

Abstract
Alzheimer's disease (AD) is defined by the concurrence of accumulation of abnormal aggregates composed of two proteins: Amyloid beta (Aβ) and tau, and of cellular changes including neurite degeneration and loss of neurons and cognitive functions. Based on their strong association with disease, genetically and pathologically, it is not surprising that there has been a focus towards developing therapies against the aggregated structures. Unfortunately, current therapies have but mild benefit. With this in mind we will focus on the relationship of synaptic plasticity with Aβ and tau protein and their role as potential targets for the development of therapeutic drugs. Finally, we will provide perspectives in developing a multifactorial strategy for AD treatment.
1. Introduction
Amyloid Beta: The Therapeutic Strategy —
Alzheimer's disease (AD) is histopathologically characterized by extraneuronal amyloid-beta protein (Aβ) deposits. Historically, Aβ has been related to cell toxicity and genetic evidence provides the basis for its proposal as the primary cause of the disease [1, 2]. Supporting these hypotheses, impairment in Aβ clearance by the central nervous system (CNS) has been reported in AD patients [3]. In cultured neurons, Aβ was shown to activate apoptotic pathways, leading to caspase activation, ultimately contributing to neurodegeneration [4]. It was also found that Aβ can activate apoptosis signal-regulating kinase (ASK1) that is required for ROS- and ER-stress-induced JNK activation and apoptosis, mainly through production of reactive oxygen species (ROS), but not through endoplasmic-reticulum-(ER-) mediated stress [2, 5]. Aβ has been found to modulate redox factor-1 that plays crucial roles in both cell death signaling pathways and DNA repair by interacting with transcription factors such as AP-1, NF-kappaB, and p53 and directly participating in the cleavage of apurinic/apyrimidinic DNA lesions, therefore affecting both the cell death signaling pathways and DNA repair [6]. Furthermore, Aβ was responsible for inducing oxidative stress, predominantly via mitochondria, which also affected cholesterol balance [7] and can cause neurotoxicity due to production of free radicals [8].
The proposed mechanism for Aβ to exert the neurotoxic effects was the assembly into Aβ plaques and oligomers [9]. This was further nurtured by the finding of a genetic component associated to the hypothesis of Aβ deposition. Mutations in the amyloid precursor protein (APP) that facilitate its cleavage to generate amyloid peptide and/or mutations in presenilin-1 (PS-1) or presenilin-2 (PS-2), that promote amyloid peptide formation and consequently Aβ deposition have also been reported [10–12]. Although the genetic component is documented, we have to mention that far less than 1% of the worldwide AD cases are based on APP or PS mutations. Indeed, in a systematic genetic study of AD patients in a Latin American population, PS and APP mutations were absent [13].
Aβ-containing plaques have been classified into subtypes, such as senile, diffuse, and neuritic [14], with diffuse plaques having little impact on cognitive function, whereas neuritic plaques are associated with cognitive decline [15]. Therefore, it was hypothesized that diffuse plaques appear during early preclinical stages of the disease with later appearance of neuritic plaques. However the “peptide to plaque” model remains debated [16]. In recent years, other stages of Aβ aggregation, beside plaques, seem to play a role in the development of AD [17]. In this regard it has been shown that the Aβ aggregation/oligomerization process is playing a central role in pathogenesis; in other words, one soluble Aβ molecule (monomer) interacts with other Aβ monomers to form dimers, oligomers, and polymers, each state of aggregations forming a potentially pathogenic entity. Indeed, different states of soluble aggregates have been strongly related to synaptic loss and cognitive impairment [18], a topic that we will discuss later. From these data, the aggregation state (i.e., the different oligomers) seems to be related to the disease. Given that, disaggregation of a plaque will lead to an increase of Aβ monomers/oligomers and therefore could cause neurotoxicity. Furthermore, inhibiting the formation of aggregates will preserve monomeric or polymeric structures and, therefore, could cause neurotoxicity. Overall, the perceived strength of the amyloid cascade hypothesis is reflected in the scientific literature, which is voluminous and dominated by experimental studies that adhere to the following statement: Aβ accumulates in the AD brain, consequently leading to neurodegeneration [19]. Although spatial distribution of increased levels of Aβ is related to AD pathology [20] and some degree of correlation with neuronal loss has been reported [21], no strong clinical correlation between plaque deposition and the degree of cognitive decline during AD has been found [22]. It is therefore in question whether the Aβ plaque is responsible for all the damage seen during the process of neurodegeneration or not.
An Alternative Point of View —
We have suggested that Aβ deposition as plaques could represent the effect rather than the cause of AD [23, 24]. Aβ aggregation may not be a harbinger of death, but rather a protective response to neuronal insult [25]. And, perhaps most contrary to current thinking, due to the fact that there is a negative correlation between Aβ deposition and oxidative damage [26], it has been proposed that diffuse amyloid plaques may be a compensatory response aimed at reducing oxidative stress [27–30]. If this hypothesis is correct, it means that Aβ deposits are simply a compensatory response, explaining the failure of therapeutic approaches simply directed to removal of Aβ plaques [31].
