosis. Consistent with the role of miR-15a and miR-16-1 as tumor suppressors, the respective genes, mir-15a and mir-16-1, are found to be deleted in more than half of B-CLL (B-cell chronic lymphocytic leukemias) and their expression was found down-regulated in ∼65% of B-CLL patients (Cimmino et al. 2005). Another tumor suppressor miRNA miR-122 targets a known promoter of metastasis, a disintegrin and metalloprotease 17 and inhibits both tumor angiogenesis and cancer cell migration/invasion (Tsai et al. 2009). miR-126 also appears to inhibit cancer cell growth, proliferation, adhesion, and invasion, and is down-regulated in colon, lung, and breast cancers (see review by Le et al. 2010).
Examples of some other oncogenic miRNAs include miR-10b, miR-372, miR-373, and examples of some other tumor suppressor miRNAs include miR-34, miR-335, miR-143, miR-145, and miR-181a/b/c (Zhang et al. 2007; Ma and Weinberg 2008).
Both genetic and epigenetic mechanisms have been described in the regulation of miRNAs, occurring at the miRNA itself or in its targets, ultimately altering the miRNA compatibility with the target sequence (Ventura and Jacks 2009). It is also important to understand that defects in genes that participate in the miRNA machinery will also lead to its down-regulation (Melo et al. 2009).
An attractive aspect of miRNA as cancer biomarkers is that they are stable in blood based bodily fluids (Mitchell et al. 2008) as well as sputum (Xie et al. 2010), and urine (Hanke et al. 2010), which offers the opportunity of developing a simple, very little to noninvasive test to detect the tumor-related alterations.
By whole genome approaches, like miRNA expression microarrays, miRNA signatures are potentially identified for different tumor types (Calin and Croce 2006; Volinia et al. 2006; Lujambio et al. 2008), and these profiling studies not only propose tumor-specific panels of altered miRNA but also validate individual targets as participants in the carcinogenic process. Once a tumor-specific miRNA signature is established, it can be used to distinguish the tumor tissue from its normal counterpart, thus making these miRNAs potentially useful cancer biomarkers. Still in relationship with metastasis, Esteller's group (Lujambio et al. 2008) identified epigenetically silenced miRNAs associated with metastasis, and validated individual candidates in xenograft models, obtaining very exciting and promising data on the therapeutic potential of this small molecules.
A recent review on miRNA profiles in breast cancer shows down-regulation of tumor suppressor miRNAs (miR-206, miR-17-5p, miR-125a, miR-125b, miR-200, let-7, miR-34, and miR-31) and the overexpression of certain oncogenic miRNAs (miR-21, miR-155, miR-10b, miR-373, and miR-520c), along with their correlation in breast cancer pathways and metastasis (O'Day and Lal 2010). Liu et al. (2011) showed in vivo that miR-34a, when injected in a mouse with prostate cancer, was capable of inhibiting already established metastasis, giving evidence for the potential of this miRNA as a therapeutic agent.
Polymorphisms in miRNAs and their target sequences has been linked to variations in drug activation and metabolism, revealing the potential use of miRNA in pharmacogenomics (reviewed in Passetti et al. 2009).
The cumulative evidence strongly suggests that miR-NAs definitely play an important role in the etiology of cancer, along with the other epigenetic changes (DNA methylation, histone deacetylation), in addition to the long-known player, the genetic changes.
The potential of epigenetic therapies
The fact that epigenetic alterations are a hallmark of cancer and are reversible by definition makes them valid targets for oncologic therapy. Many clinical trials using novel epigenetics-based therapies against cancer are ongoing.
Tumors that display tumor suppressor genes targeted by epigenetic inactivation, such as promoter DNA hypermethylation or histone deacetylation, can certainly benefit from therapeutic agents that can reverse those modifications. Different classes of pharmacological inhibitors that have been developed are discussed below.
DNMT inhibitors
The DNMT inhibitors are widely used in in vitro in research. They are mainly cytosine analogs: 5-aza-cytidine and 5-aza-2′-deoxycytidine. As nucleoside analogs, they are converted to deoxynucleotide triphosphates inside the cell and then incorporated into the DNA during replication in the original C positions. They attach to the DNMT enzymes blocking their activity (Jones and Taylor 1980), which results in heritable demethylated DNA. In order for these inhibitors to be active, replications have to occur (Goffin and Eisenhauer 2002). Both drugs have been approved by the US Food and Drug Administration (FDA) for use in the treatment of a hematologic malignancy: myelodysplastic syndrome, after successful results in clinical trials (Gal-Yam et al. 2008).
