

Abstract
Quorum sensing, a bacterial cell–cell communication process, controls biofilm formation and virulence factor production in Vibrio cholerae, a human pathogen that causes the disease cholera. The major V. cholerae autoinducer is (S)-3-hydroxytridecan-4-one (CAI-1). A membrane bound two-component sensor histidine kinase called CqsS detects CAI-1, and the CqsS → LuxU → LuxO phosphorelay cascade transduces the information encoded in CAI-1 into the cell. Because the CAI-1 ligand is known and because the signalling circuit is simple, consisting of only three proteins, this system is ideal for analysing ligand regulation of a sensor histidine kinase. Here we reconstitute the CqsS → LuxU → LuxO phosphorylation cascade in vitro. We find that CAI-1 inhibits the initial auto-phosphorylation of CqsS whereas subsequent phosphotransfer steps and CqsS phosphatase activity are not CAI-1-controlled. CAI-1 binding to CqsS causes a conformational change that renders His194 in CqsS inaccessible to the CqsS catalytic domain. CqsS mutants with altered ligand detection specificities are faithfully controlled by their corresponding modified ligands in vitro. Likewise, pairing of agonists and antagonists allows in vitro assessment of their opposing activities. Our data are consistent with a two-state model for ligand control of histidine kinases.
Introduction
Two-component systems (TCS) are ubiquitous in bacteria and are used to adapt to environmental changes (Krell et al., 2010). A typical TCS system consists of a sensor histidine kinase and a response regulator (Casino et al., 2010). Sensor histidine kinases usually contain N-terminal transmembrane sensing domains, dimerization histidine phosphotransfer (DHp) domains and C-terminal catalytic ATP-binding (CA) domains (Dutta et al., 1999). In response to environmental signals, the CA domain catalyses the phosphorylation of a specific histidine residue in the DHp domain using ATP as the phospho-donor. This phosphoryl group is subsequently transferred to a particular aspartate residue in the receiver domain of the cognate response regulator, regulating its DNA binding activity, enzymatic activity or protein–protein interactions (Lupas and Stock, 1989; Weiss et al., 1992; Takai et al., 2006; Yoshida et al., 2006; Petrova and Sauer, 2009; Paul et al., 2011). Histidine kinases can also possess a phosphatase activity that removes the phosphoryl group from the response regulator (Igo et al., 1989; Yoshida et al., 2002; Huynh et al., 2010). Some of these sensory systems, called phosphorelay systems, include additional modules that exist as separate proteins or are contained within the sensor histidine kinases (Hoch, 2000). In phosphorelay systems, the phosphorylated residues are denoted H1 → D1 → H2 → D2. Following phosphorylation on the sensor histidine kinase (H1) and subsequent phosphotransfer to a response regulator module (D1), in a phosphorelay system, the phosphoryl group is sequentially transferred to a histidine on a histidine-containing phosphotransfer (HPt) module (H2), and finally to an aspartate on another response regulator module (D2). Presumably, the additional sites of phosphorylation increase the potential points at which regulation can occur or enable signal integration from multiple sensory pathways (Hoch, 2000).
