

Abstract
The post-translational modification of proteins is critical for the spatial and temporal regulation of signalling cascades. This is especially important in the CNS where the processes affecting differentiation, growth, targeting and communication between neurones are highly complex and very tightly regulated. In recent years it has emerged that modification of proteins by members of the SUMO (small ubiquitin-related modifier) family of proteins play key roles in neuronal function. SUMOylation involves the covalent conjugation of a member of the SUMO family to lysine residues in target proteins. Multiple nuclear and perinuclear SUMOylation targets have been reported to be involved in nuclear organisation and transcriptional regulation. In addition, a growing number of extranuclear SUMO substrates have been identified that can have important acute effects on neuronal function. The SUMOylation of both intra- and extranuclear proteins have been implicated in a diverse array of processes that have far-reaching implications for neuronal function and pathophysiology. Here we review the current understanding of the targets and consequences of protein SUMOylation in the brain and examine its established and potential involvement in a wide range of neurological and neurodegenerative diseases.
Keywords: SUMO, Ubc9, Ubiquitin, SENP, Post-translational modification, Neuronal function
1. Introduction
Post-translational modification is the enzymatic attachment of chemical groups, lipids, sugars, or polypeptides to a protein after its primary synthesis. These modifications are of critical importance because they can dictate protein folding, distribution, stability, activity and function. Post-translational modifications are also integral components of the signalling cascades that enable cells to efficiently and reversibly respond to extracellular stimuli. This is especially important in the CNS where the processes affecting synaptic communication between neurones are highly complex and very tightly regulated.
1.1. What is SUMOylation?
SUMO (small ubiquitin-related modifier) is a 97-amino acid protein that can be covalently attached to lysine residues on target proteins via an enzymatic cascade analogous to the ubiquitin pathway. SUMOylation of the target protein requires an E1 (SUMO-activating), E2 (SUMO-specific conjugating, Ubc9), and, in most cases, E3 (SUMO ligase, e.g., PIAS3) enzymes (for detailed reviews of the machinery and biochemistry of SUMO modification, see Geiss-Friedlander and Melchior, 2007; Hay, 2005; Hilgarth et al., 2004; Johnson, 2004; Muller et al., 2001; Watts, 2004). There are four reported SUMO paralogues, SUMO-1 to SUMO-4 (Bohren et al., 2004; Lapenta et al., 1997), present in mammals. In their conjugatable forms, SUMO-2 and SUMO-3 differ only in three N-terminal residues. All SUMO paralogues are conjugated to substrate proteins by the same enzymatic machinery (Tatham et al., 2001) but they can modify distinct targets (Saitoh and Hinchey, 2000), although some proteins can be SUMOylated by either SUMO-1 or SUMO-2/3 (e.g., Hardeland et al., 2002; Hofmann et al., 2000). SUMO-4 remains enigmatic since it has a restricted tissue distribution and may be unable to SUMOylate substrate proteins due to a proline residue that appears to prevent its maturation to an prevent its maturation to a conjugatable form (Owerbach et al., 2005).
Following SUMO activation by the E1 enzyme, a heterodimer of SAE1 and SAE2 in mammals (Gong et al., 1999), SUMO proteins are passed to the active site cysteine of the sole SUMO-specific conjugating enzyme, Ubc9. Ubc9, either alone or in combination with a number of reported SUMO E3 enzymes, then catalyses conjugation of SUMO to the lysine residue in the target protein (Fig. 1). SUMO conjugation often occurs at a consensus sequence in target proteins, designated ψKxD/E, where ψ is a large hydrophobic residue, K is the target lysine and D/E are acidic residues (Rodriguez et al., 2001; Sampson et al., 2001). The consensus sequence is directly bound by Ubc9 (Sampson et al., 2001), however it must be noted that not all proteins containing this consensus are SUMOylated, and a growing number of proteins are being reported to be modified at non-consensus lysine residues.
Fig. 1.
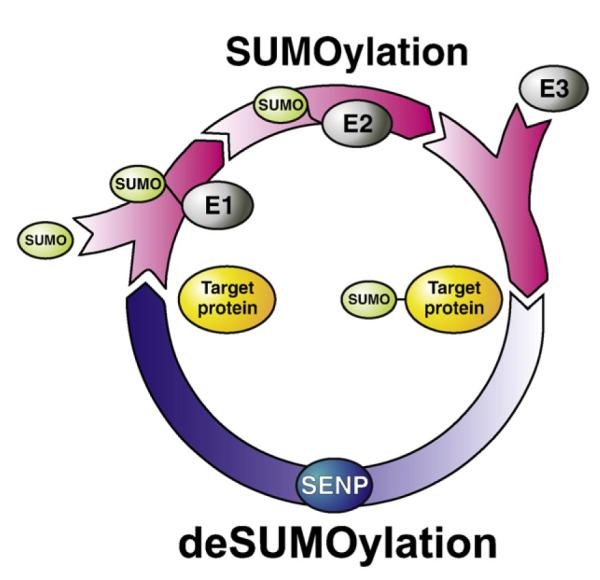
SUMO conjugation. SUMO proteins are produced as immature pre-proteins that are first cleaved C-terminally by the SENP proteins to allow their conjugation to substrate proteins (not shown). SUMO proteins are then ‘activated’ for conjugation by formation of a thioester bond between the C-terminus of SUMO and the active site cysteine of the E1 complex in an ATP-dependent process. SUMO is then passed to the active site cysteine of the sole reported SUMO-specific E2 enzyme, Ubc9. Ubc9, either alone or in conjunction with one of a number of reported E3 enzymes, then conjugates SUMO to the lysine residue in the substrate protein, altering the properties of the substrate. SUMOylation can subsequently be reversed, again by the activity of the SENP enzyme.
Despite being a covalent protein modification, SUMOylation is readily reversible due to the protease activity of SUMO-specific deSUMOylation enzymes, SENPs. These enzymes exhibit specificity between the SUMO paralogues and have distinct subnuclear and subcellular localisation patterns. There are six SENPs, designated SENP1-3 and SENP5-7 in mammals (for reviews, see Mukhopadhyay and Dasso, 2007; Yeh, 2009). SENP proteins themselves are coming under increasing scrutiny as cell regulators (Kim and Baek, 2009).
1.2. What does SUMOylation do?
SUMOylation is essential in eukaryotic cells and knockdown or deletion of Ubc9, a critical component of the SUMOylation pathway, is fatal for mammalian cells (Hayashi et al., 2002; Nacerddine et al., 2005). The balance between Ubc9-mediated conjugation and SENP-mediated deconjugation determines the SUMOylation state of a specific protein and the mechanisms underlying regulation of these processes is an area of very active research.
A major function of SUMOylation is to inhibit, modify or enable protein–protein interactions (Geiss-Friedlander and Melchior, 2007). Thus, the attachment of SUMO can prevent binding of interacting proteins or modify interactions by altering binding kinetics. In addition, a growing number of proteins have been reported to bind SUMO non-covalently, via SUMO interaction motifs (SIMs), suggesting that SUMOylation of a target protein can act to promote interactions with proteins that contain SIMs (Minty et al., 2000; Song et al., 2004). Numerous neuronal and synaptic proteins appear to contain SIMs and how these potential SUMO-interacting proteins mediate the consequences of protein SUMOylation at the synapse promises to be an exciting avenue for future research.
The functional consequences of SUMO attachment vary greatly depending on the substrate and the cell type and in many cases remain only poorly understood (Meulmeester and Melchior, 2008). The majority of SUMOylated proteins that have been identified so far are either localised to the nucleus or involved in the nuclear trafficking of cytosolic proteins (for reviews see Geiss-Friedlander and Melchior, 2007; Hay, 2005; Johnson, 2004; Muller et al., 2001; Seeler and Dejean, 2003). Since the first identification of SUMOylation as a modification of the nuclear pore component RanGAP1 (Mahajan et al., 1997; Matunis et al., 1996), several hundred confirmed and putative SUMOylation substrates have been identified, including numerous proteins abundant in or exclusive to neurons, many of which are extranuclear (Hietakangas et al., 2006; Yang et al., 2006).
2. SUMO roles in the transcriptional regulation of neuronal development and function
It is well-established that protein SUMOylation and deSUMOylation perform critical regulatory roles in the nucleus. Protein substrates include many transcription factors and co-factors, nuclear pore proteins, nuclear structural proteins and proteins that maintain genome integrity. Some specific examples particularly important in neurons are discussed below but for more comprehensive reviews see (Heun, 2007; Johnson, 2004; Palancade and Doye, 2008; Seeler and Dejean, 2003).
2.1. Developmental profile and localisation of Ubc9 and potential roles in neuronal differentiation and plasticity
Ubc9 mRNA and protein expression are spatio-temporally regulated during rat brain development (Watanabe et al., 2008). The highest levels of Ubc9 expression and SUMO-1 conjugation occur early in development. In particular, there is high expression of Ubc9 in the proliferating neuronal stem cells in the ventricular zone and in differentiated but immature embryonic neurons, consistent with SUMOylation participating in neuronal differentiation and proliferation. In the adult brain, Ubc9 is most abundant in dentate granular neurons and pyramidal neurons in the hippocampus and large pyramidal neurons in the cerebral cortex, suggesting possible roles for Ubc9 and SUMOylation in synaptic and neuronal plasticity. Although not directly determined, it has been inferred that key targets for SUMOylation in stem cells and immature embryonic neurons were likely to be transcription factors. Likely candidates include topoisomerases I and II (Lee and Bachant, 2009; Mao et al., 2000a,b), which are enzymes essential for DNA replication and transcriptional regulation that are highly expressed in neuronal stem cells (Li and Wang, 1998), the tumour suppressor protein p53, a known SUMO substrate which plays a critical role in the differentiation and apoptosis of neurons and oligodendrocytes (Eizenberg et al., 1996; Tedeschi et al., 2009) (for reviews see Melchior and Hengst, 2002; Stehmeier and Muller, 2009) and, as discussed below, members of the MEF2 family of transcription factors, which are SUMO substrates implicated in the regulation of neuronal differentiation. Considerable work remains to be done to identify which nuclear proteins are targets for SUMOylation during neuronal differentiation and development. Nonetheless, the observation that neuronal stem cells show high levels of the essential SUMOylation enzyme Ubc9 indicates that protein SUMOylation is required for embryonic development (Nacerddine et al., 2005) and the fact that proteins that regulate transcription are SUMO substrates suggest that SUMOylation is a key regulator of these processes.
2.2. SUMO regulation of nuclear Cajal bodies
Another example of specialised nuclear SUMOylation in neurons is the localisation of SUMO-1 and Ubc9 in Cajal bodies (CBs), which are nuclear organelles involved in the maturation of small nuclear ribonucleoproteins required for the processing of pre-mRNAs. CBs are prominent in mammalian neurons and their abundance correlates with cell size and transcriptional activity (Berciano et al., 2007). CBs are highly enriched in the proteins coilin and survival of motor neuron (SMN) protein (Carvalho et al., 1999), which are both putative substrates of SUMO-1. SUMO-1 and Ubc9 colocalise in a subset of CBs in undifferentiated neuron-like UR61 cells, but not in cells that have been treated with dexamethasone, an inducer of UR61 differentiation. Further, SUMO-1 associates with CBs in rat trigeminal ganglion neurons subjected to osmotic stress, but not in unstressed cells. The SUMO-1-positive CBs contain coilin and small nuclear ribonucleoproteins, suggesting that they are actively involved in pre-mRNA processing and it has been proposed that SUMOylation of coilin and/or SMN might play a role in the molecular reassembly of CBs during the neuronal stress–response (Navascues et al., 2008). While the exact role of SUMO-1 recruitment to CBs during the early stages of differentiation or during neuronal stress is unclear, these results may suggest a role for SUMOylation in the transcriptional reprogramming and nuclear reorganisation associated with neuronal differentiation and response to stress.