2. Aβ: The Therapeutic Target
Presently Aβ is one of the main therapeutic targets. In fact, immunization with Aβ was successful at removing Aβ from the brain. Imaging studies in AD patients showed that immunization with Aβ decreased amyloid plaques in the brain; however, this had no effect on cognition [32]. In mouse models, immunization has cleared small deposits and diffuses Aβ surrounding fibrillar cores [33]. But beyond removing Aβ, immunization has failed to clearly improve cognition in patients [34]. Active vaccination with Aβ in patients with mild-to-moderate AD in a phase II trial showed CNS inflammatory response [35], that was blamed for the failure. However, the inflammatory response has not been definitively proven to be the cause for the failure. Despite this outcome, new vaccines are presently in trial: Eli Lilly's solanezumab and Janssen and Pfizer's bapineuzumab (originally developed by the Dublin-based company Elan), are using monoclonal antibodies that work with the immune system, binding to amyloid-β and helping to clear accumulated amyloid-β peptides in the brain. Both are being tested in phase III trials on thousands of participants with mild-to-moderate Alzheimer's disease [36]. Following the hypothesis of preventing aggregates, β-secretase and γ-secretase inhibitors have also been used therapeutically, but with mixed results due to inhibition of other vital pathways, for example, the Notch pathway [37]. Although the main goal is to block the production of Aβ [37], γ-secretase inhibitors also block the proteolytic processing and function of Notch, which is essential for brain morphogenesis, making γ-secretase a worthwhile but difficult target for intervention [38]. In sum, the therapeutic outcome of current Aβ-related trials is rather discouraging. While there are without a doubt multiple factors that could explain a negative outcome of these trials, the above hypothesis raises the possibility that Aβ plaques are not a viable primary therapeutic target. Overall, there is little debate about Aβ being involved in AD pathology, but a strong debate about whether it is an appropriate therapeutic target.
3. Tau Protein: The Therapeutic Approach
Tau is an axonal protein that regulates microtubule stability [39]. According to current AD hypotheses, (a) tau becomes abnormally phosphorylated, (b) dissociates from microtubules and, (c) aggregates into neurofibrillary tangles (NFTs) [40, 41]. Tau has at least 45 phosphorylation sites, most of them are located in the proline-rich region (P-region) (residues 172–251) and the C-terminal tail region (C-region) (residues 368–441) [42]. Tau phosphorylation at both of these regions affects its capacity to interact with microtubules [43]. In terms of AD development, the phosphorylation sites located in the C-terminal region seem to play an interesting role. Phosphorylation at Ser262 selectively impairs binding of tau to microtubules [44]. Phosphorylation at Ser202 enhances tau polymerization; phosphorylation at the two neighbouring sites Ser202-Thr205 makes filament formation more sensitive to small changes in tau concentration [45]. Taking these data together, it seems that multiple phosphorylation events of tau, rather than just singular phosphorylation, play a crucial role during AD-related tau pathology [46, 47].
The abnormal phosphorylation of tau during AD is either related to an increase in kinase activity (glycogen synthase kinase 3β, cyclin-dependent kinase-5, p42/44 MAP kinase, p38 MAPK, stress-activated protein kinases, mitotic protein kinases) and/or a decrease in phosphatase activity (protein phosphatases 1, 2a, 2b) [48–52]. Certainly one kinase known to be of importance to tau phosphorylation is GSK3β [53]. In terms of phosphatase deregulation, it has been shown that protein phosphatases PP1, PP2A, PP2B, and PP5 dephosphorylate tau in vitro at sites Ser199, Ser202, Thr205, Thr212, Ser214, Ser235, Ser262, Ser396, Ser404, and Ser409 [54]. Of all phosphatases, PP2A was found to be the strongest tau-related phosphatase [54].
4. Therapeutic Strategy
Tau phosphorylation is established as a major factor during AD with a therapeutic focus on its kinases and phosphatases [55, 56]. Recently, it was reported that sodium selenate was able to reduce tau phosphorylation by stabilizing PP2A, therefore, mitigating tau pathology in transgenic AD models [57]. Targeting PP2B, several neuroleptics (such as chlorpromazine, trifluoperazine, and clozapine) have also been suggested for AD treatment [58]. In an approach, parallel to prevent Aβ plaque formation, drugs that prevent tau aggregation have also been developed [59, 60]. At the end, the answer is quite the same: no proven success has yet emerged from these approaches.
