

Abstract
It is difficult to determine a chemical inhibitor’s binding site in multi-protein mixtures, particularly when high-resolution structural studies are not straightforward. Building upon previous research involving photo-crosslinking and the use of mixtures of stable isotopes, we report a method, Stable Isotope Labeled Inhibitors for Crosslinking (SILIC), for mapping a small molecule inhibitor’s binding site in its target protein. In SILIC, structure-activity relationship data is used to design inhibitor analogs that incorporate a photo-crosslinking group along with either natural or ‘heavy’ stable isotopes. An equimolar mixture of these inhibitor analogs is crosslinked to the target protein to yield a robust signature for identifying inhibitor-modified peptide fragments in complex mass spectrometry data. As a proof of concept, we applied this approach to an ATP-competitive inhibitor of kinesin-5, a widely conserved motor protein required for cell division and an anti-cancer drug target. This crosslinking analysis, along with mutagenesis studies, suggests that the inhibitor binds at an allosteric site in the motor protein.
Bioactive small molecules can be important chemotherapeutic drugs as well as valuable tools to elucidate the cellular functions of their target proteins.1,2 In both contexts, the value of the small molecule can be limited by a lack of understanding of its mechanism of inhibition and mode of target protein binding. Without these data, it can be difficult to improve potency, evaluate specificity, and fully explain cellular phenotypes resulting from drug treatment. A precise understanding of how a bioactive molecule interacts with its target can address these issues,3 but in many cases a crystal of the drug bound to the protein is difficult to obtain. Moreover, when the small molecule’s target is part of a multi-protein complex, analyzing the mechanism of inhibition using structural approaches can be challenging.
Photo-crosslinking of small molecules to proteins has been used to trap drug-target protein interactions in complex protein mixtures.4-6 Identifying drug targets and mapping drug binding sites after photo-crosslinking typically relies on systematic mass spectrometry based analyses of digested protein fragments to identify those with a small molecule adduct.7,8 While there are examples of the successful use of this approach, the general applicability of the method has been limited as crosslinking is often sub-stoichiometric,9 and the different possible inhibitor-peptide adducts can be difficult to detect in complex mass spectra. One strategy to address this involves generating inhibitor analogs with an affinity tag for capturing the inhibitor-peptide adducts.10 In many cases, however, the inhibitor’s dual modifications, for photocrosslinking and affinity-capture, can alter the compound’s mechanism of action. As an alternative approach to identify inhibitor-protein adducts within complex mass spectra, inhibitor analogs can be generated such that they carry a unique isotope pattern.11,12 The incorporation of natural and ‘heavy’ stable isotopes into a benzophenone photo-crosslinker moiety appended to the inhibitor of interest has been shown to aid the identification of its target in a proof-of-concept study.13 However, the method is not likely to be useful for mapping an inhibitor’s binding site. This is, in large part, due to the crosslinking group being incorporated via a linker, so that it is a significant distance from the functional groups that are likely to make key contacts with the target’s binding site.
Here, building on these studies, we have developed a method, named Stable Isotope Labeled Inhibitors for Crosslinking (SILIC), for mapping small molecule-protein binding sites. In the first step of this approach we incorporate a photo-crosslinking group (e.g. azide) into the inhibitor of interest (Figure 1), guided by available structure-activity relationship (SAR) data. The photo-crosslinking group is appended at a site that does not change the inhibitor’s mechanism of action, but is in the closest proximity possible to the inhibitor’s activity-conferring functionality, so as to increase the probability that crosslinks are at, or near, the protein’s inhibitor-binding pocket. Natural and heavy isotope inhibitor analogs, which have a mass difference of a few daltons but otherwise identical physical properties, are then generated. The multi-protein complex to be analyzed is then incubated with a 1:1 mixture of natural and heavy inhibitor. After photo-crosslinking and protein digestion, the resulting mixture of peptide fragments is separated by HPLC and analyzed using high-resolution mass spectrometry. The resulting mass spectra can comprise thousands of peaks and the peptide-inhibitor adduct is likely to be of low abundance due to sub-stoichiometric labeling. The peptide-inhibitor adduct is identified when a pair of peptides that co-elute in the LC have the expected mass difference and essentially equal signal intensity. Finally, guided by these data, site-directed mutagenesis experiments can be designed to further examine the inhibitor-binding sites identified by SILIC.