2.3. SUMOylation and rod photoreceptor development
While the expression pattern of Ubc9 and the regulated localisation of SUMO at CBs hints at an important role for SUMOylation in neuronal development and differentiation, a specific example has emerged recently in the case of retinal rod photoreceptor neurons. For rod photoreceptors to differentiate correctly, rod-specific genes must be activated while cone-specific genes must be repressed. Central to this process is the transcription factor NR2E3, which is SUMOylated in a PIAS3-dependent manner (Onishi et al., 2009). PIAS3 is highly expressed in the mouse retina during the period of rod photoreceptor development (Blackshaw et al., 2004) and overexpression of PIAS3 during retinal development increases the number of cells with a rod-like morphology that express the rod-specific photopigment rhodopsin (Onishi et al., 2009). PIAS3 directly mediates the SUMOylation of NR2E3. Mice lacking NR2E3 show an abnormal retinal phenotype characterised by excessive numbers of cone-like cells that could be rescued by wild-type NR2E3, but not with a SUMOylation-deficient mutant. However, retinal expression of rhodopsin was normal in cells rescued with non-SUMOylatable NR2E3, suggesting NR2E3 SUMOylation does not play a role in the induction of rod-specific genes. Nonetheless, general inhibition of SUMOylation in the retina via overexpression of the SUMOylation inhibitor Gam1 lead to a phenotype of excessive cone production, and deficient production of rhodopsin, suggesting that SUMOylation of some unknown protein other than NR2E3 functions in the promotion of rod-specific gene expression (Onishi et al., 2009). Thus, while SUMOylation of NR2E3 appears to be important for the repression of cone-specific genes, SUMOylation per se is required for the activation of rod-specific genes. These results indicate that SUMOylation can play complex positive and negative regulatory roles in the differentiation of neuronal subtypes.
2.4. SUMO regulation of synapse formation via MEF2A
In neurons, synapse formation involves contact between pre- and post-synaptic membranes. In the cerebellum, dendrites from granule neurons develop dendritic claws that contact mossy fibre terminals to form synapses. The family of myocyte enhancer factor 2 (MEF2) transcription factors are critical regulators of dendritic claw formation. MEF2 family members suppress synapse number in an activity- and calcineurin-dependent manner. Neuronal activation leads to calcineurin activation and dephosphorylation of MEF2. Dephosphorylated (active) MEF2 then leads to the transcription of various genes that restrict synapse number, such as ARC and synGAP (Flavell et al., 2006).
SUMOylation of one of the MEF2 family members, MEF2A, strongly influences synapse formation through a phospho-regulated SUMO-acetyl switch (Shalizi et al., 2006). Early in differentiation, SUMOylation at lysine 403 represses MEF2A. Neuronal activity leads to the activation of calcineurin and dephosphorylation of the nearby serine S408. This favours deSUMOylation of K403 and the replacement of SUMO with an acetyl group, leading to MEF2A activation and the inhibition of synapse formation. This phosphorylation-dependent switch between an inhibitory modification (SUMOylation) and an activating modification (acetylation) on MEF2a provides an elegant illustration of the interplay between multiple post-translational modifications tightly regulating a complex physiological process. Further support for a role of MEF2A SUMOylation in synapse development comes from the observation that PIASx functions as an E3 for MEF2A in vivo. Overexpression or knockdown of PIASx was shown to significantly enhance or inhibit dendritic claw formation, respectively, and this effect was dependent on the presence of MEF2A, consistent with its role in the suppression of MEF2A (Shalizi et al., 2007).
2.5. SUMO regulation of circadian rhythms via BMAL1
Melanopsin-expressing retinal ganglion cells perceive light for circadian regulation (Hankins et al., 2008). These neurons project to an area of the brain known as the suprachiasmatic nucleus (SCN), which contains the ‘master oscillator’ of the circadian clock, and entrains the circadian behaviour of other tissues (Reppert and Weaver, 2002). One critical regulator of the circadian clock is the transcription factor component BMAL1 which, together with its heterodimerisation partner CLOCK, provides positive feedback for the circadian oscillator. SUMOylation regulates the timing of the circadian cycle through mediating the stability of BMAL1 (Cardone et al., 2005; Lee et al., 2008). BMAL1 can be SUMOylated by both SUMO-1 and SUMO-2 at a single consensus lysine residue, K259, in a manner that requires CLOCK. SUMOylation of BMAL1 occurs periodically, peaking 9 h into the circadian cycle. In addition, while wild-type BMAL1 shows rhythmic oscillations in protein levels, the non-SUMOylatable mutant does not, and shows significantly increased protein stability in comparison to the wild-type, suggesting that SUMOylation regulates BMAL1 turnover (Cardone et al., 2005; Lee et al., 2008).
While it is evident that SUMO regulation of BMAL1 stability is critical to the mammalian circadian cycle, exactly how this regulation occurs is currently unclear. It has been reported that BMAL1 SUMOylation, predominantly by chains of SUMO-2/3, is a prerequisite for BMAL1 ubiquitination and subsequent degradation by the proteasome (Lee et al., 2008). Indeed, SUMO-2-mediated control of ubiquitination is emerging as a recurring theme in protein degradation (for review see Geoffroy and Hay, 2009). However, decreased promoter occupancy of the non-SUMOylatable BMAL1 mutant was also observed, suggesting that a SUMO-mediated decrease in BMAL1 transcriptional activity may cause the increase in BMAL1 stability through decreased production of the BMAL1-destabilising proteins directly under the control of BMAL1. Interestingly, BMAL1 stability is also regulated by GSK3β-mediated phosphorylation (Sahar et al. 2010). GSK3β is itself a SUMO substrate (Eun Jeoung et al., 2008), indicating that SUMOylation may play a yet more complex role in the regulation of the circadian system. Thus, while further work is required, it is clear that SUMOylation plays a central role in governing crucial transcriptional and protein interaction pathways involved in circadian regulation.
3. SUMO modulation of kinase signalling pathways
Phosphorylation dynamics play critical roles in multiple cellular signalling pathways and a number of kinases, phosphatases and scaffolding proteins involved in the activation of these pathways have been reported to be targets of SUMO conjugation.
3.1. Focal adhesion kinase (FAK)
FAK regulates the maturation and turnover of focal adhesions (Mitra et al., 2005), contacts between the extracellular matrix and the cytoskeleton. FAK is a SUMO substrate and its SUMOylation is associated with increased levels of autophosphorylation (Kadare et al., 2003) that leads to binding of Src, which, in turn, further phosphorylates FAK resulting in full activation (Mitra et al., 2005). Further, the observation that SUMOylated FAK is enriched in the nuclear fraction suggests that SUMOylation may also regulate FAK nuclear shuttling in addition to enhancing its activation.
3.2. Glycogen synthase kinase 3β (GSK3β)
GSK3β is a proline-directed serine/threonine kinase that was initially identified for its role in phosphorylating and inactivating glycogen synthase. GSK3β has been well-characterised for its role in energy metabolism, however, it also plays important roles in the brain and has been implicated in the pathogenesis of Alzheimer’s disease, schizophrenia and bipolar disorders (Bhat et al., 2004; Eldar-Finkelman, 2002; Kozlovsky et al., 2002). In addition, GSK3β has been reported to play a crucial role in dictating the balance between LTP and LTD in hippocampal slice preparations (Peineau et al., 2007). GSK3β is SUMOylated at a single non-consensus lysine in the central region required for its interaction with the scaffolding protein axin, itself a SUMO substrate (Eun Jeoung et al., 2008; Rui et al., 2002). Wild-type GSK3β shows both a nuclear and cytosolic distribution but the non-SUMOylatable point mutant is excluded from the nucleus, indicating that SUMOylation may play a role in the subcellular localisation of GSK3β. In addition, the non-SUMOylatable mutant displays decreased protein stability and enhanced phosphorylation activity against tau, a prototypical GSK3β substrate (Eun Jeoung et al., 2008). Based on these initial observations, future work may reveal an important role for GSK3β SUMOylation in synaptic plasticity and neurodegenerative diseases such as Alzheimer’s.
3.3. Protein tyrosine phosphatase 1B (PTP1B)
PTP1B is an endoplasmic reticulum-associated protein phosphatase that down-regulates multiple signalling pathways via dephosphorylation of receptor tyrosine kinases (Dube and Tremblay, 2005). PTP1B is highly expressed in the brain and is a critical negative regulator of leptin signalling (Bence et al., 2006). Leptin acts on the hypothalamus to decrease feeding behaviour and promote energy expenditure (Ahima and Flier, 2000). PTP1B SUMOylation occurs in fibroblasts in response to insulin signalling and down-regulates its catalytic activity and expression levels (Dadke et al., 2007). SUMOylation of PTP1B in the brain has not yet been shown but an intriguing possibility is that it may play a role in the hypothalamic regulation of feeding behaviour and body mass.
3.4. Axin
Axin acts to cluster proteins required for the activation of both c-Jun N-terminal kinase (JNK) and Wnt signalling in neurons. JNK is activated by various surface receptors and signalling pathways while Wnt activation plays a major role in axon guidance. Axin is SUMOylated at two atypical SUMO consensus lysines at its extreme C-terminus (Rui et al., 2002). A SUMOylation-deficient form of axin is unable to interact with MEKK, a critical enzyme in the activation of JNK, resulting in an incapacity to activate the JNK pathway whereas its roles in the activation of the Wnt pathway are unaffected (Rui et al., 2002) suggesting a selective effect of SUMOylation on JNK signalling.
From these examples it seems clear that SUMOylation can directly influence phosphorylation cascades, modifying both the enzymes directly involved in these pathways, as well as selectively affecting pathways via actions on multifunctional scaffolding proteins.
4. SUMOylation at mitochondria
In eukaryotic cells, mitochondrial number is regulated by a balance between mitochondrial fusion and fission events and appropriate regulation of these dynamics are essential to meet the energy needs of the cell (Chan, 2006; McBride et al., 2006). For example, in both cultured neurons and hippocampal slices, neuronal activity is linked to an increase in mitochondrial number and inhibiting mitochondrial fission severely reduces the number of dendritic protrusions, highlighting a role for mitochondrial dynamics in the maintenance of synapse number (Li et al., 2004). Immunoblots of mitochondrial fractions from COS-7 cells probed with anti-SUMO antibodies have suggested the presence of multiple mitochondrial SUMO substrates yet to be defined (Harder et al., 2004). Further, the mitochondria-enriched fraction of brain lysate shows a large number of SUMO-1 and SUMO-2/3 reactive bands upon Western blotting (Martin et al., 2007a).
4.1. Dynamin-related protein 1 (DRP1)
The mitochondrial GTPase DRP1 is required for the membrane scission step during mitochondrial fission (Smirnova et al., 2001). Knockout mice lacking DRP1 die before birth and exhibit defective forebrain development. Cultured DRP1−/− neurons show defects in synapse formation, highlighting a critical role for proper mitochondrial dynamics in neurons (Ishihara et al., 2009).
DRP1 is SUMOylated and this modification was initially reported to stabilise the protein, ultimately leading to an increase in the active pool of DRP1 present on the surface of mitochondria, where it is presumably able to perform its role in mitochondrial fission (Harder et al., 2004). In addition, increased SUMOylation was associated with DRP1 nucleotide binding. Although a more recent study has confirmed DRP1 SUMOylation and determined the SUMOylation sites, the functional role has been questioned (Figueroa-Romero et al., 2009). These workers found that mutagenesis of the SUMOylatable lysines in DRP1 had no effect on protein stability and that its recruitment to mitochondria under both basal conditions and after treatment with staurosporine, an inducer of apoptosis, was normal (Figueroa-Romero et al., 2009). In contrast to the previous study that reported a dominant-negative DRP1 mutant showed extremely low levels of Ubc9 and SUMO-1 binding (Harder et al., 2004), Figueroa-Romero et al. found that this dominant-negative DRP1 exhibited enhanced SUMOylation (Figueroa-Romero et al., 2009). While these apparent differences remain to be fully resolved it is relevant that the SUMOylation of DRP1 has also been reported to be enhanced by the pro-apoptotic protein Bax (Wasiak et al., 2007), potentially implicating SUMOylation in the induction of apoptosis. Intriguingly, the apoptotic proteases caspase-7 and -8 have also been reported to be targets of SUMOylation (Besnault-Mascard et al., 2005; Hayashi et al., 2006).
4.2. Mitochondrial-associated protein ligase (MAPL)
A novel mitochondrial SUMO E3 ligase, mitochondrial-associated protein ligase (MAPL) has recently been identified (Braschi et al., 2009). MAPL was initially identified as a novel RING-finger domain-containing mitochondrial outer membrane protein involved in mitochondrial fragmentation (Neuspiel et al., 2008). Proteins containing RING domains often function as ubiquitin or SUMO ligases and MAPL was shown to stimulate DRP1 SUMOylation in in vitro. Further, shRNA-mediated silencing of MAPL significantly reduced levels of mitochondrial SUMOylation, without any observed effect on ubiquitin conjugation. It has been suggested that the fragmented phenotype of mitochondria observed upon the overexpression of MAPL may result from the proposed SUMO-mediated stabilisation of DRP1 (Braschi et al., 2009). However, given the mixed reports on the roles of DRP1 SUMOylation, further work is required. In addition, identification of the other mitochondrial proteins that show reduced SUMOylation following MAPL knockdown will shed light on the further roles played by SUMOylation at mitochondria.