4.1. Synaptic Plasticity: The Link
4.1.1. Synaptic Plasticity and Kinases
Synaptic plasticity has been proposed to play a central role in brain capacity to incorporate transient experiences into persistent memory traces. Synaptic transmission can be enhanced (long-term potentiation, LTP) or depressed (long-term depression, LTD) by activity, and these changes can range from seconds to hours and days [61, 62]. Importantly, the affected intracellular pathways leading to LTP or LTD activation involve several kinases, such as GSK3β, SRC family tyrosine kinases, protein kinase A, protein kinase C, and, in particular, Ca2+/calmodulin-dependent protein kinase II [63, 64], which are known to play a role in AD. Given the importance of synaptic plasticity, it is not surprising that these phenomena could be affected during neurodegeneration and AD [65]. Presently, there is growing evidence that Aβ and tau are involved in synaptic dysfunction [66, 67]. Reports had placed Aβ close to synaptic terminals [68]. Indeed, Aβ was found to enhance N-methyl-D-aspartate (NMDA) receptor function by direct interaction [69]. Aβ was also found responsible for changes in presynaptic mechanism at the CA1–CA3 synapse of pyramidal neurons in the hippocampus [70] and for functional deficits in the mossy fibre pathway [71]. Furthermore, Aβ leads to decreased mitochondria in dendrites that resulted in the reduction in the number of spines or synapses [72, 73] and has been related to neuritic degeneration [74]. In the context of synaptic plasticity, Aβ leads to impairment of LTP [75] and facilitates LTD [76, 77]. Of note, Aβ has been suggested to exacerbate synaptic mitochondrial alterations including increased oxidative stress, decreased respiration, and compromised calcium handling capacity [78], all of them having an impact on synaptic plasticity. Overall, there is growing evidence showing that Aβ is related to changes in synaptic function.
4.1.2. Phosphorylation as the Link
The critical question to raise at this point is how Aβ and tau are interconnected during the disease. Although the relationship between these two proteins remains vague, data has lent support to the hypothesis that phosphorylation of tau protein could be the key linking mechanism. Ten years ago it was found that Aβ fibrils accelerate the formation of abnormally phosphorylated neurofibrillary tangles (NFTs) in a tau transgenic mouse [79]. The following years, it was reported that Aβ could induce tau phosphorylation and toxicity in cultured septal cholinergic neurons [80]. More recently, it has been shown that Aβ oligomers cause abnormal tau phosphorylation and morphology changes of spines by missorting of endogenous tau into dendrites [81]. Finally, natural Aβ isolated from AD brains is sufficient to induce AD-type tau phosphorylation and, consequently, neuritic dystrophy [74]. Concerning synaptic plasticity, it has been found that the absence of tau protein inhibits the impairment of LTP and neurotoxicity caused by Aβ [82] and targeting tau by immunotherapy prevents cognitive decline in a tangle mouse model [83]. Additionally, recent data show that tau protein has a dendritic function in targeting the Src kinase Fyn to postsynaptic NMDA receptors [84]. Summarizing these data, it appears that synaptic failure and neurotoxicity induced by Aβ require tau phosphorylation. Not surprisingly, elimination of tau has been suggested as a therapeutic target in order to ameliorate disease progression [85]. However, neuronal alterations that underlie symptoms of AD are not exclusively due to a direct toxic effect of Aβ [24]. Furthermore, we do not know the molecular mechanisms underlying the complex changes of synaptic plasticity during AD. Just recently, we have found that tau protein has a physiological function at the synaptic terminal that is regulated by tau phosphorylation (unpublished data). Therefore, simply getting rid of tau protein as some have suggested could adversely affect the equilibrium of different forms of synaptic plasticity. The question remains: is synaptic plasticity the link between Aβ and tau? Clearly, the potency to look at AD from a synaptic perspective is that it integrates Aβ and tau in a functional concept.