Figure 1.
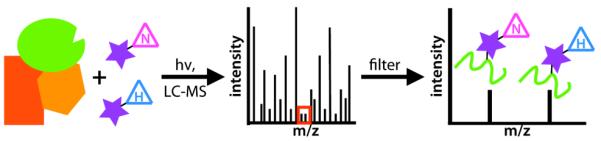
Schematic for SILIC. Inhibitors, with a photocrosslinking substituent and natural (N) or heavy (H) isotopes, are mixed with a complex of proteins, including the target (green). After UV-crosslinking, protein digestion, and LC-MS one obtains complex mass spectra that can be filtered based upon a ‘signature’ of peaks with the expected mass difference and equal signal intensities.
As a proof of concept, we focused on compound 1, an inhibitor of kinesin-5 (Figure 2a).14 Kinesins, which comprise a family of over 40 proteins, are motor proteins that move cargo along microtubules, polymers of the cytoskeletal protein tubulin.15,16 The kinesin-5 family is required for the assembly of the microtubule-based apparatus necessary for cell division.17 Inhibitors of kinesin-5 have provided valuable insight into mechanisms of cell division and have entered clinical trials as anti-cancer drugs.18,19 Kinesin-5 inhibitors that are in clinical trials, and have been used for cytological experiments, bind an allosteric site not conserved in other kinesins.20,21 These inhibitors are not competitive with respect to ATP.22 Recently, ATP-competitive inhibitors of kinesin-5 have been reported, including compound 1.14,23 As the ATP-binding site is the most conserved feature in kinesins, the possibility arises that these inhibitors may provide valuable starting points for developing new inhibitors for other kinesins. However, the binding site of 1 in kinesin-5 is not known and structural data has been difficult to obtain.14
Figure 2.
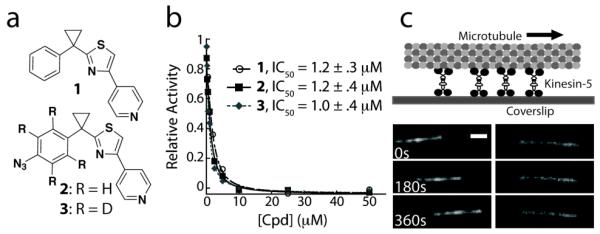
(a) Chemical structure of an ATP-competitive kinesin-5 inhibitor and analogs generated for SILIC. (b) Potency of compounds 1, 2 and 3 in inhibiting kinesin-5 activity, as examined using a steady-state microtubule-stimulated ATP hydrolysis assay. (c) In the presence of MgATP (1 mM) and DMSO, homotetrameric kinesin-5 drives microtubule gliding at an average rate of 19.6 ± 4.5 nm/s (left panel, n > 25). 1 (50nM) inhibits this activity (right panel, n > 30). Scale bar is 2μm.