We are interested in phosphorelay systems involved in quorum sensing (QS). QS is a bacterial cell–cell communication process that relies on the production, release and detection of extracellular signalling molecules called autoinducers (Ng and Bassler, 2009). This process enables populations of bacteria to synchronize gene expression in response to fluctuations in population density. QS ensures that collective behaviours such as bioluminescence, biofilm formation and virulence factor production are only performed at cell densities that enable them to be effective (Bassler et al., 1994; Miller et al., 2002; Greenberg, 2003). In Vibrio cholerae, two QS autoinducer/phosphorelay pathways, CAI-1/CqsS and AI-2/LuxPQ converge to regulate gene expression (Miller et al., 2002). The CAI-1/CqsS system, the focus of this study, is the major QS system in V. cholerae. The autoinducer CAI-1 is (S)-3-hydroxytridecan-4-one (Higgins et al., 2007; Wei et al., 2011). Detection of and response to CAI-1 relies on a phosphorelay system in which CqsS is the receptor for CAI-1 (Miller et al., 2002). The membrane bound sensor histidine kinase CqsS contains the typical transmembrane/sensing domain, DHp domain, and CA domain (H1). In addition, CqsS contains a C-terminal receiver domain (D1). At low cell density, when the concentration of CAI-1 is below the threshold for detection, CqsS functions as a kinase and auto-phosphorylation on His194 (H1) occurs. Following phosphotransfer to Asp618 (D1) on the CqsS receiver domain, the phosphoryl group is transferred to His58 on LuxU (H2), an HPt protein. Phosphorylated LuxU, in turn, transfers the phosphoryl group to Asp47 on the response regulator LuxO (D2) (Freeman and Bassler, 1999a, b). Phosphorylated LuxO activates the transcription of genes encoding four small regulatory RNAs Qrr1-4 (Lenz et al., 2004), which promote the translation of aphA and inhibit the translation of hapR (Lenz et al., 2004; Rutherford et al., 2011). AphA and HapR are the master quorum-sensing regulators in V. cholerae at low and high cell densities respectively (Fig. 1, left). Thus, at low cell density, AphA protein is made and HapR is not. AphA controls the regulon of genes involved in individual cell behaviours. At high cell density, when CAI-1 has accumulated, CqsS binds to CAI-1. This event switches CqsS to a phosphatase and reverses the flow of phosphate through the circuit resulting in dephosphorylation of LuxU and LuxO. Transcription of qrr1-4 ceases, which frees the hapR mRNA for translation and terminates the activation of translation of aphA mRNA. Thus, at high cell density, HapR protein is produced and AphA protein is not. HapR regulates downstream target genes that underpin group behaviours (Fig. 1, right). Traits controlled by QS in V. cholerae include biofilm formation and virulence factor production (Zhu et al., 2002; Hammer and Bassler, 2003).
Fig. 1.
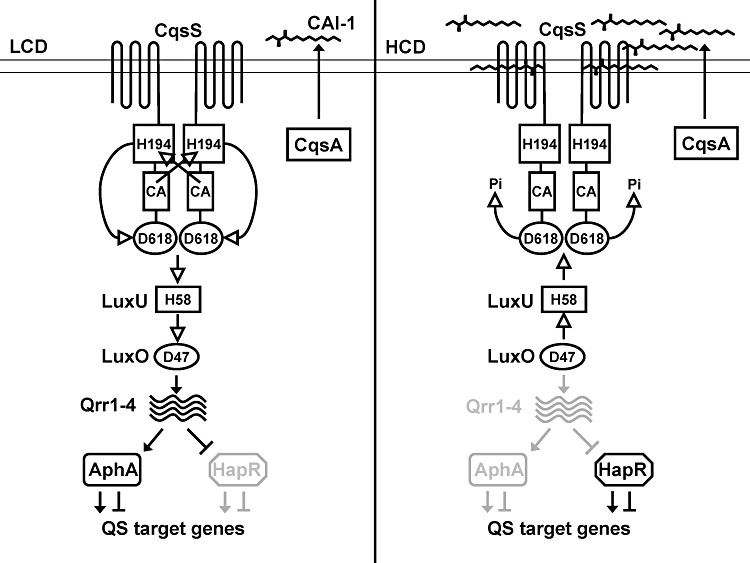
The V. cholerae CqsS/CAI-1 quorum-sensing phosphorelay system. CqsA synthesizes CAI-1, which is (S)-3-hydroxytridecan-4-one and CqsS is the CAI-1 receptor. At low cell density (LCD, left), when CAI-1 concentration is below the detection limit, CqsS functions as a kinase. Following auto-phosphorylation at His194, the phosphoryl group is transferred to Asp618 on the CqsS receiver domain. The next transfer is to His58 on LuxU. LuxU, in turn, transfers the phosphoryl group to Asp47 on LuxO. Once phosphorylated, LuxO activates the transcription of genes encoding four small regulatory RNAs called Qrr1-4. Qrr1-4 activates the translation of AphA and represses the translation of HapR. AphA regulates genes that are beneficial for individual behaviours. At high cell density (HCD, right), CAI-1 accumulates, binds CqsS, and switches CqsS to a phosphatase. Phospho-flow is reversed and LuxO is dephosphorylated. Qrr1-4 are not produced, and HapR protein is translated. HapR regulates genes that promote group behaviours. Open arrows denote phospho-flow and closed arrows denote gene regulation and CAI-1 synthesis. We note that the trans phosphorylation of CqsS His194 is shown for simplicity based on data from other TCS systems. We do not exclude the possibility of cis phosphorylation. In this study, the reverse phosphate flow from LuxU to CqsS that occurs at high cell density is termed the CqsS phosphatase activity for continuity with previous reports. We note that the canonical definition of phosphatase activity in a two-component system refers to that of a histidine kinase targeting the aspartyl-phosphate on the partner response regulator.