5. Conclusion and Perspectives
Pathogenesis of AD comprises neurodegeneration in the hippocampal area of brain that is critically involved in learning and memory. Presently, synaptic plasticity (LTP and LTD), the process by which synapses modulate their connections with other neurons, seems to be playing an important role in response to injury and disease. But more importantly, emerging evidence suggests that synaptic dysfunction beside neuronal death is leading to cognitive failure associated with AD. In fact, parts of AD pathogenesis could be explained by a loss of synaptic plasticity. In this regard, a growing amount of data is showing that Aβ and tau protein are both necessary to cause changes in synaptic plasticity. However, the question remains: should either Aβ or tau be the therapeutic targets? The concept that aggregation of Aβ and tau is deleterious to cells and amenable to therapeutic molecules may be too simplistic. Instead, upstream and downstream targets have to emerge as therapeutic options. In this context, synaptic plasticity modulators could be an interesting target. The most important aspect of this approach is that synaptic plasticity links Aβ and tau to the synaptic terminal. In this regard, some molecules have already been tested. Memantine, for example, is a partial antagonist of NMDA-receptor function, approved for moderate-to-severe Alzheimer's disease (AD) treatment within the USA and Europe (under the brand name Namenda (Forest), Axura and Akatinol (Merz), and Ebixa and Abixa (Lundbeck)), has some promise [86, 87]. Memantine, in its current therapeutic form, only slows down the neuronal degeneration process, but does not improve cognitive function [88]. However, Memantine might be beneficial to modulate the NMDA-receptor response in earlier stages of AD, where synaptic plasticity rather than neurotoxicity is playing a role. Furthermore, another target for therapeutic intervention would be Fyn kinase, a member of the Src kinase family, which is intricately involved with potentiating NMDA-receptor-dependent transmission [89]. Fyn kinase is receptive to changes in intracellular tau and extracellular Aβ at the synaptic terminal [84, 90]. Therefore, drugs that directly regulate Fyn activity might be beneficial. Like Fyn, there are other targets critically involved during synaptic plasticity and AD, as previously mentioned. One such target is GSK3β, which is necessary for the induction of synaptic LTD, while at the same time inhibits LTP [91].
In summary, it appears that the balance in synaptic plasticity during AD is tipped toward the induction of LTD. Hence, drugs that enhance LTP in combination with drugs that reduce induction of LTD might be of great value to treat AD. This therapeutic approach (i.e., simultaneously targeting critical check points for synaptic plasticity) will hopefully improve memory formation during AD (Figure 1). In sum, it is becoming all the clearer that approaches that focus on removing the pathological manifestations of AD might miss the intended outcome. Therefore, working with the biology of AD offers new hope for effective therapeutics.
Figure 1.
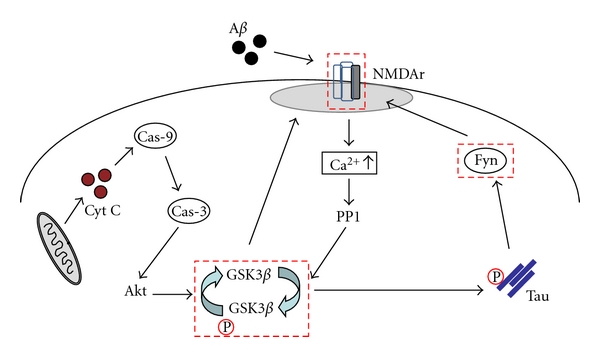
The role of GSK3β and Fyn during AD-related neurodegeneration and memory formation, along with NMDA receptor, makes them important therapeutic targets (red square). Impairment of hippocampal LTP by Aβ is through direct interaction with NMDA receptor. Calcium (Ca2+) enters via NMDA receptors and this leads to activation of protein phosphatase 1 (PP1), a key enzyme in synaptically induced LTD. PP1 can dephosphorylate GSK3β that determines whether NMDA receptor activation induces LTD or inhibits LTD. Aβ leads to decreased mitochondria and oxidative injury that promotes the release of cytochrome C (Cyt C) that may activate caspase-9 and caspase-3, which can cleave Akt, resulting in GSK3β activation. GSK3β under the control of Akt and PP1, is a critical determinant of the direction of NMDA receptor-dependent plasticity. The active GSK3β isoforms critically contribute to neurodegeneration by hyperphosphorylation of tau which deregulates Fyn activity and consequently affects NMDA receptor response.
Conflict of Interests
G. Perry is, or has been, a paid consultant for and/or owns equity or stock options in Neurotez Pharmaceuticals, Panacea Pharmaceuticals, Takeda Pharmaceuticals, and Voyager Pharmaceuticals. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the paper apart from those disclosed.
Acknowledgments
Work in the authors' laboratories is supported by the Alzheimer's Association, USA; Alzheimer Society, Canada; CIHR, Canada; GRSNC, Québec, Canada.