4.3. Mitochondrial actions of SENP5
Protein regulation by SUMOylation constitutes a delicate balance between regulated SUMOylation and deSUMOylation of substrate proteins. This is exemplified by the observation that the SUMO protease SENP5 can regulate mitochondrial fission, partially through its ability to deSUMOylate DRP1. These actions appear to be cell cycle-regulated since SENP5 translocates from the nucleoli to mitochondria at the G2/M transition, prior to mitosis (Zunino et al., 2009). For appropriate cell division to occur, mitochondria must proliferate and segregate between the two daughter cells. SENP5 translocation from the nucleus leads to a dramatic reduction in mitochondrial SUMOylation which, in turn, increases the labile pool of DRP1 available for mitochondrial fragmentation to occur, likely through both direct deSUMOylation of DRP1 and indirect effects (Zunino et al., 2009). While the exact role of SENP5 in regulating mitochondrial number in post-mitotic neurons is unclear, the current data are consistent with a nucleus-to-mitochondria signalling pathway regulating the activity-dependent increase in mitochondrial number reported previously (Li et al., 2004).
5. SUMOylation and axonal mRNA trafficking
Local protein synthesis in axons is critical for growth-cone guidance in developing neurons, and for axonal regeneration and synaptic plasticity in adult neurons (Giuditta et al., 2002; Martin, 2004). SUMOylation can directly influence these processes through modification of the mRNA-binding protein La (van Niekerk et al., 2007). La binds multiple axonal mRNAs and facilitates their axonal trafficking from the cell soma while protecting them from exonuclease activity (Wolin and Cedervall, 2002). Fast axonal transport relies on the motor proteins dynein (generally for retrograde transport) and kinesin (for anterograde transport; Hirokawa and Takemura, 2005). La is SUMOylated at a single lysine residue in both cultured mouse dorsal root ganglion (DRG) neurons and isolated mouse sciatic nerve and overexpressed SUMO-1 and SUMO-2/3 can modify La in cultured DRG neurons (van Niekerk et al 2007), although the abundance of SUMO-1 versus SUMO-2/3-modified La under endogenous conditions has not yet been addressed. Non-SUMOylated La binds kinesin but not dynein, conversely when SUMOylated, La binds only to dynein. Indeed, in cultured DRG neurons, overexpressed GFP-La shows both anterograde and retrograde movement while non-SUMOylatable La shows only anterograde movement (van Niekerk et al., 2007). Thus, the SUMOylation status of La dictates the direction of its axonal transport and therefore also enables it to act as’ a shuttle carrying multiple loads of mRNA. Although more work is required to determine the interaction sites between SUMO-modified La and dynein, these results suggest that SUMO-mediated motor-protein switching could serve as a general regulator of bi-directional transport along neuronal processes.
6. SUMOylation in synaptic function and neuronal activity
It is becoming increasingly well-documented that activity-dependent SUMOylation of transcription factors and of synaptic proteins plays important regulatory roles in synapse formation and neuronal morphology (Fig. 2).
Fig. 2.

SUMOylation and neuronal function. SUMOylation has been reported to regulate various aspects of neuronal function and morphology. Shown are a number of these processes, along with identified SUMO substrates involved in them. See text for more details. Abbreviations: MEF2A, myocyte enhancer factor 2A; CASK, calcium/calmodulin-dependent serine kinase; CB1, cannabinoid receptor 1; mGluR, metabotropic glutamate receptor; Drp1, dynamin-related protein 1.
6.1. CASK
CASK is a synaptic scaffold protein that binds various post-synaptic receptors and is required for the formation of dendritic spines onto which more than 90% of excitatory synapses form (Nimchinsky et al., 2002). CASK has recently been identified as a SUMO substrate (Chao et al., 2008). The function of CASK in spine morphogenesis requires its interaction with protein 4.1. Fusion of SUMO to CASK significantly reduces its interaction with protein 4.1 and expression of this fusion protein in neurons causes a decrease in spine size and density (Chao et al., 2008) highlighting SUMOylation as a potential regulator of dendritic spine morphology.
6.2. Presynaptic neurotransmitter release
Recently, SUMOylation of pre-synaptic proteins has been shown to regulate glutamate release from nerve terminals. Enhancing pre-synaptic SUMOylation by inclusion of conjugatable SUMO-1 in synaptosomes led to a decrease in K+-evoked glutamate release whereas inclusion of SENP1 catalytic domain enhanced neurotransmitter release (Feligioni et al., 2009). These data suggest SUMOylation may be playing a negative role in activity-dependent glutamate release. However, this effect may be stimulus-dependent since inclusion of SUMO enhanced glutamate release stimulated by kainate, whereas SENP1 decreased kainate-evoked glutamate release (Feligioni et al., 2009). Thus, SUMOylation can act to either enhance or reduce glutamate release depending on the stimulus, likely through the SUMOylation or deSUMOylation of distinct subsets of proteins leading to different effects on neurotransmitter release.
6.3. Post-synaptic ionotropic glutamate receptors
Kainate receptors (KARs) are tetrameric glutamate-gated ion channels composed of combinations of the subunits GluK1, GluK2, GluK3, GluK4 and GluK5 (formerly known as GluR5, GluR6, GluR7, KA-1 and KA-2, respectively; Pinheiro and Mulle, 2006). Kainate receptors are expressed widely in the nervous system and have various roles in regulating neuronal responses – acting pre-synaptically to modulate neurotransmitter release, post-synaptically to contribute to the post-synaptic response and by regulating neuronal excitability via the inhibition of K+ channels (for reviews see Jaskolski et al., 2005; Pinheiro and Mulle, 2006).
The kainate receptor subunit GluK2 binds both Ubc9 and the E3 enzyme PIAS3 and is SUMOylated at a single C-terminal lysine residue (Martin et al., 2007a). SUMOylation is stimulated by kainate treatment and is required for agonist-induced endocytosis of GluK2-containing kainate receptors but not for NMDA-induced KAR endocytosis. Further, at mossy fibre synapses, infusion of SUMO-1 caused a run down in kainate receptor-mediated excitatory post-synaptic currents (EPSCs) but had no effect on AMPA receptor-mediated EPSCs, indicating that SUMO can play an important and specific role in the regulation of synaptic transmission.
6.4. Activity-dependence of protein SUMOylation
While the SUMOylation of given substrates can be specifically regulated, global levels of SUMOylation in neurons can be modulated in an activity-dependent manner, potentially placing changes in protein SUMOylation central to the coordination of neuronal signalling.
AMPA stimulation of synaptosomes induced an increase in pre-synaptic SUMOylation, and this increase was concomitant with an increased association of Ubc9 with the pre-synaptic active zone, suggesting activity-dependent relocation of Ubc9 may act to regulate synaptic SUMOylation levels. Conversely, kainate treatment was shown to lead to a decrease in pre-synaptic SUMOylation, and SUMOylation levels were unchanged in response to NMDA (Feligioni et al., 2009). These results appear to suggest that levels of SUMOylation at the pre-synaptic active zone can be tightly regulated in an activity-dependent manner that varies depending on the stimulus. In conjunction with the observation that enhancing or inhibiting pre-synaptic SUMOylation can have robust effects on pre-synaptic neurotransmitter release, these data hint at activity-dependent SUMOylation of multiple substrates regulating a complex physiological process.
The observation that levels of SUMOylation can be dramatically altered by physiological neuronal activity has been further supported by data indicating that depolarization of SHSY5Y neuroblastoma cells with KCl enhances global levels of SUMO conjugation (Lu et al., 2009). In SHSY5Y cells stably transfected with either SUMO-1, SUMO-2 or SUMO-3, treatment with KCl for 1 h (but not for shorter time periods) was shown to induce a robust increase in conjugation of each of the SUMO proteins. In addition, chelation of extracellular calcium with EGTA largely blocked this effect, as did the calmodulin inhibitor cyclosporin A, suggesting that calcium influx acting through the calcium sensor calmodulin is at least partially responsible for the global increase in SUMOylation (Lu et al., 2009). How these activity-dependent stimuli and calcium-dependent signalling pathways induce changes in global SUMOylation remain to be determined.
7. Plasma membrane receptors, channels and transporters
The discovery that multiple classes of membrane protein are SUMO substrates represented a significant advance. In addition to glutamate receptors, several other classes of neuronal membrane protein have been reported to be SUMOylated, strongly implicating SUMOylation in the control of neuronal excitability, synaptic transmission and glucose transport.
7.1. Potassium channels
7.1.1. K2P channels
K2P channels are potassium-selective plasma membrane leak channels (Plant et al., 2005). K2P1 mRNA has been shown to be expressed strongly in heart, brain and kidney (Orias et al., 1997); however, this channel had been reported to be inactive, even when expressed at the cell surface (Plant et al., 2005). It has been proposed that this inactivity is due to SUMO conjugation to K274 of the channel, which acts to block the pore (Rajan et al., 2005). Immunoprecipitation experiments indicated that K2P1 could be SUMO-modified in Xenopus oocytes, and overexpression of a lysine point mutant that cannot be SUMOylated generated an active, pH-sensitive potassium channel in COS-7 cells. Indeed, overexpression of wild-type K2P1 along with SENP-1, to enhance the number of deSUMOylated channels, produced more active channels.
However, SUMOylation of native K2P1 channels has not been established and the trigger(s) for SUMOylation or deSUMOylation of the channel remain elusive. Indeed, more recent reports have questioned whether K2P1 channels are bona fide SUMO substrates (Feliciangeli et al., 2007, 2010). These authors were unable to detect SUMOylation of K2P1 channels, and reported that mutation of the proposed SUMOylatable lysine to arginine (instead of a negatively charged glutamic acid, as used in the previous report) had no effect on channel functions. They suggest that the observed effects of the K274E mutation may thus be due to the change in charge rather than the prevention of SUMOylation. Further work will be required to determine whether K2P1 is a genuine SUMO substrate and to isolate the functional consequences of this modification.
7.1.2. Kv1.5 channels
Another potassium channel, the voltage-gated Kv1.5, has also been reported to be a SUMO substrate (Benson et al., 2007). Kv1.5 appears to be modified by all three SUMO paralogues at two cytosolic lysine residues both in vivo and in vitro, and disruption of SUMOylation either by mutation of the acceptor lysines or by overexpression of the deSUMOylating enzyme SENP2 caused a hyperpolarizing shift in the voltage-dependence of steady-state inactivation, indicating SUMOylation has a role in fine-tuning channel function.
These reports of SUMOylation mediated changes in potassium channel function, coupled with the observation that multiple potassium channel subunits present in the brain also contain SUMOylation consensus motifs, suggest that one important role of SUMOylation in the brain may be the regulation of ion fluxes and neuronal excitability.
7.2. Glucose transporters
Glucose enters cells via the actions of a family of specific glucose transporters. The glucose transporters GLUT1 and GLUT4 are highly expressed in the brain and they were the first membrane proteins to be shown to be SUMOylated (Giorgino et al., 2000). Overexpression of Ubc9 caused a 65% decrease in GLUT1 levels, but an 8-fold increase in GLUT4 expression. GLUT1 is responsible for the majority of basal glucose transport whereas GLUT4 is transported to the surface in response to insulin to increase glucose uptake (Bryant et al., 2002). Thus, these results suggest that SUMO decreases the basal transport of glucose, but enhances the insulin-responsiveness of glucose transport into adipocytes. However, data on how SUMOylation differentially affects GLUT1 versus GLUT4 has not been published, nor has any explanation been offered as to how SUMOylation of these two transporters is regulated and under what conditions SUMO modification occurs to alter the insulin-responsiveness of adipocytes, if this occurs at all under physiological conditions. Indeed, more recent evidence suggests that the increase in GLUT4 stability versus GLUT1 can be reproduced with an inactive point mutant of Ubc9, implying this phenomenon may be due to a role of Ubc9 distinct from SUMO conjugation (Liu et al., 2007). Further investigation is required to determine whether these transporters are genuine SUMO substrates. Nonetheless, given the abundant expression of GLUT1 and GLUT4 in neurons, an intriguing possibility is that SUMOylation of these targets could represent an important regulator of glucose transport in the brain.
7.3. Type I TGF-β receptor
Transforming growth factor-β (TGF-β) is a proinflammatory cytokine that controls proliferation and cellular differentiation. The TGF peptide acts on a family of TGF receptor proteins that are highly expressed in the brain by both neurons and microglia, and has been implicated in several neurodegenerative disorders, including Alzheimer’s, Parkinson’s, multiple sclerosis and ischemia (Flanders et al., 1998; Pratt and McPherson, 1997).