Elegant studies of bacterial TCSs have revealed their importance and many of their functions (Casino et al., 2009; Martin et al., 2009). However, biochemical studies of membrane bound sensor histidine kinases like CqsS have been plagued by the following issues. First, the identities of the ligands for most TCSs are not known. Thus, it has not been possible to study ligand regulation of these sensory circuits in vitro. Second, structural studies of membrane bound proteins are notoriously difficult and therefore structural analyses of TCS have been limited to sensors with well-defined periplasmic domains (Neiditch et al., 2005; 2006; Cheung and Hendrickson, 2010; Tajima et al., 2011). The CqsS pathway provides an excellent opportunity for detailed biochemical studies because the ligand CAI-1 is known, CAI-1 is membrane permeable making it possible to study the receptor–ligand interactions in membrane vesicles, and because the circuit is simple; only three proteins are involved, CqsS, LuxU and LuxO. In this report, we reconstitute the complete CqsS QS phosphorylation cascade with full-length membrane bound CqsS receptor and purified LuxU and LuxO and we show that the circuit is controlled by CAI-1. Phosphorylation of His194 on CqsS (H1), rather than phosphotransfer or phosphatase activity is the step that CAI-1 controls. CAI-1 functions by rendering His194 inaccessible to the CA domain. We also characterize antagonist action using this in vitro system. The signal transduction events we quantified using this reconstituted system can be explained by a two-state theoretical framework for histidine kinases.
Results
Reconstitution of the CqsS phosphorylation cascade in vitro
In order to investigate how CAI-1 binding regulates QS signalling events, we undertook a biochemical approach to reconstitute the CqsS → LuxU → LuxO circuit in vitro. We cloned and overexpressed the CqsS receptor in recombinant Escherichia coli, and examined in vitro auto-phosphorylation of CqsS (H1) using inverted membrane vesicles and [γ-32P] ATP. To verify the prediction that His194 in the DHp domain is the site of phosphorylation, we constructed a CqsS H194Q mutant (called CqsS His-). In our assay, wild-type CqsS exhibits auto-phosphorylation; however, CqsS His- does not (Fig. 2A, top row). Several conserved glycine residues in the CA domain are predicted to be critical for ATP binding. To test this assumption, the mutant CqsS G379A/G381A (called CqsS Cat-) was constructed and it too is incapable of auto-phosphorylation (Fig. 2A, top row). Finally, and also as expected, when the predicted phosphoryl receiving aspartate residue Asp618 in the attached receiver domain (D1) is mutated to Asn, the resulting mutant CqsS D618N (CqsS Asp-) remains capable of auto-phosphorylation (Fig. 2A, top row).
Fig. 2.