Abbreviations
- AD:
Alzheimer's disease
- Aβ:
Amyloid-beta protein
- CNS:
Central nervous system
- ROS:
Reactive oxygen species
- APP:
Amyloid precursor protein
- ER:
Endoplasmic reticulum
- PS-1:
Presenilin-1, or
- PS-2:
Presenilin-2
- NFTs:
Neurofibrillary tangles
- LTP:
Long-term potentiation
- LTD:
Long-term depression
- PP1:
Protein phosphatase 1
- NMDA:
N-methyl-D-aspartate receptor
References
- 1.Lambert J-C, Amouyel P. Genetics of Alzheimer's disease: new evidences for an old hypothesis? Current Opinion in Genetics and Development. 2011;21(3):295–301. doi: 10.1016/j.gde.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Kadowaki H, Nishitoh H, Urano F, et al. Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death and Differentiation. 2005;12(1):19–24. doi: 10.1038/sj.cdd.4401528. [DOI] [PubMed] [Google Scholar]
- 3.Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science. 2010;330(6012):p. 1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ding H, Matthews TA, Johnson GVW. Site-specific phosphorylation and caspase cleavage differentially impact tau-microtubule interactions and tau aggregation. Journal of Biological Chemistry. 2006;281(28):19107–19114. doi: 10.1074/jbc.M511697200. [DOI] [PubMed] [Google Scholar]
- 5.Hashimoto Y, Niikura T, Chiba T, et al. The cytoplasmic domain of Alzheimer’s amyloid-β protein precursor causes sustained apoptosis signal-regulating kinase 1/c-Jun NH 2-terminal kinase-mediated neurotoxic signal via dimerization. Journal of Pharmacology and Experimental Therapeutics. 2003;306(3):889–902. doi: 10.1124/jpet.103.051383. [DOI] [PubMed] [Google Scholar]
- 6.Tan Z, Shi L, Schreiber SS. Differential expression of redox factor-1 associated with beta-amyloid-mediated neurotoxicity. Open Neuroscience Journal. 2009;3:26–34. doi: 10.2174/1874082000903010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colell A, Fernández A, Fernández-Checa JC. Mitochondria, cholesterol and amyloid β peptide: a dangerous trio in Alzheimer disease. Journal of Bioenergetics and Biomembranes. 2009;41(5):417–423. doi: 10.1007/s10863-009-9242-6. [DOI] [PubMed] [Google Scholar]
- 8.Harris ME, Hensley K, Butterfield DA, Leedle RA, Carney JM. Direct evidence of oxidative injury produced by the Alzheimer’s β-amyloid peptide (1-40) in cultured hippocampal neurons. Experimental Neurology. 1995;131(2):193–202. doi: 10.1016/0014-4886(95)90041-1. [DOI] [PubMed] [Google Scholar]
- 9.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. Journal of Neuroscience. 1993;13(4):1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borchelt DR, Lee MK, Gonzales V, et al. Accumulation of proteolytic fragments of mutant presenilin 1 and accelerated amyloid deposition are co-regulated in transgenic mice. Neurobiology of Aging. 2002;23(2):171–177. doi: 10.1016/s0197-4580(01)00280-9. [DOI] [PubMed] [Google Scholar]
- 11.Borchelt DR, Ratovitski T, Van Lare J, et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19(4):939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 12.Campion D, Flaman JM, Brice A, et al. Mutations of the presenilin I gene in families with early-onset Alzheimer’s disease. Human Molecular Genetics. 1995;4(12):2373–2377. doi: 10.1093/hmg/4.12.2373. [DOI] [PubMed] [Google Scholar]
- 13.Arango D, et al. Systematic genetic study of Alzheimer disease in Latin America: mutation frequencies of the amyloid β precursor protein and presenilin genes in Colombia. American Journal of Medical Genetics. 2001;103(2):138–143. doi: 10.1002/1096-8628(20011001)103:2<138::aid-ajmg1529>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 14.D’Andrea MR, Nagele RG. Morphologically distinct types of amyloid plaques point the way to a better understanding of Alzheimer’s disease pathogenesis. Biotechnic and Histochemistry. 2010;85(2):133–147. doi: 10.3109/10520290903389445. [DOI] [PubMed] [Google Scholar]
- 15.Kokubo H, et al. Amyloid Beta annular protofibrils in cell processes and synapses accumulate with aging and Alzheimer-associated genetic modification. International Journal of Alzheimer's Disease. 2009;2009:7 pages. doi: 10.4061/2009/689285. Article ID 689285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu JW, Breydo L, Isas JM, et al. Fibrillar oligomers nucleate the oligomerization of monomeric amyloid β but do not seed fibril formation. Journal of Biological Chemistry. 2010;285(9):6071–6079. doi: 10.1074/jbc.M109.069542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miñano-Molina AJ, España J, Martín E, et al. Soluble oligomers of amyloid-β peptide disrupt membrane trafficking of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor contributing to early synapse dysfunction. Journal of Biological Chemistry. 2011;286(31):27311–27321. doi: 10.1074/jbc.M111.227504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lue LF, Kuo YM, Roher AE, et al. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. American Journal of Pathology. 