Another cytosine analog, 1-β-d-ribofuranosyl-2(1H)-pyrimidinone, presented advantages over the two mentioned above because it is very stable and thus suitable for oral administration, less toxic and showed higher selectivity for cancer cell (Cheng et al. 2004). Although very promising at first, high levels of this drug were necessary to achieve the desired effect, which diminishes its potential of clinical use. Together with other cytosine analogs, the clinical trial results did not live up to the pilot results (Goffin and Eisenhauer 2002).
Other types of DNMT inhibitors have been developed and they include non-nucleoside inhibitors (Segura-Pacheco et al. 2003) and agents that target one specific DNMT enzyme (e.g. with antisense oligonucleotides) (Yan et al. 2003).
HDAC inhibitors
HDAC inhibitors will act on the genes known to be regulated by histone deacetylation. They have shown antitumor, growth inhibitory, pro-apoptotic, and pro-differentiation properties (Minucci and Pelicci 2006). A very promising aspect of these compounds is that tumor cells are more sensitive to them than normal cells (Johnstone 2002). A variety of naturally occurring or synthetic HDAC inhibitors have been characterized for their antitumor activities in preclinical studies (Acharya et al. 2005).
In research labs, probably the most widely used HDAC inhibitor is trichostatin A (TSA), but it showed limited use in the clinic due to its high toxicity (Vigushin et al. 2001).
Suberoylannilide hydroxamic acid (SAHA) is the first HDAC inhibitor approved by US FDA for the treatment of T-cell cutaneous lymphoma (Gal-Yam et al. 2008). Many more HDAC inhibitors are being tested in xenograft models, and at least a dozen HDAC inhibitors are in various stages of clinical development (Cang et al. 2009).
Another important histone modification is histone methylation. Histone methylation machinery has also been considered as a potential target for epigenetic drugs. Unfortunately, so far, only in vitro results have been obtained, and their real clinical use is yet to be assessed (Gal-Yam et al. 2008). In vitro data suggest that 3-deazaneplanocin A depletes Polycomb group components, inhibits histone H3K27 methylation, and induces selective apoptotic cell death in breast cancer cells (Tan et al. 2007). By targeting the enzyme that is responsible for removing methylation from H3K4, polyamine analogs were successful in inhibiting it and consequently up-regulating silenced genes in a cancer cell line (Huang et al. 2007).
Combination of DNMT inhibitors and HDAC inhibitors
In the tumorigenesis process, there are interactions of apparently independent pathways and mechanisms. It is a well-accepted paradigm that the epigenetic machinery acts using all its players during cancer progression. It is intuitive to think about combining drugs that target different pathways and/or mechanisms to assess their effects. Epigenetic drugs have been combined with each other as well as with conventional chemotherapeutic agents.
Over a decade ago, Cameron et al. (1999) showed synergistic effects on transcriptional activation of HDAC inhibitors and DNMT inhibitors. As for clinical trials, using such combinations, initial results have been promising (Issa 2007). Similar exciting results have also been observed initially for coupling the epigenetic drugs with conventional chemotherapy (Issa 2007).
There are promising in vitro and in vivo data concerning epigenetic therapeutic potential, but except for a few FDA-approved drugs, those are still in early development. Overall, toxicity has been a serious drawback to the use of such drugs in clinics. Therefore, molecular characteristics that might help overcome these undesirable aspects in a potential drug candidate are being taken into consideration, and all efforts are being made toward developing specific and effective epigenetic therapeutic agents. Nevertheless, since the epigenetic machinery is very active and has antagonistic effects in different classes of genes, it is inevitable that blocking one mechanism may benefit a class of genes but stimulate another category of cancer-related genes, although the present good results of some demethylating agents contradict this idea (Gal-Yam et al. 2008). For that reason, the future lies on developing more specific drugs targeting pathways as well as on individual genes, even classes of genes.
Concluding remarks and future perspectives
Cancer epigenetics provides us a vision “above and beyond” the dogma of genotype (DNA sequence) determining the phenotype without intervention from any other major players. It allows scientists to picture the complexity of interactions existing from the genetic code to the cellular fate. The understanding that the mechanisms can be reversible gives the process a range of flexibility not imagined before. This fascinating fexibility of biological characteristics is also very attractive for clinical cancer management and monitoring aspects.