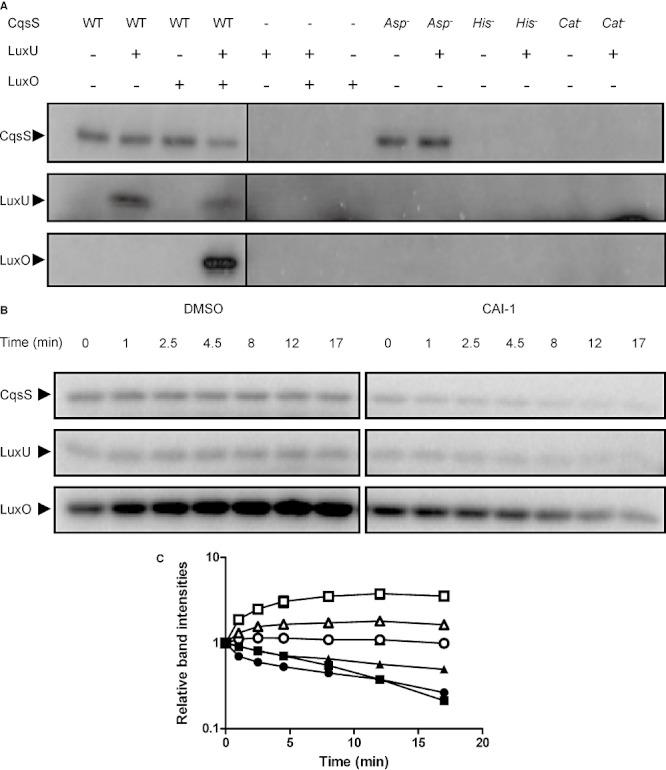
In vitro reconstitution of the CqsS → LuxU → LuxO phosphorylation cascade. A. Auto-phosphorylation of CqsS and phosphotransfer to LuxU and to LuxO were assayed with wild-type CqsS, CqsS His-, CqsS Cat- and CqsS Asp-. All phosphorylated proteins were run on the same gel and extracted. Only the relevant regions are shown for simplicity. B. A phosphorylation reaction was carried out with CqsS, LuxU and LuxO for 1 min and divided in half. One half was supplemented with DMSO (left) and the other with 500 µM CAI-1 (right). Samples were taken at the indicated time points. The top, middle and bottom rows show the phosphorylated CqsS, LuxU and LuxO proteins respectively. C. Experiments in (B) were performed in duplicate. Band intensities for CqsS∼P (circles), LuxU∼P (triangles) and LuxO∼P (squares) when DMSO was added (open symbols) and when CAI-1 was added (closed symbols) were normalized to the time zero level of each protein.
Genetic analyses imply that, at low cell density, CqsS is a kinase that transfers the phosphoryl group to the HPt protein LuxU, and LuxU, in turn, transfers the phosphoryl group to the response regulator LuxO. To examine this in vitro, LuxU and LuxO were overexpressed and purified from E. coli. LuxU phosphorylation occurred when both CqsS and LuxU proteins were present and no LuxU phosphorylation occurred in the absence of CqsS (Fig. 2A, middle row). Neither the CqsS His- nor the CqsS Cat- protein could transfer phosphoryl groups to LuxU. Likewise, the CqsS Asp- mutant, although active for auto-phosphorylation, does not transfer the phosphoryl group to LuxU, presumably due to the participation of Asp618 in the phosphorelay shuttle (Freeman et al., 2000) (Fig. 2A, middle row). We next turned our attention to LuxO. LuxO phosphorylation was observed only when wild-type CqsS, LuxU and LuxO proteins were present together (Fig. 2A, bottom row and Fig. S1). Thus, CqsS cannot transfer phosphoryl groups to LuxO directly; LuxU is required. Exactly analogous to what we found for LuxU, defects in any of the key CqsS residues (CqsS His-, CqsS Cat- and CqsS Asp-) eliminated phosphotransfer to LuxO (Fig. S1).