To map the binding site of 1, we first analyzed its mechanism of action. We find that 1 inhibits steady state ATP hydrolysis by human kinesin-5’s ATPase domain (residues 1-368, expressed in bacteria) 25-times more potently when microtubules, the motor protein’s tracks, are present in the reaction (IC50: 1 + microtubules + kinesin-5 = 1.2 ± .3 μM; 1 + kinesin-5 = 30 ± 7 μM; Figure 2b, Table S1). We next examined inhibition of kinesin-5 driven microtubule gliding. These assays require protein constructs that are larger than those consisting of the monomeric ATPase domain. We generated full-length homotetrameric kinesin-5 (Xenopus laevis, expressed in insect cells as published previously24) and found that it drives microtubule gliding at 19.6 ± 4.5 nm/s (1 mM MgATP). Remarkably, even at 50 nM of 1, the motor activity was completely inhibited (Figure 2c), suggesting that tightly bound motor protein-microtubule complexes are formed in the presence of 1 and these complexes act as ‘brakes’ against other active motor protein molecules to stop microtubule motion.25 Together, these ATPase and microtubule gliding assay data suggest that the binding mode of 1 to kinesin-5 should be examined in the presence of microtubules. Analyzing the inhibitor-kinesin interaction in this context provided an interesting case for developing and applying our approach.
Guided by available SAR data,14 we designed analogs of 1, which contain an azide substituent on the phenyl ring as a photoreactive group and have natural (hydrogen, H; compound 2) and heavy (deuterium, D; compound 3) isotopes (Figure 2a). Synthesis of compound 2 was based on the published procedure for 1.14 To incorporate deuterium atoms that generate a 4 Da mass difference compared with 2, toluene-d8 was used as a starting material to synthesize the substituted phenyl ring moiety in 3 (see Supporting Information). Notably, the introduction of the crosslinking group in 1 did not affect the potency of the compounds and, as expected, the natural and heavy analogs have similar activities against kinesin-5 (Figure 2b, IC50: 2 = 1.2 ± .4 μM; 3 = 1.0 ± .4 μM). A 1:1 mixture of 2 and 3 (3 μM each) was incubated with kinesin-5 ATPase domain and microtubules. After UV irradiation at 254 nm for 30 min, the reaction mixture was resolved by SDS-PAGE. Following in-gel digestion, the peptide mixture was separated and analyzed by LC-MS/MS. Computer-based analysis (MaxQuant26) was used to efficiently detect peaks with a mass difference of 4 Da that eluted at the same time from the LC column. In independent experiments (n=3), we found equal intensity peaks corresponding to a single peptide (Figure 3a), indicating that this was likely to be the major inhibitor-peptide adduct. Further analysis by MS/MS identified this peptide as a fragment corresponding to Ser120–Arg138 of kinesin-5 and the crosslinking site of the inhibitors is very likely at one of three amino acid residues (Tyr125, Thr126, or Trp127) (Figure 3b).
Figure 3.
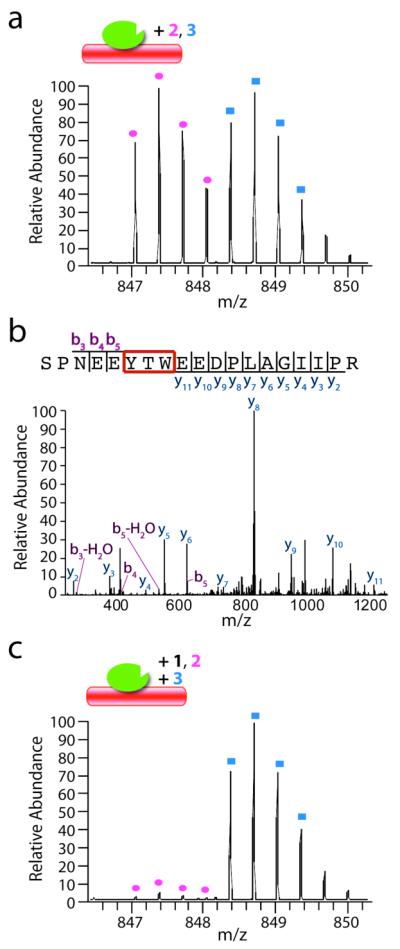
(a) A representative mass spectrum of the ‘signature’ peaks that denote peptide fragments of kinesin-5 crosslinked by equimolar 2 (pink circles) and 3 (blue squares) in the presence of microtubules. (b) The MS/MS spectrum and summary of fragmented ions of the crosslinked peptide. (c) When 2 (3 μM) is crosslinked in the presence of 1 (45 μM), there is near complete loss of crosslinked peptide, relative to that observed in the presence of 3 (3 μM) alone. Cartoon shows kinesin-5 (green) and microtubules (red) with crosslinking compounds.