In oncology, important issues that need to be addressed are:
population screening for risk assessment;
early disease detection;
prognosis prediction;
therapeutic targets;
therapeutic response monitoring;
patient stratification;
reversion of malignant phenotype.
The recognition of the epigenetic machinery participation in carcinogenesis, modestly exemplified in this review and largely described in the last decades cancer research literature, allows us to contemplate the chance of dealing with the aforementioned oncologic issues in new ways that may have a significant impact in the therapy and management of cancer as a disease.
Going forward, we foresee a multitude of possibilities that need to evolve from academic exercises to valuable tools for clinical practice.
Well-characterized cohorts are needed for the validation of the candidate epigenetic biomarkers and drugs. Examples of difficulties lay on the heterogeneity of cancer samples; on the existence of not fully understood “cancer-specific epigenetic changes” in normal samples, which does not allow biomarker-based tests to present the high specificity and sensitivity needed to enter clinical routine. New efficient technologies that will represent enhanced specificity and sensitivity of biomarker detection as well as epigenetic target therapies with less toxicity are needed in this field.
Nowadays, there are several companies specialized in the development of epigenetic alteration-based assays: Oncomethylome Sciences, Epigenomics AG, Sequenom, Exact Sciences, Orion Genomics, Rubicon Genomics (Zhu and Yao 2009; Costa 2010a). Nevertheless, there are only a few epigenetic drugs approved so far for use in cancer patients. The anticipated new tools need to meet stricter standards to become usable oncologic tests. A great deal of data is generated with promising drug molecules, or biomarkers, but very few of these translate into reliable candidate molecules that can be used in the clinical setting (Sidransky 2002).
With all the accumulated knowledge about the role of DNA methylation alterations, histones modifications, DNA packaging processes, miRNAs expression, and interactions in carcinogenesis, the design of more rational interventions to target/detect epigenetic/epigenomic changes is expected in the not-so-distant future. The development of these epigenetic/epigenomic alteration-based tests and therapies is expected to complement the science of genomics in making new inroads to effective personalized medicine.
Acknowledgments
This work was funded by the National Institute of Dental and Craniofacial Research (NIDCR) SPORE program in Head and Neck Cancer Research grant number: P50 DE019032; and National Institute of Health (NIH), National Cancer Institute (NCI), Early Detection Research Network (EDRN) grant number: U01-CA084986. The funding agencies had no role in the design of the study, data collection, analysis, interpretation of the results, preparation of the manuscript, or the decision to submit the manuscript for publication. Under a licensing agreement between Oncomethylome Sciences, SA and the Johns Hopkins University, D.S. is entitled to a share of royalty received by the University upon sales of diagnostic products described in this article. D.S. owns Oncomethylome Sciences, SA stock, which is subject to certain restrictions under University policy. D.S. is a paid consultant to Oncomethylome Sciences, SA and is a paid member of the company's Scientific Advisory Board.
Footnotes
Declaration of interest: The Johns Hopkins University in accordance with its conflict of interest policies is managing the terms of this agreement.