Type I TGF-β receptor Type I TGF-β receptor (TβRI) has recently been reported to be a substrate for SUMOylation (Kang et al., 2008). TGF-β induces the heterodimerisation of the receptor serine/threonine kinases TβRI and type II TGF-β receptor (TβRII), which leads to the TβRII-mediated phosphorylation of TβRI and downstream signalling through phosphorylation of Smad proteins. Smad proteins are divided into three families: receptor activated (R-Smads, Smad1, 2, 3, 5, 8); co-mediator Smad (Smad4) and inhibitory Smads (Smad6 and 7). TβRI activation through phosphorylation by TβRII leads to the TβRI-mediated phosphorylation of the R-Smads, Smad2 and Smad3. The active, phosphorylated Smads then homo-trimerize and complex with the co-mediator Smad4, leading to nuclear translocation and regulation of transcription. Inhibitory Smads function by competing with R-Smads for binding to either the receptor or Smad4 (for review see Shi and Massague, 2003).
TGF-βI is SUMOylated predominantly at a non-consensus lysine residue located at the amino-terminal boundary of its kinase domain, and SUMOylation appears to require the kinase domains of both TβRI and TβRII (Kang et al., 2008). SUMOylation is involved in the regulation of TβRI-mediated activation of Smad2 and Smad3, possibly by enhancement of R-Smad binding to the receptor. Supplementation of TβRI−/− fibroblasts with wild-type TβRI led to a robust phosphorylation of Smad2/3 and increased Smad2/3-mediated transcription whereas replacement with a non-SUMOylatable mutant significantly attenuated TβRI-mediated signalling. Interestingly, a mutation in TβRI (S387Y) has been associated with metastasis of breast and head-and-neck cancers, when compared with primary tumours (Chen et al., 1998, 2001). This site lies close to the SUMOylated lysine (K389) and SUMOylation levels of the S387Y mutant were significantly decreased. Indeed, similar to the non-SUMOylatable mutant, TGF-β-mediated Smad3 activation was also reduced in this mutant (Kang et al., 2008). Together, these results suggest that the S387Y mutant potentially enhances metastasis by mediating downregulation of TGF-β signalling through inhibition of TβRI SUMOylation. Future work will determine how exactly SUMOylation of TβRI promotes R-Smad association and downstream signalling.
7.4. G protein-coupled receptors (GPCRs)
GPCRs are a large family of integral seven transmembrane domain proteins. More than 90% of all known GPCRs are expressed in the brain where they participate in all functions of the nervous system (Vassilatis et al., 2003).
7.4.1. Cannabinoid receptors
CB1 cannabinoid receptors are among the most widely expressed GPCRs in the brain. It has been reported that the CB1 receptor is a SUMO substrate and that the receptor is rapidly deSUMOylated in response to the agonist delta(9)-tetrahydrocannabinol (Δ9-THC) (Gowran et al., 2009). This would infer that, unusually for SUMO substrates, the CB1 receptor is basally SUMOylated. CB1 receptors activate multiple signalling pathways and the same group report that Δ9-THC evokes deSUMOylation of p53, an effect blocked by AM251 an inverse agonist at the CB1 cannabinoid receptor. p53 is a tumour suppressor protein that plays a key role in cell cycle arrest or apoptosis in response to a variety of stressors (Levine, 1997). Post-translational modifications of p53 allow cells to rapidly activate a unique set of responses to diverse stress stimuli (Lavin and Gueven, 2006). In unstressed cells, p53 is maintained at low levels through targeted degradation mediated by murine double minute 2 (Mdm2) (Appella and Anderson, 2001). SUMO-1 conjugates to the C-terminus of p53 and has positive effects on p53 transactivation (Gostissa et al., 1999; Rodriguez et al., 1999) and decreases the strength of the interaction between p53 and Mdm2 (Carter et al., 2007). Although it needs further validation, SUMOylation of CB1 may play roles in a diverse range of neuronal processes, not least through the control of p53 stability and activity.
7.4.2. Metabotropic glutamate receptors
Group III metabotropic glutamate (mGlu) receptors are expressed widely throughout the brain and predominantly function as pre-synaptic autoreceptors for glutamate, acting to modulate pre-synaptic release. mGlu8 has been proposed as a potential SUMO substrate based on the observation that its intracellular C-terminus can bind Ubc9 and the E3 enzyme PIAS1 and that GST-ct-mGlu8 can be SUMOylated by SUMO-1 when overexpressed in HEK293 cells (Tang et al., 2005). In fact, the C-termini of all of the group III mGlu receptors interact with PIAS1 (Tang et al., 2005) and can be modified by SUMO-1 in vitro (Wilkinson et al., 2008). However, while the observation that they may be SUMO substrates places them as potential targets mediating the previously reported SUMO-regulation of pre-synaptic release (Feligioni et al., 2009) there has, to date, been no report of the SUMOylation of endogenous mGlu receptors in neurons.
8. Regulation of G protein-coupled receptor signalling
In their inactive state, GPCRs are bound to heterotrimeric G proteins comprising a GDP-bound Gα subunit, and Gβγ subunits. Upon stimulation, the Gα subunits exchange their bound GDP for GTP and dissociate from the Gβγ subunits to perform multiple effector functions, depending on the bound Gα subunit (for review see Pierce et al., 2002). Intriguingly, in addition to directly modifying GPCRs themselves, SUMOylation has also been implicated in the regulation of proteins that modulate the activity of the released Gα and Gβγ subunits.
8.1. Regulator or G protein signalling (RGS) proteins
RGS proteins are GTPase activating proteins (GAPs) that stimulate the GTPase activity of Gα subunits, facilitating their return to the GDP-bound ‘off’ state (Berman and Gilman, 1998). RGS proteins are almost exclusively expressed in the brain (Mao et al., 2004), and a role for the SUMOylation of RGS1 and RGS2 has been reported in the regulation of μ-opioid receptor signalling. RGS1 and RGS2 can be modified by SUMO-1 and SUMO-2/3, and upon μ-opioid receptor activation, SUMOylation RGS proteins bind strongly both to the receptor and to Gα subunits, mediating desensitisation of the receptors (Rodriguez-Munoz et al., 2007).
8.2. Phosducin
Phosducin is a phosphoprotein highly expressed in the outer and inner segments of the rod cell layers in the retina and in the pineal gland. It modulates the phototransduction cascade by sequestering the released Gβγ subunits of the retinal G protein transducin and inhibiting their ability to activate effector pathways (Bauer et al., 1992). SUMOylation of phosducin appears to function both in promoting the stability of phosducin via reducing its ubiquitination and also by decreasing its capability to bind Gβγ subunits (Klenk et al., 2006).
9. SUMOylation substrates and neurodegeneration
Since SUMOylation plays critical roles in neuronal signalling, excitability and development, it is unsurprising that dysregulation of the SUMO pathway has been implicated in neurodegenerative disorders (for recent reviews see Anderson et al., 2009; Dorval and Fraser, 2007; Martin et al., 2007b). Many of these neurodegenerative diseases are characterised by the formation of nuclear and/or cytosolic inclusions, thought to arise from aberrant protein trafficking or degradation. In a growing number of disorders, these inclusions have been shown to stain positive for SUMO, and a growing number of disease-associated SUMO substrates have been identified (Table 1).
Table 1.
SUMOylation and neurodegenerative disorders.
| Implicated SUMO substrates |
References | ||
|---|---|---|---|
| Neuronal intranuclear inclusion disorder | HDAC4 | McFadden et al. (2005), Pountney et al. (2003), Takahashi-Fujigasaki et al. (2006) | |
| PML | |||
| RanGAP1 | |||
| NSF* | |||
| Ubc18-1* | |||
| Dynamin-1* | |||
| HSP90* | |||
| Polyglutamine diseases | Huntington’s | Huntingtin | Steffan et al. (2004) |
| SBMA | Androgen receptor |
Poukka et al. (2000) | |
| DRPLA | Unknown | Terashima et al. (2002) | |
| SCA Type I | Ataxin-1 | Riley et al. (2005), Ryu et al. (2010) | |
| SCA Type VII | Ataxin-7 | Janer et al. (2010) | |
| Alzheimer’s disease | APP | Gocke et al. (2005), Zhang and Sarge (2008) | |
| Tau | Dorval and Fraser (2006) | ||
| α-Synucleinopathies | Parkinson’s | α-Synuclein | Dorval and Fraser (2006) |
| DJ-1 | Shinbo et al. 2006 | ||
| MSA | α-Synuclein | Dorval and Fraser (2006) | |
| DLB | α-Synuclein | Dorval and Fraser (2006) | |
| Hypoxia, ischemia and cellular stress | Various unknown |
Cimarosti et al. (2008), Yang et al. (2008a,b) |
SUMOylation has been associated with multiple neurological disorders and a number of disease-associated proteins have been reported to be SUMO substrates. Proteins marked with an asterisk (*) have been isolated from SUMO-positive inclusions but have not been directly shown to be SUMOylated.
Abbreviations: SBMA, spinal and bulbar muscular atrophy; DRPLA, dentatorubral-pallidoluysian atrophy; SCA, spinocerebellar ataxia; APP, amyloid precursor protein; MSA, multiple system atrophy; DLB, demetia with Lewy bodies.
9.1. Nuclear inclusion disorder
Neuronal intranuclear inclusion disorder (NIID) is a rare disease of both the central and peripheral nervous systems characterised by extensive neuronal intranuclear inclusions (Sung et al., 1980). The disease predominantly manifests as ataxia and movement disorders in younger patients and as dementia in adult-onset cases, suggesting NIID may comprise a family of pathologically distinct neuropathies. However, inclusions stain strongly for SUMO reactivity in familial (Pountney et al., 2003; Takahashi-Fujigasaki et al., 2006), juvenile (McFadden et al., 2005) and sporadic (Takahashi-Fujigasaki et al., 2006) forms of the disease raising the possibility that the various forms may share a may share a similar mechanism. In addition, the known SUMO substrates HDAC4 and PML are present in intranuclear inclusions associated with both sporadic and familial NIID, and RanGAP1, a highly SUMOylated nuclear pore component, has been found in inclusions from a patient with familial NIID, suggesting these inclusions may at least in part represent aggregation of SUMOylated proteins (Takahashi-Fujigasaki et al., 2006). SUMO-1 immunocapture from tissue from patients with familial NIID followed by mass spectrometry identified NSF, Unc18-1, dynamin-1 and HSP90 and these proteins were also observed in NIID inclusions by immunohistochemistry (Pountney et al., 2008). However, although these proteins immunoprecipitate with SUMO-1, it is currently unclear whether they are directly modified by SUMO or represent SUMO binding proteins. For example, dynamin-1 has previously been reported to bind SUMO-1 (Mishra et al., 2004). Nonetheless, the presence of NSF, Unc18-1 and dynamin-1, proteins well-characterised for their roles in membrane trafficking, in the inclusions is perhaps suggestive that defects in the secretory or lysosomal pathways may contribute to the formation of nuclear inclusions associated with NIID.
9.2. Polyglutamine disorders
Polyglutamine (polyQ) disorders arise from a toxic stretch of polyglutamine (CAG) repeats in disease-specific genes, resulting in neuronal inclusions composed of the affected protein (for reviews see Gatchel and Zoghbi, 2005; Zoghbi and Orr, 2000). PolyQ disorders are dominantly inherited disorders, which vary in their age of onset depending on the number of polyQ repeats in the disease-associated protein. Interestingly, inclusions associated with the polyQ disorders Huntington’s, spinal and bulbar muscular atrophy (SBMA), dentatorubral-pallidoluysian atrophy (DRPLA), and spinocerebellar ataxias (SCA) have all been reported to stain strongly for SUMO reactivity (for review see Ueda et al., 2002).
9.2.1. Huntington’s disease
The most well-characterised polyQ disorder, Huntington’s, is characterised by a polyQ repeat in the N-terminal domain of the Huntingtin (Htt) protein (Gil and Rego, 2008). Htt can be both SUMOylated and ubiquitinated at a lysine residue in its N-terminus (Steffan et al., 2004). In a Drosophila model of Huntington’s, SUMOylation exacerbates disease pathology, possibly by leading to a decrease in mutant Htt aggregation resulting in an increase in the cytotoxic soluble form, whereas ubiquitination decreases pathogenicity, suggesting a likely interplay between SUMO and ubiquitin in the regulation of Htt stability. Recent work has also highlighted the role of Htt SUMOylation in mutant Htt-mediated cytotoxicity, and has offered an explanation for the localised neurodegeneration characteristic of Huntington’s disease. Rhes (Ras homolog enriched in striatum) has been identified as a SUMO E3 which binds to and enhances SUMOylation of mutant Htt, but not wild-type Htt. In both heterologous cells and striatal neurons, overexpression of mutant Htt does not lead to cytotoxicity in the absence of Rhes, suggesting that Rhes-mediated SUMOylation of mutant Htt was leading to Htt-mediated toxicity (Subramaniam et al., 2009). Indeed, while Htt is ubiquitously expressed, Rhes is located only in the striatum, potentially explaining the striatal-specific nature of the neurodegeneration associated with Huntington’s disease.