We next examined whether excess 1 can suppress the crosslinking of 2 (or 3) to kinesin-5. For this experiment, we crosslinked kinesin-5 with 2 (3 μM) in the presence of excess 1 (45 μM) and, in parallel, crosslinked kinesin-5 with 3 (3 μM) alone. These samples were then mixed and processed for mass spectrometry analysis. This protocol provided a quantitative readout of the competition, as only one set of peaks in the mass spectrum should be suppressed. As shown in Figure 3c, 1 competes with 2, suggesting that 1 also binds at a site proximal to residues Tyr125, Thr126, and Trp127.
In the three-dimensional structure of the kinesin-5 ATPase domain (Figure 4a, PDB: 2WOG27), residues Y125, T126, and W127 map to a portion of loop-5. This loop is part of the allosteric binding site of other kinesin-5 inhibitors,21,27 such as S-Trityl-L-cyteine (STLC, IC50 = 1 μM, steady-state ATPase assay without microtubules28). As these inhibitors bind this pocket in the absence of microtubules, we examined if 1 did the same. To test this, we repeated the crosslinking and competition experiments in the absence of microtubules and found the same single crosslinking site (Figure S1). Furthermore, we found that addition of excess STLC (50 μM) prevented crosslinking of 2 (5 μM) to kinesin-5, relative to the reference sample (kinesin-5 crosslinked to 3, Figure 4b). Together, these data suggest that 1, like STLC, binds in an allosteric site.
Figure 4.
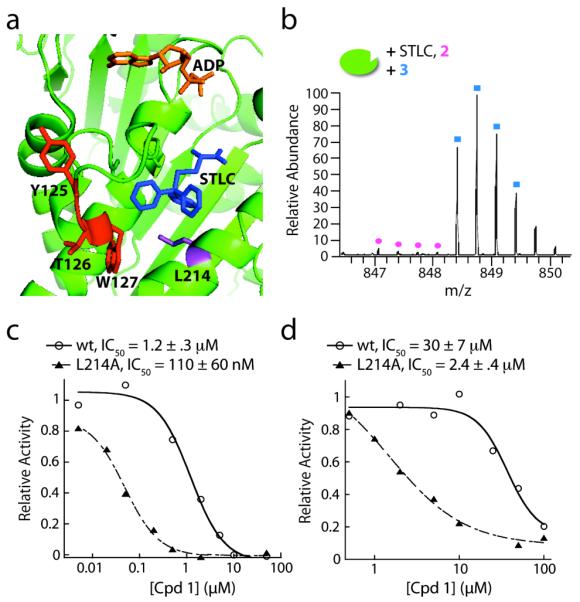
(a) The kinesin-5 ATPase domain (green, PDB: 2WOG27) in complex with ADP (orange) and STLC (blue). The crosslinked residues Y125, T126, and W127 are shown in red, while residue L214 is in purple. (b) Crosslinking of 2 (5μM) to kinesin-5 in the presence of STLC (50μM). (c) Representative curve of dose-dependent inhibition of steady-state microtubule-stimulated ATP hydrolysis by wildtype (wt) kinesin-5 and the L214A mutant protein. (d) Representative curve of dose-dependent inhibition of steady-state ATP hydrolysis by wildtype (wt) kinesin-5 and the L214A mutant protein, in the absence of microtubules. IC50 values are averages ± s.d. (n = 3).