References
- Acharya MR, Sparreboom A, Venitz J, Figg WD. Rational development of histone deacetylase inhibitors as anticancer agents: a review. Mol Pharmacol. 2005;68:917–932. doi: 10.1124/mol.105.014167. [DOI] [PubMed] [Google Scholar]
- Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–196. [PubMed] [Google Scholar]
- Baylin SB, Ohm JE. Epigenetic gene silencing in cancer—a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- Bedford MT, van Helden PD. Hypomethylation of DNA in pathological conditions of the human prostate. Cancer Res. 1987;47:5274–5276. [PubMed] [Google Scholar]
- Bhasin M, Reinherz EL, Reche PA. Recognition and classification of histones using support vector machine. J Comput Biol. 2006;13:102–112. doi: 10.1089/cmb.2006.13.102. [DOI] [PubMed] [Google Scholar]
- Bird A. The essentials of DNA methylation. Cell. 1992;70:5–8. doi: 10.1016/0092-8674(92)90526-i. [DOI] [PubMed] [Google Scholar]
- Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer. 2010;17:F19–F36. doi: 10.1677/ERC-09-0184. [DOI] [PubMed] [Google Scholar]
- Brait M, Begum S, Carvalho AL, Dasgupta S, Vettore AL, Czerniak B, Caballero OL, Westra WH, Sidransky D, Hoque MO. Aberrant promoter methylation of multiple genes during pathogenesis of bladder cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:2786–2794. doi: 10.1158/1055-9965.EPI-08-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueckner B, Stresemann C, Kuner R, Mund C, Musch T, Meister M, Sültmann H, Lyko F. The human let-7a-3 locus contains an epigenetically regulated microRNA gene with oncogenic function. Cancer Res. 2007;67:1419–1423. doi: 10.1158/0008-5472.CAN-06-4074. [DOI] [PubMed] [Google Scholar]
- Cairns P. Gene methylation and early detection of genitourinary cancer: the road ahead. Nat Rev Cancer. 2007;7:531–543. doi: 10.1038/nrc2170. [DOI] [PubMed] [Google Scholar]
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- Cameron EE, Bachman KE, Myöhänen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- Cang S, Ma Y, Liu D. New clinical developments in histone deacetylase inhibitors for epigenetic therapy of cancer. J Hematol Oncol. 2009;2:22. doi: 10.1186/1756-8722-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Chang SS, Jiang WW, Smith I, Poeta LM, Begum S, Glazer C, Shan S, Westra W, Sidransky D, Califano JA. MicroRNA alterations in head and neck squamous cell carcinoma. Int J Cancer. 2008;123:2791–2797. doi: 10.1002/ijc.23831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JC, Yoo CB, Weisenberger DJ, Chuang J, Wozniak C, Liang G, Marquez VE, Greer S, Orntoft TF, Tykjaer T, Jones PA. Preferential response of cancer cells to zebularine. Cancer Cell. 2004;6:151–158. doi: 10.1016/j.ccr.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG. Covalent histone modifications— miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- Costa FF. Epigenomics in cancer management. Cancer Manag Res. 2010a;2:255–265. doi: 10.2147/CMR.S7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa FF. Non-coding RNAs: meet thy masters. Bioessays. 2010b;32:599–608. doi: 10.1002/bies.200900112. [DOI] [PubMed] [Google Scholar]
- Costello JF, Frühwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomäki P, Lang JC, Schuller DE, Yu L, Bloomfield CD, Caligiuri MA, Yates A, Nishikawa R, Su Huang H, Petrelli NJ, Zhang X, O'Dorisio MS, Held WA, Cavenee WK, Plass C. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–138. doi: 10.1038/72785. [DOI] [PubMed] [Google Scholar]
- Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, Fuks F, Lo Coco F, Kouzarides T, Nervi C, Minucci S, Pelicci PG. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science. 2002;295:1079–1082. doi: 10.1126/science.1065173. [DOI] [PubMed] [Google Scholar]
- Ehrlich M. DNA hypomethylation, cancer, the immunodeficiency, centromeric region instability, facial anomalies syndrome and chromosomal rearrangements. J Nutr. 2002;132:2424S–2429S. doi: 10.1093/jn/132.8.2424S. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Jiang G, Fiala E, Dome JS, Yu MC, Long TI, Youn B, Sohn OS, Widschwendter M, Tomlinson GE, Chintagumpala M, Champagne M, Parham D, Liang G, Malik K, Laird PW. Hypomethylation and hypermethylation of DNA in Wilms tumors. Oncogene. 2002;21:6694–6702. doi: 10.1038/sj.onc.1205890. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16(Spec No 1):R50–R59. doi: 10.1093/hmg/ddm018. [DOI] [PubMed] [Google Scholar]