9.2.2. Spinal and bulbar muscular atrophy (SBMA)
SBMA is an X-linked neuromuscular disorder caused by a polyQ repeat in the androgen receptor (AR), a transcription factor which responds to the presence of androgenic hormones (Katsuno et al., 2006). PolyQ AR can be modified by both ubiquitin and SUMO at the same lysine residue (Poukka et al., 2000) and SUMOylation represses AR-dependent transcription (Nishida and Yasuda, 2002; Takahashi et al., 2001). In a Drosophila model, polyQ AR aggregates to form both nuclear and cytosolic inclusions that are enhanced upon prevention of both SUMOylation and ubiquitination (Chan et al., 2002) indicating that, like Huntington’s, the SUMO and ubiquitin pathways may both be involved in the advancement of the disease. Indeed, recent data have also indicated that SUMOylation may be protective against polyQ AR aggregates. In HeLa cells, overexpression of SUMO-3 leads to decreased polyQ AR aggregation, an effect that is dependent on direct modification of AR by SUMO (Mukherjee et al., 2009). SUMOylated polyQ AR shows increased solubility and overexpressed SUMO-3 is not associated with polyQ AR aggregates. Thus, it is conceivable that enhancement of polyQ AR SUMOylation could represent a potential therapeutic strategy against SBMA.
9.2.3. Dentatorubral-pallidoluysian atrophy (DRPLA)
A polyQ expansion in the atrophin-1 protein leads to DRPLA, a neuropathological condition characterised by dementia, epilepsy and involuntary movement (Schilling et al., 1999; Yazawa et al., 1995). While the function of atrophin-1 is currently unknown, coexpression of polyQ atrophin-1 with SUMO-1 leads to a significant increase in nuclear inclusions and subsequent cell death, whereas coexpression of a non-conjugatable form of SUMO-1 actually decreases inclusion formation (Terashima et al., 2002). These data suggest that SUMO conjugation plays a significant role in the formation of nuclear inclusions associated with DRPLA.
9.2.4. Spinocerebellar ataxias (SCAs)
SCAs are progressive neurodegenerative disorders, which result in atrophy of cerebellar Purkinje cells. A polyQ expansion in the ataxin-1 protein results in type I SCA. Ataxin-1 can be SUMOylated on at least five lysine residues, and the presence of the polyQ expansion significantly decreases ataxin-1 SUMOylation (Riley et al., 2005). Overexpression of SUMO-1 and/or Ubc9 appears to increase aggregation of polyQ ataxin-1 (Ryu et al., 2010). Enhancement of ataxin-1 SUMOylation by hydrogen peroxide treatment also leads to polyQ ataxin-1 aggregation yet has little effect on wild-type ataxin-1, further suggesting that SUMOylation of the polyQ mutant augments its pathogenicity (Ryu et al., 2010). Interestingly, blockade of ataxin-1 phosphorylation through mutation of S776 or inhibition of the JNK pathway prevented the oxidative stress-induced SUMOylation of both wild-type and polyQ ataxin and reduced the formation of polyQ ataxin-1 aggregates, suggesting interplay between phosphorylation and SUMOylation may regulate polyQ ataxin-1 aggregation.
Recent work has also revealed a potential role for SUMOylation in the pathogenesis of type 7 SCA. This disease is characterised by a polyQ expansion in the ataxin-7 gene. Ataxin-7 has been reported to be modified by both SUMO-1 and SUMO-2, and in both cell culture models of SCA7 and tissue from SCA7 patients, ataxin-7 inclusions also stain positively for SUMO-1 and SUMO-2 (Janer et al., 2010). In contrast to ataxin-1, the SUMOylation status of ataxin-7 appears to be unchanged by the presence of the polyQ expansion, and SUMOylation of ataxin-7 appears to reduce the propensity of ataxin-7 to form aggregates, reducing the toxicity of polyQ ataxin-7 (Janer et al., 2010) and suggesting a potential avenue of therapeutic intervention in SCA7 may involve enhancing the SUMOylation of polyQ ataxin-7.
9.3. Alzheimer’s disease
Alzheimer’s disease (AD) is an age-dependent neurodegenerative disorder characterised by progressive dementia. Patients exhibit neuronal plaques composed of amyloid-β (Aβ), a cleavage product of amyloid precursor protein (APP), and neurofibrillary plaques composed of hyperphosphorylated tau. Interestingly, both APP and tau may be SUMO substrates (Dorval and Fraser, 2006; Gocke et al., 2005; Zhang and Sarge, 2008). Exogenous SUMO-3 has been reported to regulate Aβ formation, however a direct role for SUMO-3 is currently unclear since SUMO-3 has been reported to both increase (Dorval et al., 2007) or decrease (Li et al., 2003) Aβ formation. In addition, the SUMO-3-mediated enhancement of Aβ formation could be replicated with a non-conjugatable form of SUMO-3 (Dorval et al., 2007), indicating that a non-covalent effect of SUMO-3 may be regulating Aβ levels.
Recent work has further addressed the role of APP SUMOylation in the generation of Aβ. Zhang and Sarge (2008) were able to detect SUMOylation of APP by SUMO-1 and SUMO-2 in both heterologous cells and mouse brain. Additionally, this modification appeared to regulate APP cleavage to Aβ – SUMOylation of APP appeared to reduce Aβ production and removal of the SUMO target lysines enhanced Aβ production (Zhang and Sarge, 2008).
Tau is a microtubule-associated protein that can be both SUMOylated and ubiquitinated in cell culture models (Dorval and Fraser, 2006). Inhibition of proteasomal degradation led to an increase in tau ubiquitination and decreased SUMOylation indicating roles for both SUMOylation, and the ubiquitin-proteasome system in tau clearance, which may be perturbed in Alzheimer’s, leading to the accumulation of tau protein.
While the majority of studies examining a role for SUMOylation in the pathogenesis of AD have focused on the roles of SUMOylation of AD-associated proteins, a genetic link between the SUMOylation system and AD has also been proposed. A single nucleotide polymorphism (SNP) in intron 7 of the Ubc9 gene is significantly associated with AD patients in the Korean population when compared to controls (Ahn et al., 2009). While it is unclear how this SNP manifests its consequences, the existence of a genetic link between AD and the SUMO pathway is intriguing.
9.4. Parkinson’s disease and other synucleinopathies
Parkinson’s disease (PD) results in the specific loss of dopaminergic neurons projecting from the substantia nigra pars compacta to the striatum and inclusions known as Lewy bodies occur in the cytosol of affected neurons. Lewy bodies stain strongly for α-synuclein, a protein reported to be SUMOylated in cultured cells (Dorval and Fraser, 2006). However, the existence of SUMO-modified α-synuclein in neurons has not yet been reported.
Lewy bodies are also a characteristic of dementia with Lewy bodies (DLB), a form of dementia characterised by delirium, visual hallucinations and Parkinsonism (Neef and Walling, 2006). α-Synuclein-positive Lewy bodies from DLB patients have been reported to stain positively for SUMO-1, although this staining was occasional and not detectable in fixed tissue (Pountney et al., 2005). α-Synuclein is also a component of the cytoplasmic inclusions in oligodendrocytes from patients with multiple system atrophy (MSA). Subdomains within these inclusions appear to stain positively for SUMO-1 but the potential pathophysiological significance of these observations have not yet been established (Pountney et al., 2005).
Importantly, non-covalent SUMO binding has also been implicated in synucleinopathies. Mutations in the ubiquitin E3 ligase parkin have been associated with familial Parkinsonism (Tan and Skipper, 2007). Parkin has been reported to interact non-covalently with SUMO-1 both in vitro and in vivo, and this interaction appears to enhance its E3 ligase activity (Um and Chung, 2006). Interestingly, a substrate of parkin is RanBP2 (Um et al., 2006), a SUMO E3 ligase. Thus, SUMO binding to parkin may enhance its ubiquitination activity, leading to the degradation of RanBP2, an intriguing example of negative interplay between the ubiquitin and SUMO systems.
Another PD-associated protein is the transcriptional regulator DJ-1. DJ-1 controls the expression of many genes associated with the cellular response to oxidative stress (Taira et al., 2004). Loss-of-function of DJ-1 has been reported to lead to PD, and it has been reported to be a SUMO substrate (Shinbo et al., 2006). While the physiological role of DJ-1 SUMOylation is unclear, SUMOylation of a PD-associated DJ-1 mutation (L166P) is enhanced (Shinbo et al., 2006), suggesting that inappropriate levels of SUMOylation may contribute to the pathogenicity of this mutant DJ-1.
9.5. Hypoxia, ischemia and cellular stress
Changes in global SUMOylation during neuronal excitation may contribute to the regulation of complex physiological processes (Feligioni et al., 2009; Lu et al., 2009). In addition, however, rapid global increases in protein SUMOylation result from excessive neuronal excitation and other forms of cellular stress. Heat shock, osmotic shock or oxidative stress elicit massive increases in SUMO-2/3 conjugation (for reviews see Agbor and Taylor, 2008; Bossis and Melchior, 2006; Tempe et al., 2008), which may constitute a protective cellular response to stress. Indeed, a massive increase in protein SUMOylation has recently been reported in models of ischemia (for reviews see Cimarosti and Henley, 2008; Yang et al., 2008a). Transient global ischemia in mice leads to massive increases in SUMO-2/3 conjugation in the cortex and hippocampus (Yang et al., 2008b). Another study also reported an increase in SUMO-1 conjugation in the cortical infarct region and the non-ischemic hippocampus in a mouse model of ischemia with reperfusion (Cimarosti et al., 2008). In this model, increases in SUMO-2/3 conjugation were restricted to the non-infarct regions. Intriguingly, levels of AMPA and kainate receptors were decreased by the ischemic event, suggesting the possibility that SUMOylation may play a role in decreasing glutamatergic signalling that may lead to excitotoxic cell death. Indeed, the observation that kainate receptor EPSCs are decreased as a result of SUMOylation of the GluK2 subunit adds credence to this possibility (Martin et al., 2007a).
In addition, direct evidence for a role of SUMOylation in the protection against ischemic cell death has been provided recently by the observation that overexpression of SUMO proteins makes various cell types more resistant to in vitro ischemia models (Ja Lee et al., 2009). Overexpression of SUMO-1 or SUMO-2 were shown to make SHSY5Y human neuroblastoma cells more resistant to oxygen and glucose depravation (OGD). Conversely, knockdown of SUMO-1, but not SUMO-2, made SHSY5Y cells more susceptible to OGD-mediated cell death. Finally, overexpression of SUMO-1 made rat cortical neurons more resistant to OGD (Ja Lee et al., 2009). While this work appears to suggest a protective role for SUMOylation in OGD, further work will be required to define the targets responsible for the effects observed.
10. Concluding remarks
Over the past decade there has been rapid progress in defining the roles of SUMOylation in neurons. It is clear that SUMOylation provides a rapid and effective system that can dynamically regulate the function, activity and lifetime of a wide variety of substrate proteins in multiple cell types. Further it seems likely that protein SUMOylation can direct or influence nearly all aspects of cell development, function and senescence. For example, nuclear SUMOylation plays key roles in the initiation and regulation of transcription, RNA processing, and nuclear integrity. Outside the nucleus SUMOylation is intimately involved in aspects of protein transport, regulation of cell metabolism and division and in multiple cell signalling pathways. Despite this wealth of accumulating data there remain many outstanding questions, foremost among which are how SUMOylation of specific targets is regulated and the physiological and pathophysiological roles of SUMOylation in neurons. Given the abundance of established and putative SUMO substrate proteins, SUMOylation likely represents a diverse, wide ranging and complex regulatory system that is critical for many or most neuronal processes including neurogenesis, differentiation and development, presynaptic release, vesicle recycling and synaptic plasticity.