These preliminary experiments establish the CqsS → LuxU → LuxO pathway, putting us in position to examine whether we could exploit having the CAI-1 ligand in hand to investigate what happens to a sensor histidine kinase when it encounters its stimulus. Genetic evidence indicates that CAI-1 binding switches CqsS from a kinase to a phosphatase, resulting in the dephosphorylation of phosphorylated LuxU (LuxU∼P) and phosphorylated LuxO (LuxO∼P). To examine if this switch can occur in vitro, we incubated CqsS, LuxU, LuxO and radioactive ATP together for 1 min. The reaction was then divided in half and either DMSO or CAI-1 was added. In the experiment in which DMSO was supplied, the CqsS∼P did not change while LuxU∼P and LuxO∼P levels increased initially and then remained constant over time, indicating continuous phospho-flow from CqsS to LuxU and to LuxO until the system reached steady state (Fig. 2B, left, Fig. 2C). However, the levels of all of the phosphorylated proteins decreased dramatically when CAI-1 was supplied (Fig. 2B, right, Fig. 2C), indicating that CAI-1 transforms CqsS from a kinase to a phosphatase. These results are not due to the instability of LuxU∼P and/or LuxO∼P because both LuxU∼P and LuxO∼P are stable (half life > 10 min) in our experimental set-up. Also, the CAI-1 molecule does not destabilize LuxU∼P or LuxO∼P (Fig. S2).
Receptor-ligand specificities are maintained in vitro
We wondered if the above result is specific to the CAI-1 ligand or if any ligand could convert recombinant CqsS from a kinase to a phosphatase. To examine this, we tested decanoic acid, the Vibrio harveyi autoinducer HAI-1, and AI-2 for the ability to convert CqsS to a phosphatase. Decanoic acid resembles the fatty acid tail of CAI-1 but lacks the α-hydroxy ketone head group. HAI-1 is the major V. harveyi autoinducer and AI-2 is the autoinducer of the V. cholerae LuxPQ QS pathway (Chen et al., 2002; Henke and Bassler, 2004). HAI-1 and AI-2, while being autoinducers, do not share structural similarity to CAI-1. None of these molecules decreased the levels of CqsS∼P or LuxU∼P (Fig. 3A), indicating that CqsS remains a kinase in the presence of these molecules, and therefore CAI-1 modulation of CqsS kinase activity is specific.
Fig. 3.
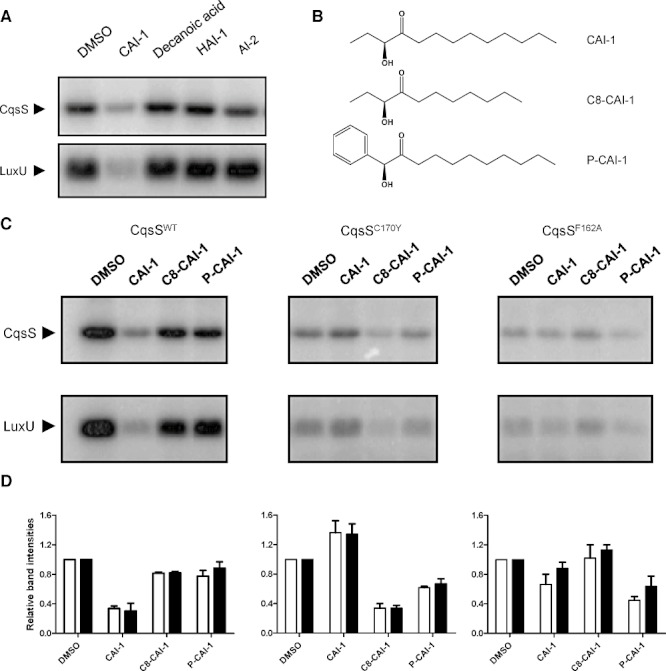
Receptor-ligand specificities are maintained in vitro. A. CqsS and LuxU phosphorylation were examined in the presence of DMSO or 100 µM CAI-1, Decanoic acid, HAI-1 (3-hydroxy-C4-HSL), or AI-2 [(2S,4S)-2-methyl-2,3,4-tetrahydroxytetrahydrofuran-borate]. Both phosphorylated proteins were run on the same gel and extracted. Only the relevant regions are shown for simplicity. B. Structures of CAI-1, C8-CAI-1 and P-CAI-1. C. CqsS auto-phosphorylation and phosphotransfer to LuxU were examined with the wild-type CqsS receptor (left), the CqsSC170Y (middle) and the CqsSF162A (right) mutant receptors in the presence of DMSO or 100 µM of CAI-1, C8-CAI-1 or P-CAI-1. Reactions were carried out for 30 s. Both phosphorylated proteins were run on the same gel and extracted. Only the relevant regions are shown for simplicity. D. Experiments in (C) were performed in duplicate and band intensities were normalized to the respective DMSO control in each set of experiments. White bars and black bars show the quantification for CqsS and LuxU respectively.