- Esteller M, Almouzni G. How epigenetics integrates nuclear functions. Workshop on epigenetics and chromatin: transcriptional regulation and beyond. EMBO Rep. 2005;6:624–628. doi: 10.1038/sj.embor.7400456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–3229. [PubMed] [Google Scholar]
- Esteller M, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Watkins DN, Issa JP, Sidransky D, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine–DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res. 2000;60:2368–2371. [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- Ferguson-Smith AC, Reik W, Surani MA. Genomic imprinting and cancer. Cancer Surv. 1990;9:487–503. [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal-Yam EN, Saito Y, Egger G, Jones PA. Cancer epigenetics: modifications, screening, and therapy. Annu Rev Med. 2008;59:267–280. doi: 10.1146/annurev.med.59.061606.095816. [DOI] [PubMed] [Google Scholar]
- Georgel PT, Hansen JC. Linker histone function in chromatin: dual mechanisms of action. Biochem Cell Biol. 2001;79:313–316. [PubMed] [Google Scholar]
- Glazer CA, Smith IM, Ochs MF, Begum S, Westra W, Chang SS, Sun W, Bhan S, Khan Z, Ahrendt S, Califano JA. Integrative discovery of epigenetically derepressed cancer testis antigens in NSCLC. PLoS ONE. 2009;4:e8189. doi: 10.1371/journal.pone.0008189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin J, Eisenhauer E. DNA methyltransferase inhibitors-state of the art. Ann Oncol. 2002;13:1699–1716. doi: 10.1093/annonc/mdf314. [DOI] [PubMed] [Google Scholar]
- Gonzalez S, Pisano DG, Serrano M. Mechanistic principles of chromatin remodeling guided by siRNAs and miRNAs. Cell Cycle. 2008;7:2601–2608. doi: 10.4161/cc.7.16.6541. [DOI] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y, Takahashi T. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005;65:9628–9632. doi: 10.1158/0008-5472.CAN-05-2352. [DOI] [PubMed] [Google Scholar]
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- Hanke M, Hoefig K, Merz H, Feller AC, Kausch I, Jocham D, Warnecke JM, Sczakiel G. A robust methodology to study urine microRNA as tumor marker: microRNA-126 and microRNA-182 are related to urinary bladder cancer. Urol Oncol. 2010;28:655–661. doi: 10.1016/j.urolonc.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- Hoque MO. DNA methylation changes in prostate cancer: current developments and future clinical implementation. Expert Rev Mol Diagn. 2009;9:243–257. doi: 10.1586/erm.09.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque MO, Kim MS, Ostrow KL, Liu J, Wisman GB, Park HL, Poeta ML, Jeronimo C, Henrique R, Lendvai A, Schuuring E, Begum S, Rosenbaum E, Ongenaert M, Yamashita K, Califano J, Westra W, van der Zee AG, Van Criekinge W, Sidransky D. Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res. 2008;68:2661–2670. doi: 10.1158/0008-5472.CAN-07-5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Greene E, Murray Stewart T, Goodwin AC, Baylin SB, Woster PM, Casero RA., Jr Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci USA. 2007;104:8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isharwal S, Miller MC, Marlow C, Makarov DV, Partin AW, Veltri RW. p300 (histone acetyltransferase) biomarker predicts prostate cancer biochemical recurrence and correlates with changes in epithelia nuclear size and shape. Prostate. 2008;68:1097–1104. doi: 10.1002/pros.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- Issa JP. DNA methylation as a therapeutic target in cancer. Clin Cancer Res. 2007;13:1634–1637. doi: 10.1158/1078-0432.CCR-06-2076. [DOI] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. A compiled and systematic reference map of nucleosome positions across the Saccharomyces cerevisiae genome. Genome Biol. 2009a;10:R109. doi: 10.1186/gb-2009-10-10-r109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009b;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- Jost JP, Bruhat A. The formation of DNA methylation patterns and the silencing of genes. Prog Nucleic Acid Res Mol Biol. 1997;57:217–248. doi: 10.1016/s0079-6603(08)60282-2. [DOI] [PubMed] [Google Scholar]
- Kacem S, Feil R. Chromatin mechanisms in genomic imprinting. Mamm Genome. 2009;20:544–556. doi: 10.1007/s00335-009-9223-4. [DOI] [PubMed] [Google Scholar]