In view of the diversity of SUMO functions it is unsurprising that dysregulation of the SUMO pathway has been closely implicated in a wide range of diseases, including neurodegenerative disorders, and this has emerged as an area of intense research interest. For example, SUMO immunoreactivity appears to be strongly associated with aberrant protein aggregates that occur in several clinically important types of neurodegeneration including Huntington’s and Alzhiemer’s diseases, and a growing number of proteins associated with these disorders are being reported to be SUMO substrates. In addition, it has been proposed that SUMOylation may participate or even mediate neuroprotective mechanisms during cell stress conditions such as ischaemia. Understanding the roles SUMO plays in these disorders could provide the basis for the potential development of novel preventative and/or therapeutic approaches that modulate the SUMOylation pathway or, perhaps more likely to be of practical application, enhance or inhibit SUMOylation of specific target proteins. Following on from the impressive progress achieved thus far we anticipate that the field will continue to advance rapidly leading to new and important insights into the physiological and pathophysiological mechanisms and consequences of protein SUMOylation in neurons.
Acknowledgments
We are grateful to the Medical Research Council, the Wellcome Trust and the European Research Council for financial support.
REFERENCES
- Agbor TA, Taylor CT. SUMO, hypoxia and the regulation of metabolism. Biochem. Soc. Trans. 2008;36:445–448. doi: 10.1042/BST0360445. [DOI] [PubMed] [Google Scholar]
- Ahima RS, Flier JS. Leptin. Annu. Rev. Physiol. 2000;62:413–437. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- Ahn K, Song JH, Kim DK, Park MH, Jo SA, Koh YH. Ubc9 gene polymorphisms and late-onset Alzheimer’s disease in the Korean population: a genetic association study. Neurosci. Lett. 2009;465:272–275. doi: 10.1016/j.neulet.2009.09.017. [DOI] [PubMed] [Google Scholar]
- Anderson DB, Wilkinson KA, Henley JM. Protein SUMOylation in neuropathological conditions. Drug News Perspect. 2009;22:255–265. doi: 10.1358/dnp.2009.22.5.1378636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 2001;268:2764–2772. doi: 10.1046/j.1432-1327.2001.02225.x. [DOI] [PubMed] [Google Scholar]
- Bauer PH, Muller S, Puzicha M, Pippig S, Obermaier B, Helmreich EJ, Lohse MJ. Phosducin is a protein kinase A-regulated G-protein regulator. Nature. 1992;358:73–76. doi: 10.1038/358073a0. [DOI] [PubMed] [Google Scholar]
- Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, Kahn BB. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006;12:917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- Benson MD, Li QJ, Kieckhafer K, Dudek D, Whorton MR, Sunahara RK, Iniguez-Lluhi JA, Martens JR. SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1. Proc. Natl. Acad. Sci. USA. 2007;104:1805–1810. doi: 10.1073/pnas.0606702104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berciano MT, Novell M, Villagra NT, Casafont I, Bengoechea R, Val-Bernal JF, Lafarga M. Cajal body number and nucleolar size correlate with the cell body mass in human sensory ganglia neurons. J. Struct. Biol. 2007;158:410–420. doi: 10.1016/j.jsb.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Berman DM, Gilman AG. Mammalian RGS proteins: barbarians at the gate. J. Biol. Chem. 1998;273:1269–1272. doi: 10.1074/jbc.273.3.1269. [DOI] [PubMed] [Google Scholar]
- Besnault-Mascard L, Leprince C, Auffredou MT, Meunier B, Bourgeade MF, Camonis J, Lorenzo HK, Vazquez A. Caspase-8 sumoylation is associated with nuclear localization. Oncogene. 2005;24:3268–3273. doi: 10.1038/sj.onc.1208448. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Budd Haeberlein SL, Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. J. Neurochem. 2004;89:1313–1317. doi: 10.1111/j.1471-4159.2004.02422.x. [DOI] [PubMed] [Google Scholar]
- Blackshaw S, Harpavat S, Trimarchi J, Cai L, Huang H, Kuo WP, Weber G, Lee K, Fraioli RE, Cho SH, Yung R, Asch E, Ohno-Machado L, Wong WH, Cepko CL. Genomic analysis of mouse retinal development. PLoS Biol. 2004;2:E247. doi: 10.1371/journal.pbio.0020247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohren KM, Nadkarni V, Song JH, Gabbay KH, Owerbach D. A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J. Biol. Chem. 2004;279:27233–27238. doi: 10.1074/jbc.M402273200. [DOI] [PubMed] [Google Scholar]
- Bossis G, Melchior F. Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol. Cell. 2006;21:349–357. doi: 10.1016/j.molcel.2005.12.019. [DOI] [PubMed] [Google Scholar]
- Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10:748–754. doi: 10.1038/embor.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell Biol. 2002;3:267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- Cardone L, Hirayama J, Giordano F, Tamaru T, Palvimo JJ, Sassone-Corsi P. Circadian clock control by SUMOylation of BMAL1. Science. 2005;309:1390–1394. doi: 10.1126/science.1110689. [DOI] [PubMed] [Google Scholar]
- Carter S, Bischof O, Dejean A, Vousden KH. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nat. Cell Biol. 2007;9:428–435. doi: 10.1038/ncb1562. [DOI] [PubMed] [Google Scholar]
- Carvalho T, Almeida F, Calapez A, Lafarga M, Berciano MT, Carmo-Fonseca M. The spinal muscular atrophy disease gene product, SMN: A link between snRNP biogenesis and the Cajal (coiled) body. J. Cell Biol. 1999;147:715–728. doi: 10.1083/jcb.147.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- Chan HY, Warrick JM, Andriola I, Merry D, Bonini NM. Genetic modulation of polyglutamine toxicity by protein conjugation pathways in Drosophila. Hum. Mol. Genet. 2002;11:2895–2904. doi: 10.1093/hmg/11.23.2895. [DOI] [PubMed] [Google Scholar]
- Chao HW, Hong CJ, Huang TN, Lin YL, Hsueh YP. SUMOylation of the MAGUK protein CASK regulates dendritic spinogenesis. J. Cell Biol. 2008;182:141–155. doi: 10.1083/jcb.200712094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Carter D, Garrigue-Antar L, Reiss M. Transforming growth factor beta type I receptor kinase mutant associated with metastatic breast cancer. Cancer Res. 1998;58:4805–4810. [PubMed] [Google Scholar]
- Chen T, Yan W, Wells RG, Rimm DL, McNiff J, Leffell D, Reiss M. Novel inactivating mutations of transforming growth factor-beta type I receptor gene in head-and-neck cancer metastases. Int. J. Cancer. 2001;93:653–661. doi: 10.1002/ijc.1381. [DOI] [PubMed] [Google Scholar]
- Cimarosti H, Henley JM. Investigating the mechanisms underlying neuronal death in ischemia using in vitro oxygen-glucose deprivation: potential involvement of protein SUMOylation. Neuroscientist. 2008;14:626–636. doi: 10.1177/1073858408322677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimarosti H, Lindberg C, Bomholt SF, Ronn LC, Henley JM. Increased protein SUMOylation following focal cerebral ischemia. Neuropharmacology. 2008;54:280–289. doi: 10.1016/j.neuropharm.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Dadke S, Cotteret S, Yip SC, Jaffer ZM, Haj F, Ivanov A, Rauscher F, III, Shuai K, Ng T, Neel BG, Chernoff J. Regulation of protein tyrosine phosphatase 1B by sumoylation. Nat. Cell Biol. 2007;9:80–85. doi: 10.1038/ncb1522. [DOI] [PubMed] [Google Scholar]
- Dorval V, Fraser PE. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J. Biol. Chem. 2006;281:9919–9924. doi: 10.1074/jbc.M510127200. [DOI] [PubMed] [Google Scholar]
- Dorval V, Fraser PE. SUMO on the road to neurodegeneration. Biochim. Biophys. Acta. 2007;1773:694–706. doi: 10.1016/j.bbamcr.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Dorval V, Mazzella MJ, Mathews PM, Hay RT, Fraser PE. Modulation of Abeta generation by small ubiquitin-like modifiers does not require conjugation to target proteins. Biochem. J. 2007;404:309–316. doi: 10.1042/BJ20061451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube N, Tremblay ML. Involvement of the small protein tyrosine phosphatases TC-PTP and PTP1B in signal transduction and diseases: from diabetes, obesity to cell cycle, and cancer. Biochim. Biophys. Acta. 2005;1754:108–117. doi: 10.1016/j.bbapap.2005.07.030. [DOI] [PubMed] [Google Scholar]
- Eizenberg O, Faber-Elman A, Gottlieb E, Oren M, Rotter V, Schwartz M. p53 plays a regulatory role in differentiation and apoptosis of central nervous system-associated cells. Mol. Cell. Biol. 1996;16:5178–5185. doi: 10.1128/mcb.16.9.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol. Med. 2002;8:126–132. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- Eun Jeoung L, Sung Hee H, Jaesun C, Sung Hwa S, Kwang Hum Y, Min Kyoung K, Tae Yoon P, Sang Sun K. Regulation of glycogen synthase kinase 3beta functions by modification of the small ubiquitin-like modifier. Open Biochem. J. 2008;2:67–76. doi: 10.2174/1874091X00802010067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciangeli S, Bendahhou S, Sandoz G, Gounon P, Reichold M, Warth R, Lazdunski M, Barhanin J, Lesage F. Does sumoylation control K2P1/TWIK1 background K+ channels? Cell. 2007;130:563–569. doi: 10.1016/j.cell.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Feliciangeli S, Tardy MP, Sandoz G, Chatelain FC, Warth R, Barhanin J, Bendahhou S, Lesage F. Potassium channel silencing by constitutive endocytosis and intracellular sequestration. J. Biol. Chem. 2010;285:4798–4805. doi: 10.1074/jbc.M109.078535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feligioni M, Nishimune A, Henley JM. Protein SUMOylation modulates calcium influx and glutamate release from presynaptic terminals. Eur. J. Neurosci. 2009;29:1348–1356. doi: 10.1111/j.1460-9568.2009.06692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa-Romero C, Iniguez-Lluhi JA, Stadler J, Chang CR, Arnoult D, Keller PJ, Hong Y, Blackstone C, Feldman EL. SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J. 2009;23:3917–3927. doi: 10.1096/fj.09-136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanders KC, Ren RF, Lippa CF. Transforming growth factor-betas in neurodegenerative disease. Prog. Neurobiol. 1998;54:71–85. doi: 10.1016/s0301-0082(97)00066-x. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 2007;8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- Geoffroy MC, Hay RT. An additional role for SUMO in ubiquitin-mediated proteolysis. Nat. Rev. Mol. Cell Biol. 2009;10:564–568. doi: 10.1038/nrm2707. [DOI] [PubMed] [Google Scholar]
- Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington’s disease. Eur. J. Neurosci. 2008;27:2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- Giorgino F, de Robertis O, Laviola L, Montrone C, Perrini S, McCowen KC, Smith RJ. The sentrin-conjugating enzyme mUbc9 interacts with GLUT4 and GLUT1 glucose transporters and regulates transporter levels in skeletal muscle cells. Proc. Natl. Acad. Sci. USA. 2000;97:1125–1130. doi: 10.1073/pnas.97.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuditta A, Kaplan BB, van Minnen J, Alvarez J, Koenig E. Axonal and presynaptic protein synthesis: new insights into the biology of the neuron. Trends Neurosci. 2002;25:400–404. doi: 10.1016/s0166-2236(02)02188-4. [DOI] [PubMed] [Google Scholar]
- Gocke CB, Yu H, Kang J. Systematic identification and analysis of mammalian small ubiquitin-like modifier substrates. J. Biol. Chem. 2005;280:5004–5012. doi: 10.1074/jbc.M411718200. [DOI] [PubMed] [Google Scholar]
- Gong L, Li B, Millas S, Yeh ET. Molecular cloning and characterization of human AOS1 and UBA2, components of the sentrin-activating enzyme complex. FEBS Lett. 1999;448:185–189. doi: 10.1016/s0014-5793(99)00367-1. [DOI] [PubMed] [Google Scholar]