1999;155(3):853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jucker M, Walker LC. Pathogenic protein seeding in alzheimer disease and other neurodegenerative disorders. Annals of Neurology. 2011;70(4):532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tosun D, Schuff N, Mathis CA, Jagust W, Weiner MW. Spatial patterns of brain amyloid-β burden and atrophy rate associations in mild cognitive impairment. Brain. 2011;134(4):1077–1088. doi: 10.1093/brain/awr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darocha-Souto B, Scotton TC, Coma M, et al. Brain oligomeric β-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. Journal of Neuropathology and Experimental Neurology. 2011;70(5):360–376. doi: 10.1097/NEN.0b013e318217a118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3):631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 23.Castellani RJ, Zhu X, Lee HG, Moreira PI, Perry G, Smith MA. Neuropathology and treatment of Alzheimer disease: did we lose the forest for the trees? Expert Review of Neurotherapeutics. 2007;7(5):473–485. doi: 10.1586/14737175.7.5.473. [DOI] [PubMed] [Google Scholar]
- 24.Mondragón-Rodríguez S, García-Sierra F, Smith MA, et al. Causes versus effects: the increasing complexities of Alzheimer’s disease pathogenesis. Expert Review of Neurotherapeutics. 2010;10(5):683–691. doi: 10.1586/ern.10.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee HG, Zhu X, Castellani RJ, Nunomura A, Perry G, Smith MA. Amyloid-β in Alzheimer disease: the null versus the alternate hypotheses. Journal of Pharmacology and Experimental Therapeutics. 2007;321(3):823–829. doi: 10.1124/jpet.106.114009. [DOI] [PubMed] [Google Scholar]
- 26.Nunomura A, Tamaoki T, Tanaka K, et al. Intraneuronal amyloid β accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiology of Disease. 2010;37(3):731–737. doi: 10.1016/j.nbd.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castellani RJ, Lee HG, Siedlak SL, et al. Reexamining Alzheimer’s disease: evidence for a protective role for amyloid-β protein precursor and amyloid-β . Journal of Alzheimer’s Disease. 2009;18(2):447–452. doi: 10.3233/JAD-2009-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA. Involvement of oxidative stress in Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 2006;65(7):631–641. doi: 10.1097/01.jnen.0000228136.58062.bf. [DOI] [PubMed] [Google Scholar]
- 29.Obrenovich ME, Joseph JA, Atwood CS, Perry G, Smith MA. Amyloid-beta: a (life) preserver for the brain. Neurobiology of Aging. 2002;23(6):1097–1099. doi: 10.1016/s0197-4580(02)00038-6. [DOI] [PubMed] [Google Scholar]
- 30.Mondragón-Rodríguez S, García-Sierra F, Mena R, Perry G, Zhu X, Smith MA. Oxidative stress and Alzheimer disease: mechanisms and therapeutic opportunities. In: Blass J, editor. Handbook of Neurochemistry and Molecular Neurobiology. Vol. 26. New York, NY, USA: Kluwer Academic Publisher; 2011. [Google Scholar]
- 31.Weller RO. Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimer's Research and Therapy. 2009;1(3):p. 6. doi: 10.1186/alzrt6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-β load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. The Lancet Neurology. 2010;9(4):363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 33.Wang A, Das P, Switzer RC, III, Golde TE, Jankowsky JL. Robust amyloid clearance in a mouse model of Alzheimer's disease provides novel insights into the mechanism of amyloid-β immunotherapy. Journal of Neuroscience. 2011;31(11):4124–4136. doi: 10.1523/JNEUROSCI.5077-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tabira T. Immunization therapy for Alzheimer disease: a comprehensive review of active immunization strategies. Tohoku Journal of Experimental Medicine. 2010;220(2):95–106. doi: 10.1620/tjem.220.95. [DOI] [PubMed] [Google Scholar]
- 35.Kuzuhara S. Treatment strategy of Alzheimer disease: pause of clinical trials of Aβ vaccine and next steps. Brain and Nerve. 2010;62(7):659–666. [PubMed] [Google Scholar]
- 36.Gravitz L. Drugs: a tangled web of targets. Nature. 2011;475(7355):S9–S11. doi: 10.1038/475S9a. [DOI] [PubMed] [Google Scholar]
- 37.Josien H. Recent advances in the development of γ-secretase inhibitors. Current Opinion in Drug Discovery and Development. 2002;5(4):513–525. [PubMed] [Google Scholar]
- 38.Louvi A, Sisodia SS, Grove EA. Presenilin 1 in migration and morphogenesis in the central nervous system. Development. 2004;131(13):3093–3105. doi: 10.1242/dev.01191. [DOI] [PubMed] [Google Scholar]
- 39.Johnson GVW, Hartigan JA. Tau protein in normal and Alzheimer’s disease brain: an update. Journal of Alzheimer’s Disease. 1999;1(4-5):329–351. doi: 10.3233/jad-1999-14-512. [DOI] [PubMed] [Google Scholar]
- 40.Farias G, et al. Mechanisms of Tau selfaggregation and neurotoxicity. Current Alzheimer Research. 2011;8(6):608–614. doi: 10.2174/156720511796717258. [DOI] [PubMed] [Google Scholar]
- 41.Johnson GVW, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. Journal of Cell Science. 2004;117(24):5721–5729. doi: 10.1242/jcs.01558. [DOI] [PubMed] [Google Scholar]