- Kim MS, Lee J, Sidransky D. DNA methylation markers in colorectal cancer. Cancer Metastasis Rev. 2010;29:181–206. doi: 10.1007/s10555-010-9207-6. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Krichevsky AM, Gabriely G. miR-21: a small multi-faceted RNA. J Cell Mol Med. 2009;13:39–53. doi: 10.1111/j.1582-4934.2008.00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Längst G, Becker PB. Nucleosome remodeling: one mechanism, many phenomena? Biochim Biophys Acta. 2004;1677:58–63. doi: 10.1016/j.bbaexp.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Le XF, Merchant O, Bast RC, Calin GA. The roles of microRNAs in the cancer invasion-metastasis cascade. Cancer Microenviron. 2010;3:137–147. doi: 10.1007/s12307-010-0037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, Anile C, Maira G, Mercatelli N, Ciafrè SA, Farace MG, Agami R. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–3708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, Wiggins JF, Bader AG, Fagin R, Brown D, Tang DG. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211–215. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Serra L, Esteller M. Proteins that bind methylated DNA and human cancer: reading the wrong words. Br J Cancer. 2008;98:1881–1885. doi: 10.1038/sj.bjc.6604374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucio-Eterovic AK, Cortez MA, Valera ET, Motta FJ, Queiroz RG, Machado HR, Carlotti CG, Jr, Neder L, Scrideli CA, Tone LG. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: class II and IV are hypoexpressed in glioblastomas. BMC Cancer. 2008;8:243. doi: 10.1186/1471-2407-8-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Lujambio A, Calin GA, Villanueva A, Ropero S, Sánchez-Céspedes M, Blanco D, Montuenga LM, Rossi S, Nicoloso MS, Faller WJ, Gallagher WM, Eccles SA, Croce CM, Esteller M. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA. 2008;105:13556–13561. doi: 10.1073/pnas.0803055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Weinberg RA. Micromanagers of malignancy: role of microRNAs in regulating metastasis. Trends Genet. 2008;24:448–456. doi: 10.1016/j.tig.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Maldonado L, Hoque MO. Epigenomics and ovarian carcinoma. Biomark Med. 2010;4:543–570. doi: 10.2217/bmm.10.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- Martinez-Garcia E, Licht JD. Deregulation of H3K27 methylation in cancer. Nat Genet. 2010;42:100–101. doi: 10.1038/ng0210-100. [DOI] [PubMed] [Google Scholar]
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008;18:1073–1083. doi: 10.1101/gr.078261.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo SA, Ropero S, Moutinho C, Aaltonen LA, Yamamoto H, Calin GA, Rossi S, Fernandez AF, Carneiro F, Oliveira C, Ferreira B, Liu CG, Villanueva A, Capella G, Schwartz S, Jr, Shiekhattar R, Esteller M. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet. 2009;41:365–370. doi: 10.1038/ng.317. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O'Briant KC, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB, Tewari M. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. 2008;105:10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momparler RL. Cancer epigenetics. Oncogene. 2003;22:6479–6483. doi: 10.1038/sj.onc.1206774. [DOI] [PubMed] [Google Scholar]
- Morey L, Brenner C, Fazi F, Villa R, Gutierrez A, Buschbeck M, Nervi C, Minucci S, Fuks F, Di Croce L. MBD3, a component of the NuRD complex, facilitates chromatin alteration and deposition of epigenetic marks. Mol Cell Biol. 2008;28:5912–5923. doi: 10.1128/MCB.00467-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;23:314–318. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Natsume A, Kondo Y, Ito M, Motomura K, Wakabayashi T, Yoshida J. Epigenetic aberrations and therapeutic implications in gliomas. Cancer Sci. 2010;101:1331–1336. doi: 10.1111/j.1349-7006.2010.01545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netto GJ, Nakai Y, Nakayama M, Jadallah S, Toubaji A, Nonomura N, Albadine R, Hicks JL, Epstein JI, Yegnasubramanian S, Nelson WG, De Marzo AM. Global DNA hypomethylation in intratubular germ cell neoplasia and seminoma, but not in nonseminomatous male germ cell tumors. Mod Pathol. 2008;21:1337–1344. doi: 10.1038/modpathol.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Day E, Lal A. MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res. 2010;12:201. doi: 10.1186/bcr2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoto S, Fujii YR. Regulation of human immunodeficiency virus 1 transcription by nef microRNA. J Gen Virol. 2005;86:751–755. doi: 10.1099/vir.0.80449-0. [DOI] [PubMed] [Google Scholar]