We next used site-directed mutagenesis to analyze whether inhibition of kinesin-5 by 1 was sensitive to changes in the allosteric pocket. A leucine-214 to alanine (L214A) mutation, in this allosteric binding site of kinesin-5, is known to suppress STLC inhibition (Figure 4a).29 Using the steady-state ATP hydrolysis assay, both with and without microtubules, we found that this mutation lead to a ten-fold increase in the potency of 1 (Figure 4c, d, Table S1). Importantly, crosslinking of 2 and 3 to kinesin-5, which had the L214A mutation, resulted in identification of the same binding site (Figure S2). In addition, we find that 1 is an ATP-competitive inhibitor of the mutant kinesin-5 (L214A) (Figure S3), indicating that the mode of inhibition is not altered by this mutation. Together, our data are consistent with 1 binding at, or close to, the site bound by other kinesin-5 inhibitors that are not ATP-competitive.
It is relatively uncommon for nucleotide-competitive inhibitors to bind at an allosteric site in an ATPase.30 Interestingly, another example of this mechanism of inhibition has been described for another kinesin-5 inhibitor, GSK-1.23 This compound has been proposed to bind to an allosteric site that is distinct from the binding site of compound 1 and STLC. Further studies with both kinesin-5 ATP-competitive inhibitors will be needed to reveal how binding at allosteric sites results in ATP-competitive inhibition that leads to a tightly bound microtubule-motor protein complex.
In summary, we demonstrate that SILIC can be used to map the binding site of an inhibitor to its target. We believe this approach should be effective in analyzing inhibitor-target interactions, particularly when structural studies are intractable. A potential limitation of the approach is that the incorporation of a photocrosslinking moiety, such as an aryl azide, may not always be feasible. However, aryl groups appear at high frequency in bioactive small molecules. In these cases, SAR studies could allow the inclusion of an azide moiety such that it does not alter the inhibitor’s mechanism of action and yet is proximal to the target’s binding site residues. In the future we wish to extend this analysis to more complex cellular contexts to examine whether cellular physiology alters the mode of drug-target interaction and mechanism of drug action.
Supplementary Material
ACKNOWLEDGMENT
We thank Brian Chait, Kelly Molloy and Yinyin Li for reagents, equipment use, technical assistance, and discussions. This work was supported by the NIH (GM65933).
Footnotes
Supporting Information Available: Details of the synthesis of compounds 2 and 3, experimental findings from crosslinking without microtubules and with the L214A mutant, ATP-competition results, experimental procedures, and complete refs 5, 8 and 23. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Peterson JR, Mitchison TJ. Chem Biol. 2002;9:1275. doi: 10.1016/s1074-5521(02)00284-3. [DOI] [PubMed] [Google Scholar]
- (2).Lampson MA, Kapoor TM. Nat Chem Biol. 2006;2:19. doi: 10.1038/nchembio757. [DOI] [PubMed] [Google Scholar]
- (3).Tarrant MK, Cole PA. Annu Rev Biochem. 2009;78:797. doi: 10.1146/annurev.biochem.78.070907.103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Shorr RG, Heald SL, Jeffs PW, Lavin TN, Strohsacker MW, Lefkowitz RJ, Caron MG. Proc Natl Acad Sci U S A. 1982;79:2778. doi: 10.1073/pnas.79.9.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Seiffert D, et al. J Biol Chem. 2000;275:34086. doi: 10.1074/jbc.M005430200. [DOI] [PubMed] [Google Scholar]