- 42.Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends in Molecular Medicine. 2009;15(3):112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Liu F, Li B, Tung EJ, Grundke-Iqbal I, Iqbal K, Gong CX. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. European Journal of Neuroscience. 2007;26(12):3429–3436. doi: 10.1111/j.1460-9568.2007.05955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischer D, Mukrasch MD, Biernat J, et al. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry. 2009;48(42):10047–10055. doi: 10.1021/bi901090m. [DOI] [PubMed] [Google Scholar]
- 45.Rankin CA, Sun Q, Gamblin TC. Pseudo-phosphorylation of tau at Ser202 and Thr205 affects tau filament formation. Molecular Brain Research. 2005;138(1):84–93. doi: 10.1016/j.molbrainres.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 46.Grundke-Iqbal I, Iqbal K, Tung YC. Abnormal phosphorylation of the microtubule-associated protein τ (tau) in Alzheimer cytoskeletal pathology. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(13):44913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alonso AD, et al. Interaction of tau isoforms with Alzheimer’s disease abnormally hyperphosphorylated tau and in vitro phosphorylation into the disease-like protein. Journal of Biological Chemistry. 2001;276(41):37967–37973. doi: 10.1074/jbc.M105365200. [DOI] [PubMed] [Google Scholar]
- 48.Qian W, et al. Activation of protein phosphatase 2B and hyperphosphorylation of Tau in Alzheimer's disease. Journal of Alzheimer's Disease. 2010;23(4):617–627. doi: 10.3233/JAD-2010-100987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Forlenza OV, Torres CA, Talib LL, et al. Increased platelet GSK3B activity in patients with mild cognitive impairment and Alzheimer’s disease. Journal of Psychiatric Research. 2011;45(2):220–224. doi: 10.1016/j.jpsychires.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 50.Hanger DP, Noble W. Functional implications of glycogen synthase kinase-3-mediated tau phosphorylation. International Journal of Alzheimer's Disease. 2011;2011:11 pages. doi: 10.4061/2011/352805. Article ID 352805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dolan PJ, Johnson GVW. The role of tau kinases in Alzheimer’s disease. Current Opinion in Drug Discovery and Development. 2010;13(5):595–603. [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson GVW, Jenkins SM. Tau protein in normal and Alzheimer’s disease brain. Journal of Alzheimer’s Disease. 1999;1(4-5):307–328. doi: 10.3233/jad-1999-14-511. [DOI] [PubMed] [Google Scholar]
- 53.Schaffer BAJ, Bertram L, Miller BL, et al. Association of GSK3B with Alzheimer disease and frontotemporal dementia. Archives of Neurology. 2008;65(10):1368–1374. doi: 10.1001/archneur.65.10.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. European Journal of Neuroscience. 2005;22(8):1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- 55.Rudrabhatla P, Pant HC. Role of protein phosphatase 2A in Alzheimer's disease. Current Alzheimer Research. 2011;8(6):623–632. doi: 10.2174/156720511796717168. [DOI] [PubMed] [Google Scholar]
- 56.Gong CX, Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Current Medicinal Chemistry. 2008;15(23):2321–2328. doi: 10.2174/092986708785909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van Eersel J, Ke YD, Liu X, et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(31):13888–13893. doi: 10.1073/pnas.1009038107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gong CX, Shaikh S, Grundke-Iqbal I, Iqbal K. Inhibition of protein phosphatase-2B (calcineurin) activity towards Alzheimer abnormally phosphorylated τ by neuroleptics. Brain Research. 1996;741(1-2):95–102. doi: 10.1016/s0006-8993(96)00904-3. [DOI] [PubMed] [Google Scholar]
- 59.Pickhardt M, Gazova Z, Von Bergen M, et al. Anthraquinones inhibit tau aggregation and dissolve Alzheimer’s paired helical filaments in vitro and in cells. Journal of Biological Chemistry. 2005;280(5):3628–3635. doi: 10.1074/jbc.M410984200. [DOI] [PubMed] [Google Scholar]
- 60.Cornejo A, Jiménez JM, Caballero L, Melo F, MacCioni RB. Fulvic acid inhibits aggregation and promotes disassembly of tau fibrils associated with alzheimer's disease. Journal of Alzheimer's Disease. 2011;27(1):143–153. doi: 10.3233/JAD-2011-110623. [DOI] [PubMed] [Google Scholar]
- 61.Howland JG, Wang YT. Chapter 8 Synaptic plasticity in learning and memory: stress effects in the hippocampus. Progress in Brain Research. 2008;169:145–158. doi: 10.1016/S0079-6123(07)00008-8. [DOI] [PubMed] [Google Scholar]
- 62.Ho VM, Lee JA, Martin KC, et al. The cell biology of synaptic plasticity. Science. 2011;334(6056):623–628. doi: 10.1126/science.1209236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nature Reviews Neuroscience. 2010;11(7):459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- 64.Appleby VJ, Corrêa SAL, Duckworth JK, et al. LTP in hippocampal neurons is associated with a CaMKII-mediated increase in GluA1 surface expression. Journal of Neurochemistry. 2011;116(4):530–543. doi: 10.1111/j.1471-4159.2010.07133.x. [DOI] [PubMed] [Google Scholar]