- Oosterhuis JW, Looijenga LH. Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer. 2005;5:210–222. doi: 10.1038/nrc1568. [DOI] [PubMed] [Google Scholar]
- Passetti F, Ferreira CG, Costa FF. The impact of microRNAs and alternative splicing in pharmacogenomics. Pharmacogenomics J. 2009;9:1–13. doi: 10.1038/tpj.2008.14. [DOI] [PubMed] [Google Scholar]
- Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- Rainier S, Johnson LA, Dobry CJ, Ping AJ, Grundy PE, Feinberg AP. Relaxation of imprinted genes in human cancer. Nature. 1993;362:747–749. doi: 10.1038/362747a0. [DOI] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Reik W, Surani MA. Cancer genetics. Genomic imprinting and embryonal tumours. Nature. 1989;338:112–113. doi: 10.1038/338112a0. [DOI] [PubMed] [Google Scholar]
- Riddihough G, Zahn LM. Epigenetics. What is epigenetics? Introduction. Science. 2010;330:611. doi: 10.1126/science.330.6004.611. [DOI] [PubMed] [Google Scholar]
- Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14:9–25. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- Riggs AD, Jones PA. 5-Methylcytosine, gene regulation, and cancer. Adv Cancer Res. 1983;40:1–30. doi: 10.1016/s0065-230x(08)60678-8. [DOI] [PubMed] [Google Scholar]
- Ropero S, Fraga MF, Ballestar E, Hamelin R, Yamamoto H, Boix-Chornet M, Caballero R, Alaminos M, Setien F, Paz MF, Herranz M, Palacios J, Arango D, Orntoft TF, Aaltonen LA, Schwartz S, Jr, Esteller M. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat Genet. 2006;38:566–569. doi: 10.1038/ng1773. [DOI] [PubMed] [Google Scholar]
- Sawan C, Vaissière T, Murr R, Herceg Z. Epigenetic drivers and genetic passengers on the road to cancer. Mutat Res. 2008;642:1–13. doi: 10.1016/j.mrfmmm.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Fondufe-Mittendorf Y, Chen L, Tåström A, Field Y, Moore IK, Wang JP, Widom J. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura-Pacheco B, Trejo-Becerril C, Perez-Cardenas E, Taja-Chayeb L, Mariscal I, Chavez A, Acuña C, Salazar AM, Lizano M, Dueñas-Gonzalez A. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin Cancer Res. 2003;9:1596–1603. [PubMed] [Google Scholar]
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky D. Emerging molecular markers of cancer. Nat Rev Cancer. 2002;2:210–219. doi: 10.1038/nrc755. [DOI] [PubMed] [Google Scholar]
- Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–625. doi: 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- Smith IM, Glazer CA, Mithani SK, Ochs MF, Sun W, Bhan S, Vostrov A, Abdullaev Z, Lobanenkov V, Gray A, Liu C, Chang SS, Ostrow KL, Westra WH, Begum S, Dhara M, Califano J. Coordinated activation of candidate proto-oncogenes and cancer testes antigens via promoter demethylation in head and neck cancer and lung cancer. PLoS ONE. 2009;4:e4961. doi: 10.1371/journal.pone.0004961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith IM, Mydlarz WK, Mithani SK, Califano JA. DNA global hypomethylation in squamous cell head and neck cancer associated with smoking, alcohol consumption and stage. Int J Cancer. 2007;121:1724–1728. doi: 10.1002/ijc.22889. [DOI] [PubMed] [Google Scholar]
- Straussman R, Nejman D, Roberts D, Steinfeld I, Blum B, Benvenisty N, Simon I, Yakhini Z, Cedar H. Developmental programming of CpG island methylation profiles in the human genome. Nat Struct Mol Biol. 2009;16:564–571. doi: 10.1038/nsmb.1594. [DOI] [PubMed] [Google Scholar]
- Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- Taby R, Issa JP. Cancer epigenetics. CA Cancer J Clin. 2010;60:376–392. doi: 10.3322/caac.20085. [DOI] [PubMed] [Google Scholar]
- Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci USA. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, Mitsudomi T, Takahashi T. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomson JP, Skene PJ, Selfridge J, Clouaire T, Guy J, Webb S, Kerr AR, Deaton A, Andrews R, James KD, Turner DJ, Illingworth R, Bird A. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464:1082–1086. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting AH, McGarvey KM, Baylin SB. The cancer epigenome—components and functional correlates. Genes Dev. 2006;20:3215–3231. doi: 10.1101/gad.1464906. [DOI] [PubMed] [Google Scholar]