In a previous report, we identified ‘gatekeeper’ residues in the CqsS transmembrane domain that constrain the CAI-1 structure allowable for agonism (i.e. conversion of CqsS from a kinase to a phosphatase) (Ng et al., 2010). Specifically, the CqsSC170Y receptor is not agonized by CAI-1. However, C8-CAI-1 (Fig. 3B), which has a carbon tail that is two carbons shorter than CAI-1, is a CqsSC170Y agonist. The CqsSF162A receptor also does not respond to CAI-1. Rather, it responds to Phenyl-CAI-1 (P-CAI-1) (Fig. 3B), a synthetic molecule with an enlarged head group compared to CAI-1. To test if these receptor-ligand specificities are maintained in vitro, inverted membranes containing the mutant CqsSC170Y and CqsSF162A receptors were prepared. CqsS phosphorylation and phosphotransfer to LuxU were examined with DMSO, CAI-1, C8-CAI-1 or P-CAI-1. Consistent with the results in Figs 2 and 3A, the wild-type receptor was converted from a kinase to a phosphatase by CAI-1, as indicated by decreased CqsS∼P and LuxU∼P compared with the DMSO control (Fig. 3C and D, left). We note that modest CqsS phosphorylation and phosphotransfer to LuxU does occur in the presence of CAI-1, indicating that there remains residual kinase activity despite ligand binding. This result is consistent with the observation that, in vivo, even at high cell density, when CAI-1 concentration is high, low levels of qrr1-4 transcripts can be detected (Rutherford et al., 2011). In contrast to CAI-1, neither C8-CAI-1 nor P-CAI-1 reduced the wild-type CqsS∼P and LuxU∼P levels compared with the DMSO control (Fig. 3C, left). These results show that, at the concentrations tested, neither C8-CAI-1 nor P-CAI-1 functions as an agonist of the wild-type CqsS receptor. At higher concentrations, C8-CAI-1 does reduce the wild-type CqsS∼P and LuxU∼P levels (see below). Thus, C8-CAI-1 is a weak agonist of CqsS, consistent with earlier genetic studies (Ng et al., 2011).
While the wild-type receptor does not respond to the CAI-1 analogues, this is not the case for the receptors containing the gate keeper mutations (Fig. 3C and D, middle and right). Whereas CqsS phosphorylation and LuxU phosphorylation are weaker overall for the mutant CqsSC170Y and CqsSF162A receptors compared with the wild-type receptor, their ligand specificities can nonetheless be examined. In the case of CqsSC170Y, CAI-1 did not significantly alter CqsSC170Y phosphorylation and LuxU phosphorylation. However, when C8-CAI-1 was added, CqsSC170Y phosphorylation and LuxU phosphorylation decreased (Fig. 3C and D, middle). Surprisingly, P-CAI-1 inhibited CqsSC170Y phosphorylation and LuxU phosphorylation but to a lesser extent than C8-CAI-1. This weak P-CAI-1 agonist activity towards the CqsSC170Y receptor has not been observed previously; however, lower concentrations were used in the earlier in vivo experiments. These results show that C8-CAI-1, but not CAI-1, functions as a strong agonist of the CqsSC170Y receptor and P-CAI-1 functions as a weak agonist. Regarding the CqsSF162A receptor, CqsSF162A phosphorylation and LuxU phosphorylation decreased only in the presence of P-CAI-1 but not when CAI-1 or C8-CAI-1 was added (Fig. 3C and D, right), indicating that CqsSF162A is specific for P-CAI-1. Thus, the specific in vivo interactions between ligands and receptor transmembrane sensing domains and their effects on signal transduction are maintained in vitro.