- Gostissa M, Hengstermann A, Fogal V, Sandy P, Schwarz SE, Scheffner M, Del Sal G. Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J. 1999;18:6462–6471. doi: 10.1093/emboj/18.22.6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowran A, Murphy CE, Campbell VA. Delta(9)-tetrahydrocannabinol regulates the p53 post-translational modifiers Murine double minute 2 and the small ubiquitin modifier protein in the rat brain. FEBS Lett. 2009;583:3412–3418. doi: 10.1016/j.febslet.2009.09.056. [DOI] [PubMed] [Google Scholar]
- Hankins MW, Peirson SN, Foster RG. Melanopsin: an exciting photopigment. Trends Neurosci. 2008;31:27–36. doi: 10.1016/j.tins.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Hardeland U, Steinacher R, Jiricny J, Schar P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002;21:1456–1464. doi: 10.1093/emboj/21.6.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr. Biol. 2004;14:340–345. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Hay RT. SUMO: a history of modification. Mol. Cell. 2005;18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Seki M, Maeda D, Wang W, Kawabe Y, Seki T, Saitoh H, Fukagawa T, Yagi H, Enomoto T. Ubc9 is essential for viability of higher eukaryotic cells. Exp. Cell Res. 2002;280:212–221. doi: 10.1006/excr.2002.5634. [DOI] [PubMed] [Google Scholar]
- Hayashi N, Shirakura H, Uehara T, Nomura Y. Relationship between SUMO-1 modification of caspase-7 and its nuclear localization in human neuronal cells. Neurosci. Lett. 2006;397:5–9. doi: 10.1016/j.neulet.2005.11.057. [DOI] [PubMed] [Google Scholar]
- Heun P. SUMOrganization of the nucleus. Curr. Opin. Cell Biol. 2007;19:350–355. doi: 10.1016/j.ceb.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Hietakangas V, Anckar J, Blomster HA, Fujimoto M, Palvimo JJ, Nakai A, Sistonen L. PDSM, a motif for phosphorylation-dependent SUMO modification. Proc. Natl. Acad. Sci. USA. 2006;103:45–50. doi: 10.1073/pnas.0503698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgarth RS, Murphy LA, Skaggs HS, Wilkerson DC, Xing H, Sarge KD. Regulation and function of SUMO modification. J. Biol. Chem. 2004;279:53899–53902. doi: 10.1074/jbc.R400021200. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci. 2005;6:201–214. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- Hofmann H, Floss S, Stamminger T. Covalent modification of the transactivator protein IE2-p86 of human cytomegalovirus by conjugation to the ubiquitin-homologous proteins SUMO-1 and hSMT3b. J. Virol. 2000;74:2510–2524. doi: 10.1128/jvi.74.6.2510-2524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Ja Lee Y, Castri P, Bembry J, Maric D, Auh S, Hallenbeck JM. SUMOylation participates in induction of ischemic tolerance. J. Neurochem. 2009;109:257–267. doi: 10.1111/j.1471-4159.2009.05957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janer A, Werner A, Takahashi-Fujigasaki J, Daret A, Fujigasaki H, Takada K, Duyckaerts C, Brice A, Dejean A, Sittler A. SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Hum. Mol. Genet. 2010;19:181–195. doi: 10.1093/hmg/ddp478. [DOI] [PubMed] [Google Scholar]
- Jaskolski F, Coussen F, Mulle C. Subcellular localization and trafficking of kainate receptors. Trends Pharmacol. Sci. 2005;26:20–26. doi: 10.1016/j.tips.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Johnson ES. Protein modification by SUMO. Annu. Rev. Biochem. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- Kadare G, Toutant M, Formstecher E, Corvol JC, Carnaud M, Boutterin MC, Girault JA. PIAS1-mediated sumoylation of focal adhesion kinase activates its autophosphorylation. J. Biol. Chem. 2003;278:47434–47440. doi: 10.1074/jbc.M308562200. [DOI] [PubMed] [Google Scholar]
- Kang JS, Saunier EF, Akhurst RJ, Derynck R. The type I TGF-beta receptor is covalently modified and regulated by sumoylation. Nat. Cell Biol. 2008;10:654–664. doi: 10.1038/ncb1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Waza M, Banno H, Suzuki K, Tanaka F, Doyu M, Sobue G. Pathogenesis, animal models and therapeutics in spinal and bulbar muscular atrophy (SBMA) Exp. Neurol. 2006;200:8–18. doi: 10.1016/j.expneurol.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Kim JH, Baek SH. Emerging roles of desumoylating enzymes. Biochim. Biophys. Acta. 2009;1792:155–162. doi: 10.1016/j.bbadis.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Klenk C, Humrich J, Quitterer U, Lohse MJ. SUMO-1 controls the protein stability and the biological function of phosducin. J. Biol. Chem. 2006;281:8357–8364. doi: 10.1074/jbc.M513703200. [DOI] [PubMed] [Google Scholar]
- Kozlovsky N, Belmaker RH, Agam G. GSK-3 and the neurodevelopmental hypothesis of schizophrenia. Eur. Neuropsychopharmacol. 2002;12:13–25. doi: 10.1016/s0924-977x(01)00131-6. [DOI] [PubMed] [Google Scholar]
- Lapenta V, Chiurazzi P, van der Spek P, Pizzuti A, Hanaoka F, Brahe C. SMT3A, a human homologue of the S. cerevisiae SMT3 gene, maps to chromosome 21qter and defines a novel gene family. Genomics. 1997;40:362–366. doi: 10.1006/geno.1996.4556. [DOI] [PubMed] [Google Scholar]
- Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006;13:941–950. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- Lee MT, Bachant J. SUMO modification of DNA topoisomerase II: trying to get a CENse of it all. DNA Repair (Amst.) 2009;8:557–568. doi: 10.1016/j.dnarep.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Lee Y, Lee MJ, Park E, Kang SH, Chung CH, Lee KH, Kim K. Dual modification of BMAL1 by SUMO2/3 and ubiquitin promotes circadian activation of the CLOCK/BMAL1 complex. Mol. Cell. Biol. 2008;28:6056–6065. doi: 10.1128/MCB.00583-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Li W, Wang JC. Mammalian DNA topoisomerase IIIalpha is essential in early embryogenesis. Proc. Natl. Acad. Sci. USA. 1998;95:1010–1013. doi: 10.1073/pnas.95.3.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang H, Wang S, Quon D, Liu YW, Cordell B. Positive and negative regulation of APP amyloidogenesis by sumoylation. Proc. Natl. Acad. Sci. USA. 2003;100:259–264. doi: 10.1073/pnas.0235361100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Liu LB, Omata W, Kojima I, Shibata H. The SUMO conjugating enzyme Ubc9 is a regulator of GLUT4 turnover and targeting to the insulin-responsive storage compartment in 3T3-L1 adipocytes. Diabetes. 2007;56:1977–1985. doi: 10.2337/db06-1100. [DOI] [PubMed] [Google Scholar]
- Lu H, Liu B, You S, Xue Q, Zhang F, Cheng J, Yu B. The activity-dependent stimuli increase SUMO modification in SHSY5Y cells. Biochem. Biophys. Res. Commun. 2009;390:872–876. doi: 10.1016/j.bbrc.2009.10.065. [DOI] [PubMed] [Google Scholar]
- Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997;88:97–107. doi: 10.1016/s0092-8674(00)81862-0. [DOI] [PubMed] [Google Scholar]
- Mao Y, Desai SD, Liu LF. SUMO-1 conjugation to human DNA topoisomerase II isozymes. J. Biol. Chem. 2000a;275:26066–26073. doi: 10.1074/jbc.M001831200. [DOI] [PubMed] [Google Scholar]
- Mao Y, Sun M, Desai SD, Liu LF. SUMO-1 conjugation to topoisomerase I: A possible repair response to topoisomerase-mediated DNA damage. Proc. Natl. Acad. Sci. USA. 2000b;97:4046–4051. doi: 10.1073/pnas.080536597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, Zhao Q, Daigle M, Ghahremani MH, Chidiac P, Albert PR. RGS17/RGSZ2, a novel regulator of Gi/o, Gz, and Gq signaling. J. Biol. Chem. 2004;279:26314–26322. doi: 10.1074/jbc.M401800200. [DOI] [PubMed] [Google Scholar]
- Martin KC. Local protein synthesis during axon guidance and synaptic plasticity. Curr. Opin. Neurobiol. 2004;14:305–310. doi: 10.1016/j.conb.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Martin S, Nishimune A, Mellor JR, Henley JM. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature. 2007a;447:321–325. doi: 10.1038/nature05736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Wilkinson KA, Nishimune A, Henley JM. Emerging extranuclear roles of protein SUMOylation in neuronal function and dysfunction. Nat.Rev.Neurosci. 2007b;8:948–959. doi: 10.1038/nrn2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matunis MJ, Coutavas E, Blobel G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 1996;135:1457–1470. doi: 10.1083/jcb.135.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr. Biol. 2006;16:R551–R560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- McFadden K, Hamilton RL, Insalaco SJ, Lavine L, Al-Mateen M, Wang G, Wiley CA. Neuronal intranuclear inclusion disease without polyglutamine inclusions in a child. J. Neuropathol. Exp. Neurol. 2005;64:545–552. doi: 10.1093/jnen/64.6.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior F, Hengst L. SUMO-1 and p53. Cell Cycle. 2002;1:245–249. [PubMed] [Google Scholar]
- Meulmeester E, Melchior F. Cell biology: SUMO. Nature. 2008;452:709–711. doi: 10.1038/452709a. [DOI] [PubMed] [Google Scholar]
- Minty A, Dumont X, Kaghad M, Caput D. Covalent modification of p73alpha by SUMO-1. Two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif. J. Biol. Chem. 2000;275:36316–36323. doi: 10.1074/jbc.M004293200. [DOI] [PubMed] [Google Scholar]
- Mishra RK, Jatiani SS, Kumar A, Simhadri VR, Hosur RV, Mittal R. Dynamin interacts with members of the sumoylation machinery. J. Biol. Chem. 2004;279:31445–31454. doi: 10.1074/jbc.M402911200. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Thomas M, Dadgar N, Lieberman AP, Iniguez-Lluhi JA. Small ubiquitin-like modifier (SUMO) modification of the androgen receptor attenuates polyglutamine-mediated aggregation. J. Biol. Chem. 2009;284:21296–21306. doi: 10.1074/jbc.M109.011494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay D, Dasso M. Modification in reverse: the SUMO proteases. Trends Biochem. Sci. 2007;32:286–295. doi: 10.1016/j.tibs.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Muller S, Hoege C, Pyrowolakis G, Jentsch S. SUMO, ubiquitin’s mysterious cousin. Nat. Rev. Mol. Cell Biol. 2001;2:202–210. doi: 10.1038/35056591. [DOI] [PubMed] [Google Scholar]
- Nacerddine K, Lehembre F, Bhaumik M, Artus J, Cohen-Tannoudji M, Babinet C, Pandolfi PP, Dejean A. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev. Cell. 2005;9:769–779. doi: 10.1016/j.devcel.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Navascues J, Bengoechea R, Tapia O, Casafont I, Berciano MT, Lafarga M. SUMO-1 transiently localizes to Cajal bodies in mammalian neurons. J. Struct. Biol. 2008;163:137–146. doi: 10.1016/j.jsb.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Neef D, Walling AD. Dementia with Lewy bodies: an emerging disease. Am. Fam. Physician. 2006;73:1223–1229. [PubMed] [Google Scholar]
- Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 2008;18:102–108. doi: 10.1016/j.cub.2007.12.038. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu. Rev. Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Nishida T, Yasuda H. PIAS1 and PIASxalpha function as SUMO-E3 ligases toward androgen receptor and repress androgen receptor-dependent transcription. J. Biol. Chem. 2002;277:41311–41317. doi: 10.1074/jbc.M206741200. [DOI] [PubMed] [Google Scholar]
- Onishi A, Peng GH, Hsu C, Alexis U, Chen S, Blackshaw S. Pias3-dependent SUMOylation directs rod photoreceptor development. Neuron. 2009;61:234–246. doi: 10.1016/j.neuron.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orias M, Velazquez H, Tung F, Lee G, Desir GV. Cloning and localization of a double-pore K channel, KCNK1: exclusive expression in distal nephron segments. Am. J. Physiol. 1997;273:F663–F666. doi: 10.1152/ajprenal.1997.273.4.F663. [DOI] [PubMed] [Google Scholar]
- Owerbach D, McKay EM, Yeh ET, Gabbay KH, Bohren KM. A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem. Biophys. Res. Commun. 2005;337:517–520. doi: 10.1016/j.bbrc.2005.09.090. [DOI] [PubMed] [Google Scholar]