- Tsai WC, Hsu PW, Lai TC, Chau GY, Lin CW, Chen CM, Lin CD, Liao YL, Wang JL, Chau YP, Hsu MT, Hsiao M, Huang HD, Tsou AP. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009;49:1571–1582. doi: 10.1002/hep.22806. [DOI] [PubMed] [Google Scholar]
- Turker MS, Bestor TH. Formation of methylation patterns in the mammalian genome. Mutat Res. 1997;386:119–130. doi: 10.1016/s1383-5742(96)00048-8. [DOI] [PubMed] [Google Scholar]
- van Haaften G, Agami R. Tumorigenicity of the miR-17-92 cluster distilled. Genes Dev. 2010;24:1–4. doi: 10.1101/gad.1887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136:586–591. doi: 10.1016/j.cell.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigushin DM, Ali S, Pace PE, Mirsaidi N, Ito K, Adcock I, Coombes RC. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin Cancer Res. 2001;7:971–976. [PubMed] [Google Scholar]
- Visone R, Russo L, Pallante P, De Martino I, Ferraro A, Leone V, Borbone E, Petrocca F, Alder H, Croce CM, Fusco A. MicroRNAs (miR)-221 and miR-222, both overexpressed in human thyroid papillary carcinomas, regulate p27Kip1 protein levels and cell cycle. Endocr Relat Cancer. 2007;14:791–798. doi: 10.1677/ERC-07-0129. [DOI] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic EA, Brown CJ, Lam WL. Epigenetics of cancer progression. Pharmacogenomics. 2008;9:215–234. doi: 10.2217/14622416.9.2.215. [DOI] [PubMed] [Google Scholar]
- Waddington CH. The epigenotype. Endeavour. 1942;1:18–20. [Google Scholar]
- Wang QZ, Xu W, Habib N, Xu R. Potential uses of microRNA in lung cancer diagnosis, prognosis, and therapy. Curr Cancer Drug Targets. 2009;9:572–594. doi: 10.2174/156800909788486731. [DOI] [PubMed] [Google Scholar]
- Wang Y, Leung FC. An evaluation of new criteria for CpG islands in the human genome as gene markers. Bioinformatics. 2004;20:1170–1177. doi: 10.1093/bioinformatics/bth059. [DOI] [PubMed] [Google Scholar]
- Weissman B, Knudsen KE. Hijacking the chromatin remodeling machinery: impact of SWI/SNF perturbations in cancer. Cancer Res. 2009;69:8223–8230. doi: 10.1158/0008-5472.CAN-09-2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff EM, Byun HM, Han HF, Sharma S, Nichols PW, Siegmund KD, Yang AS, Jones PA, Liang G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010;6:e1000917. doi: 10.1371/journal.pgen.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurdinger T, Costa FF. Molecular therapy in the microRNA era. Pharmacogenomics J. 2007;7:297–304. doi: 10.1038/sj.tpj.6500429. [DOI] [PubMed] [Google Scholar]
- Xie Y, Todd NW, Liu Z, Zhan M, Fang H, Peng H, Alattar M, Deepak J, Stass SA, Jiang F. Altered miRNA expression in sputum for diagnosis of non-small cell lung cancer. Lung Cancer. 2010;67:170–176. doi: 10.1016/j.lungcan.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Nass SJ, Smith D, Nelson WG, Herman JG, Davidson NE. Specific inhibition of DNMT1 by antisense oligonucleotides induces re-expression of estrogen receptor-alpha (ER) in ER-negative human breast cancer cell lines. Cancer Biol Ther. 2003;2:552–556. doi: 10.4161/cbt.2.5.469. [DOI] [PubMed] [Google Scholar]
- Yu J, Yu J, Rhodes DR, Tomlins SA, Cao X, Chen G, Mehra R, Wang X, Ghosh D, Shah RB, Varambally S, Pienta KJ, Chinnaiyan AM. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. 2007;67:10657–10663. doi: 10.1158/0008-5472.CAN-07-2498. [DOI] [PubMed] [Google Scholar]
- Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. Genome-scale identifcation of nucleosome positions in S. cerevisiae. Science. 2005;309:626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302:1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- Zhu J, Yao X. Use of DNA methylation for cancer detection: promises and challenges. Int J Biochem Cell Biol. 2009;41:147–154. doi: 10.1016/j.biocel.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Zhu P, Martin E, Mengwasser J, Schlag P, Janssen KP, Göttlicher M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 2004;5:455–463. doi: 10.1016/s1535-6108(04)00114-x. [DOI] [PubMed] [Google Scholar]
- Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39:61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]