- (6).Chen JK, Taipale J, Cooper MK, Beachy PA. Genes Dev. 2002;16:2743. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Al-Mawsawi LQ, Fikkert V, Dayam R, Witvrouw M, Burke TR, Jr., Borchers CH, Neamati N. Proc Natl Acad Sci U S A. 2006;103:10080. doi: 10.1073/pnas.0511254103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wood KW, et al. Proc Natl Acad Sci U S A. 2010;107:5839. doi: 10.1073/pnas.0915068107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hermanson GT. Bioconjugate Techniques. 2nd Edition Academic Press, Inc; 2008. [Google Scholar]
- (10).Salisbury CM, Cravatt BF. Proc Natl Acad Sci U S A. 2007;104:1171. doi: 10.1073/pnas.0608659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kelleher NL, Nicewonger RB, Begley TP, McLafferty FW. J Biol Chem. 1997;272:32215. doi: 10.1074/jbc.272.51.32215. [DOI] [PubMed] [Google Scholar]
- (12).Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Nature. 2010;468:790. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lamos SM, Krusemark CJ, McGee CJ, Scalf M, Smith LM, Belshaw PJ. Angew Chem Int Ed Engl. 2006;45:4329. doi: 10.1002/anie.200600743. [DOI] [PubMed] [Google Scholar]
- (14).Rickert KW, Schaber M, Torrent M, Neilson LA, Tasber ES, Garbaccio R, Coleman PJ, Harvey D, Zhang Y, Yang Y, Marshall G, Lee L, Walsh ES, Hamilton K, Buser CA. Arch Biochem Biophys. 2008;469:220. doi: 10.1016/j.abb.2007.10.016. [DOI] [PubMed] [Google Scholar]
- (15).Miki H, Okada Y, Hirokawa N. Trends Cell Biol. 2005;15:467. doi: 10.1016/j.tcb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- (16).Vale RD, Milligan RA. Science. 2000;288:88. doi: 10.1126/science.288.5463.88. [DOI] [PubMed] [Google Scholar]
- (17).Sharp DJ, Rogers GC, Scholey JM. Nature. 2000;407:41. doi: 10.1038/35024000. [DOI] [PubMed] [Google Scholar]
- (18).Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Science. 1999;286:971. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- (19).Duhl DM, Renhowe PA. Curr Opin Drug Discov Devel. 2005;8:431. [PubMed] [Google Scholar]
- (20).Maliga Z, Kapoor TM, Mitchison TJ. Chem Biol. 2002;9:989. doi: 10.1016/s1074-5521(02)00212-0. [DOI] [PubMed] [Google Scholar]
- (21).Yan Y, Sardana V, Xu B, Homnick C, Halczenko W, Buser CA, Schaber M, Hartman GD, Huber HE, Kuo LC. J Mol Biol. 2004;335:547. doi: 10.1016/j.jmb.2003.10.074. [DOI] [PubMed] [Google Scholar]
- (22).Cochran JC, Gilbert SP. Biochemistry. 2005;44:16633. doi: 10.1021/bi051724w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Luo L, et al. Nat Chem Biol. 2007;3:722. doi: 10.1038/nchembio.2007.34. [DOI] [PubMed] [Google Scholar]
- (24).Weinger JS, Qiu M, Yang G, Kapoor TM. Curr Biol. 2011;21:154. doi: 10.1016/j.cub.2010.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Groen AC, Needleman D, Brangwynne C, Gradinaru C, Fowler B, Mazitschek R, Mitchison TJ. J Cell Sci. 2008;121:2293. doi: 10.1242/jcs.024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cox J, Mann M. Nat Biotechnol. 2008;26:1367. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- (27).Kaan HY, Ulaganathan V, Hackney DD, Kozielski F. Biochem J. 2010;425:55. doi: 10.1042/BJ20091207. [DOI] [PubMed] [Google Scholar]
- (28).DeBonis S, Skoufias DA, Lebeau L, Lopez R, Robin G, Margolis RL, Wade RH, Kozielski F. Mol Cancer Ther. 2004;3:1079. [PubMed] [Google Scholar]
- (29).Brier S, Lemaire D, DeBonis S, Forest E, Kozielski F. J Mol Biol. 2006;360:360. doi: 10.1016/j.jmb.2006.04.062. [DOI] [PubMed] [Google Scholar]
- (30).Zhang J, Yang PL, Gray NS. Nat Rev Cancer. 2009;9:28. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.