- Palancade B, Doye V. Sumoylating and desumoylating enzymes at nuclear pores: underpinning their unexpected duties? Trends Cell Biol. 2008;18:174–183. doi: 10.1016/j.tcb.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Pinheiro P, Mulle C. Kainate receptors. Cell Tissue Res. 2006;326:457–482. doi: 10.1007/s00441-006-0265-6. [DOI] [PubMed] [Google Scholar]
- Plant LD, Rajan S, Goldstein SA. K2P channels and their protein partners. Curr. Opin. Neurobiol. 2005;15:326–333. doi: 10.1016/j.conb.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Poukka H, Karvonen U, Janne OA, Palvimo JJ. Covalent modification of the androgen receptor by small ubiquitin-like modifier 1 (SUMO-1) Proc. Natl. Acad. Sci. USA. 2000;97:14145–14150. doi: 10.1073/pnas.97.26.14145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pountney DL, Huang Y, Burns RJ, Haan E, Thompson PD, Blumbergs PC, Gai WP. SUMO-1 marks the nuclear inclusions in familial neuronal intranuclear inclusion disease. Exp. Neurol. 2003;184:436–446. doi: 10.1016/j.expneurol.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Pountney DL, Chegini F, Shen X, Blumbergs PC, Gai WP. SUMO-1 marks subdomains within glial cytoplasmic inclusions of multiple system atrophy. Neurosci. Lett. 2005;381:74–79. doi: 10.1016/j.neulet.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Pountney DL, Raftery MJ, Chegini F, Blumbergs PC, Gai WP. NSF, Unc-18-1, dynamin-1 and HSP90 are inclusion body components in neuronal intranuclear inclusion disease identified by anti-SUMO-1-immunocapture. Acta Neuropathol. 2008;116:603–614. doi: 10.1007/s00401-008-0437-4. [DOI] [PubMed] [Google Scholar]
- Pratt BM, McPherson JM. TGF-beta in the central nervous system: potential roles in ischemic injury and neurodegenerative diseases. Cytokine Growth Factor Rev. 1997;8:267–292. doi: 10.1016/s1359-6101(97)00018-x. [DOI] [PubMed] [Google Scholar]
- Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SA. Sumoylation silences the plasma membrane leak K + channel K2P1. Cell. 2005;121:37–47. doi: 10.1016/j.cell.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- Riley BE, Zoghbi HY, Orr HT. SUMOylation of the polyglutamine repeat protein, ataxin-1, is dependent on a functional nuclear localization signal. J. Biol. Chem. 2005;280:21942–21948. doi: 10.1074/jbc.M501677200. [DOI] [PubMed] [Google Scholar]
- Rodriguez MS, Desterro JM, Lain S, Midgley CA, Lane DP, Hay RT. SUMO-1 modification activates the transcriptional response of p53. EMBO J. 1999;18:6455–6461. doi: 10.1093/emboj/18.22.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MS, Dargemont C, Hay RT. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 2001;276:12654–12659. doi: 10.1074/jbc.M009476200. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Munoz M, Bermudez D, Sanchez-Blazquez P, Garzon J. Sumoylated RGS-Rz proteins act as scaffolds for Mu-opioid receptors and G-protein complexes in mouse brain. Neuropsychopharmacology. 2007;32:842–850. doi: 10.1038/sj.npp.1301184. [DOI] [PubMed] [Google Scholar]
- Rui HL, Fan E, Zhou HM, Xu Z, Zhang Y, Lin SC. SUMO-1 modification of the C-terminal KVEKVD of Axin is required for JNK activation but has no effect on Wnt signaling. J. Biol. Chem. 2002;277:42981–42986. doi: 10.1074/jbc.M208099200. [DOI] [PubMed] [Google Scholar]
- Ryu J, Cho S, Park BC, Lee do H. Oxidative stress-enhanced SUMOylation and aggregation of ataxin-1: Implication of JNK pathway. Biochem. Biophys. Res. Commun. 2010;393:280–285. doi: 10.1016/j.bbrc.2010.01.122. [DOI] [PubMed] [Google Scholar]
- Sahar S, Zocchi L, Kinoshita C, Borrelli E, Sassone-Corsi P. Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PLoS ONE. 2010;5:e8561. doi: 10.1371/journal.pone.0008561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh H, Hinchey J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 2000;275:6252–6258. doi: 10.1074/jbc.275.9.6252. [DOI] [PubMed] [Google Scholar]
- Sampson DA, Wang M, Matunis MJ. The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J. Biol. Chem. 2001;276:21664–21669. doi: 10.1074/jbc.M100006200. [DOI] [PubMed] [Google Scholar]
- Schilling G, Wood JD, Duan K, Slunt HH, Gonzales V, Yamada M, Cooper JK, Margolis RL, Jenkins NA, Copeland NG, Takahashi H, Tsuji S, Price DL, Borchelt DR, Ross CA. Nuclear accumulation of truncated atrophin-1 fragments in a transgenic mouse model of DRPLA. Neuron. 1999;24:275–286. doi: 10.1016/s0896-6273(00)80839-9. [DOI] [PubMed] [Google Scholar]
- Seeler JS, Dejean A. Nuclear and unclear functions of SUMO. Nat. Rev. Mol. Cell Biol. 2003;4:690–699. doi: 10.1038/nrm1200. [DOI] [PubMed] [Google Scholar]
- Shalizi A, Gaudilliere B, Yuan Z, Stegmuller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006;311:1012–1017. doi: 10.1126/science.1122513. [DOI] [PubMed] [Google Scholar]
- Shalizi A, Bilimoria PM, Stegmuller J, Gaudilliere B, Yang Y, Shuai K, Bonni A. PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. J. Neurosci. 2007;27:10037–10046. doi: 10.1523/JNEUROSCI.0361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shinbo Y, Niki T, Taira T, Ooe H, Takahashi-Niki K, Maita C, Seino C, Iguchi-Ariga SM, Ariga H. Proper SUMO-1 conjugation is essential to DJ-1 to exert its full activities. Cell Death Differ. 2006;13:96–108. doi: 10.1038/sj.cdd.4401704. [DOI] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Durrin LK, Wilkinson TA, Krontiris TG, Chen Y. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. USA. 2004;101:14373–14378. doi: 10.1073/pnas.0403498101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- Stehmeier P, Muller S. Regulation of p53 family members by the ubiquitin-like SUMO system. DNA Repair (Amst.) 2009;8:491–498. doi: 10.1016/j.dnarep.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science. 2009;324:1327–1330. doi: 10.1126/science.1172871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JH, Ramirez-Lassepas M, Mastri AR, Larkin SM. An unusual degenerative disorder of neurons associated with a novel intranuclear hyaline inclusion (neuronal intranuclear hyaline inclusion disease). A clinicopathological study of a case. J. Neuropathol. Exp. Neurol. 1980;39:107–130. doi: 10.1097/00005072-198003000-00001. [DOI] [PubMed] [Google Scholar]
- Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004;5:213–218. doi: 10.1038/sj.embor.7400074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Taira T, Niki T, Seino C, Iguchi-Ariga SM, Ariga H. DJ-1 positively regulates the androgen receptor by impairing the binding of PIASx alpha to the receptor. J. Biol. Chem. 2001;276:37556–37563. doi: 10.1074/jbc.M101730200. [DOI] [PubMed] [Google Scholar]
- Takahashi-Fujigasaki J, Arai K, Funata N, Fujigasaki H. SUMOylation substrates in neuronal intranuclear inclusion disease. Neuropathol. Appl. Neurobiol. 2006;32:92–100. doi: 10.1111/j.1365-2990.2005.00705.x. [DOI] [PubMed] [Google Scholar]
- Tan EK, Skipper LM. Pathogenic mutations in Parkinson disease. Hum. Mutat. 2007;28:641–653. doi: 10.1002/humu.20507. [DOI] [PubMed] [Google Scholar]
- Tang Z, El Far O, Betz H, Scheschonka A. Pias1 interaction and sumoylation of metabotropic glutamate receptor 8. J. Biol. Chem. 2005;280:38153–38159. doi: 10.1074/jbc.M508168200. [DOI] [PubMed] [Google Scholar]
- Tatham MH, Jaffray E, Vaughan OA, Desterro JM, Botting CH, Naismith JH, Hay RT. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001;276:35368–35374. doi: 10.1074/jbc.M104214200. [DOI] [PubMed] [Google Scholar]
- Tedeschi A, Nguyen T, Steele SU, Feil S, Naumann U, Feil R, Di Giovanni S. The tumor suppressor p53 transcriptionally regulates cGKI expression during neuronal maturation and is required for cGMP-dependent growth cone collapse. J. Neurosci. 2009;29:15155–15160. doi: 10.1523/JNEUROSCI.4416-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempe D, Piechaczyk M, Bossis G. SUMO under stress. Biochem. Soc. Trans. 2008;36:874–878. doi: 10.1042/BST0360874. [DOI] [PubMed] [Google Scholar]
- Terashima T, Kawai H, Fujitani M, Maeda K, Yasuda H. SUMO-1 co-localized with mutant atrophin-1 with expanded polyglutamines accelerates intranuclear aggregation and cell death. NeuroReport. 2002;13:2359–2364. doi: 10.1097/00001756-200212030-00038. [DOI] [PubMed] [Google Scholar]
- Ueda H, Goto J, Hashida H, Lin X, Oyanagi K, Kawano H, Zoghbi HY, Kanazawa I, Okazawa H. Enhanced SUMOylation in polyglutamine diseases. Biochem. Biophys. Res. Commun. 2002;293:307–313. doi: 10.1016/S0006-291X(02)00211-5. [DOI] [PubMed] [Google Scholar]
- Um JW, Chung KC. Functional modulation of parkin through physical interaction with SUMO-1. J. Neurosci. Res. 2006;84:1543–1554. doi: 10.1002/jnr.21041. [DOI] [PubMed] [Google Scholar]
- Um JW, Min DS, Rhim H, Kim J, Paik SR, Chung KC. Parkin ubiquitinates and promotes the degradation of RanBP2. J. Biol. Chem. 2006;281:3595–3603. doi: 10.1074/jbc.M504994200. [DOI] [PubMed] [Google Scholar]
- van Niekerk EA, Willis DE, Chang JH, Reumann K, Heise T, Twiss JL. Sumoylation in axons triggers retrograde transport of the RNA-binding protein La. Proc. Natl. Acad. Sci. USA. 2007;104:12913–12918. doi: 10.1073/pnas.0611562104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilatis DK, Hohmann JG, Zeng H, Li F, Ranchalis JE, Mortrud MT, Brown A, Rodriguez SS, Weller JR, Wright AC, Bergmann JE, Gaitanaris GA. The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. USA. 2003;100:4903–4908. doi: 10.1073/pnas.0230374100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Takahashi K, Tomizawa K, Mizusawa H, Takahashi H. Developmental regulation of Ubc9 in the rat nervous system. Acta Biochim. Pol. 2008;55:681–686. [PubMed] [Google Scholar]
- Watts FZ. SUMO modification of proteins other than transcription factors. Semin. Cell Dev. Biol. 2004;15:211–220. doi: 10.1016/j.semcdb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Nishimune A, Henley JM. Analysis of SUMO-1 modification of neuronal proteins containing consensus SUMOylation motifs. Neurosci. Lett. 2008;436:239–244. doi: 10.1016/j.neulet.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin SL, Cedervall T. The La protein. Annu. Rev. Biochem. 2002;71:375–403. doi: 10.1146/annurev.biochem.71.090501.150003. [DOI] [PubMed] [Google Scholar]
- Yang SH, Galanis A, Witty J, Sharrocks AD. An extended consensus motif enhances the specificity of substrate modification by SUMO. EMBO J. 2006;25:5083–5093. doi: 10.1038/sj.emboj.7601383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Sheng H, Homi HM, Warner DS, Paschen W. Cerebral ischemia/stroke and small ubiquitin-like modifier (SUMO) conjugation–a new target for therapeutic intervention? J. Neurochem. 2008a;106:989–999. doi: 10.1111/j.1471-4159.2008.05404.x. [DOI] [PubMed] [Google Scholar]
- Yang W, Sheng H, Warner DS, Paschen W. Transient focal cerebral ischemia induces a dramatic activation of small ubiquitin-like modifier conjugation. J. Cereb. Blood Flow Metab. 2008b;28:892–896. doi: 10.1038/sj.jcbfm.9600601. [DOI] [PubMed] [Google Scholar]
- Yazawa I, Nukina N, Hashida H, Goto J, Yamada M, Kanazawa I. Abnormal gene product identified in hereditary dentatorubral-pallidoluysian atrophy (DRPLA) brain. Nat. Genet. 1995;10:99–103. doi: 10.1038/ng0595-99. [DOI] [PubMed] [Google Scholar]
- Yeh ET. SUMOylation and De-SUMOylation: wrestling with life’s processes. J. Biol. Chem. 2009;284:8223–8227. doi: 10.1074/jbc.R800050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Sarge KD. Sumoylation of amyloid precursor protein negatively regulates Abeta aggregate levels. Biochem. Biophys. Res. Commun. 2008;374:673–678. doi: 10.1016/j.bbrc.2008.07.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- Zunino R, Braschi E, Xu L, McBride HM. Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J. Biol. Chem. 2009;284:17783–17795. doi: 10.1074/jbc.M901902200. [DOI] [PMC free article] [PubMed] [Google Scholar]