- 65.VanGuilder HD, Farley JA, Yan H, et al. Hippocampal dysregulation of synaptic plasticity-associated proteins with age-related cognitive decline. Neurobiology of Disease. 2011;43(1):201–212. doi: 10.1016/j.nbd.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nature Neuroscience. 2010;13(7):812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 68.Pham E, Crews L, Ubhi K, et al. Progressive accumulation of amyloid-β oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS Journal. 2010;277(14):3051–3067. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoe HS, Fu Z, Makarova A, et al. The effects of amyloid precursor protein on postsynaptic composition and activity. Journal of Biological Chemistry. 2009;284(13):8495–8506. doi: 10.1074/jbc.M900141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang H, Gong B, Liu S, et al. Synaptic fatigue is more pronounced in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Current Alzheimer Research. 2005;2(2):137–140. doi: 10.2174/1567205053585936. [DOI] [PubMed] [Google Scholar]
- 71.Witton J, Brown JT, Jones MW, Randall AD. Altered synaptic plasticity in the mossy fibre pathway of transgenic mice expressing mutant amyloid precursor protein. Molecular Brain. 2010;3(1, article 32) doi: 10.1186/1756-6606-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang X, Su B, Lee HG, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. Journal of Neuroscience. 2009;29(28):9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nature Neuroscience. 2010;13(2):190–196. doi: 10.1038/nn.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(14):5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 76.Hsieh H, Boehm J, Sato C, et al. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron. 2006;52(5):831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cheng L, Yin WJ, Zhang JF, Qi JS. Amyloid β-protein fragments 25-35 and 31-35 potentiate long-term depression in hippocampal CA1 region of rats in vivo. Synapse. 2009;63(3):206–214. doi: 10.1002/syn.20599. [DOI] [PubMed] [Google Scholar]
- 78.Du H, Guo L, Yan SS. Synaptic mitochondrial pathology in Alzheimer's disease. Antioxidants and Redox Signaling. doi: 10.1089/ars.2011.4277. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Götz J, Chen F, Van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science. 2001;293(5534):1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 80.Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S. Amyloid β peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience. 2002;115(1):201–211. doi: 10.1016/s0306-4522(02)00404-9. [DOI] [PubMed] [Google Scholar]
- 81.Zempel H, Thies E, Mandelkow E, Mandelkow EM. Aβ oligomers cause localized Ca2+ elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. Journal of Neuroscience. 2010;30(36):11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shipton OA, Leitz JR, Dworzak J, et al. Tau protein is required for amyloid β-induced impairment of hippocampal long-term potentiation. Journal of Neuroscience. 2011;31(5):1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. Journal of Neuroscience. 2010;30(49):16559–16566. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ittner LM, Ke YD, Delerue F, et al. Dendritic function of tau mediates amyloid-β toxicity in alzheimer’s disease mouse models. Cell. 2010;142(3):387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 85.Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 86.Olivares D, et al. N-Methyl D-Aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer's disease, vascular dementia and Parkinson's disease. doi: 10.2174/156720512801322564. Current Alzheimer Research. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Winblad B, Gauthier S, Åström D, Stender K. Memantine benefits functional abilities in moderate to severe Alzheimer’s disease. Journal of Nutrition, Health and Aging. 2010;14(9):770–774. doi: 10.1007/s12603-010-0122-x. [DOI] [PubMed] [Google Scholar]
- 88.Weiner MW, Sadowsky C, Saxton J, et al. Magnetic resonance imaging and neuropsychological results from a trial of memantine in Alzheimer's disease. Alzheimer's and Dementia. 2011;7(4):425–435. doi: 10.1016/j.jalz.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 89.Abe T, Matsumura S, Katano T, et al. Fyn kinase-mediated phosphorylation of NMDA receptor NR2B subunit at Tyr1472 is essential for maintenance of neuropathic pain. European Journal of Neuroscience. 2005;22(6):1445–1454. doi: 10.1111/j.1460-9568.2005.04340.x. [DOI] [PubMed] [Google Scholar]
- 90.Roberson ED, Halabisky B, Yoo JW, et al. Amyloid-β/fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of alzheimer's disease. Journal of Neuroscience. 2011;31(2):700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peineau S, Taghibiglou C, Bradley C, et al. LTP inhibits LTD in the hippocampus via regulation of GSK3β . Neuron. 2007;53(5):703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]