CqsS antagonism occurs in vitro
We wondered if we could adapt our in vitro system to study antagonism of the CqsS receptor. In vivo, P-CAI-1 antagonizes the CAI-1 activity of wild-type CqsS. When both CAI-1 and P-CAI-1 were added to membranes containing wild-type CqsS, phosphorylation of CqsS and LuxU were stronger than when CAI-1 was present alone. This result shows that the CAI-1 agonist activity is antagonized by P-CAI-1 (Fig. 4A and B, left). To confirm that antagonism in vitro applies to more than one pair of molecules and a single receptor, we expanded our investigation to the CqsSC170Y receptor. As a reminder, the CqsSC170Y receptor is agonized by C8-CAI-1 and antagonized by CAI-1 (Ng et al., 2010). This holds true in vitro: phosphorylation of CqsSC170Y and LuxU is significantly stronger in the presence of both CAI-1 and C8-CAI-1 compared with C8-CAI-1 alone (Fig. 4A and B, middle).
Fig. 4.
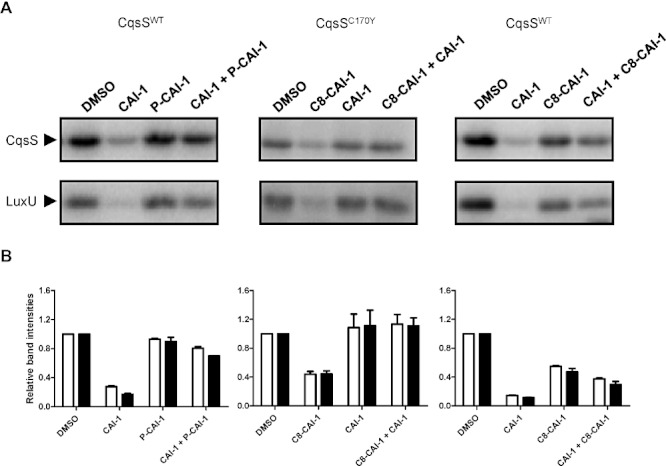
CqsS antagonism occurs in vitro. Antagonism was examined with the CqsS wild-type receptor (left and right) and the CqsSC170Y mutant receptor (middle). Reactions containing LuxU and either wild-type CqsS or CqsSC170Y were supplemented with the ligand(s) indicated above each lane. Agonists were present at 10 µM while antagonists were present at 100 µM (left and middle) or 500 µM (right). Reactions were carried out for 30 s. Both phosphorylated proteins were run on the same gel and extracted. Only the relevant regions are shown for simplicity. B. Experiments in (A) were performed in duplicate and band intensities were normalized to the respective DMSO control in each set of experiments. White bars and black bars show the quantification for CqsS and LuxU respectively.
We have previously proposed a theoretical two-state model for histidine kinases (Swem et al., 2008). In our model, the kinase domain of a receptor like CqsS exists in two states, a ‘kinase on’ state and a ‘kinase off’ state (Ng et al., 2010). Agonists, like CAI-1, preferentially bind and stabilize the ‘kinase off’ state. Because CqsS also possesses phosphatase activity, it appears to be a phosphatase in the presence of an agonist such as CAI-1. A weak agonist also prefers to bind and stabilize the ‘kinase off’ state. However, a weak agonist exhibits reduced preference for the ‘kinase off’ state compared with a strong agonist. The two-state model predicts that when a weak agonist and a strong agonist are present together, the weak agonist will functionally behave as an antagonist due to competition for the ligand binding site. In such a scenario, the receptor will have a higher probability to be in the ‘kinase on’ state when both molecules are present, compared with when the stronger agonist is present alone. Having CqsS functioning in vitro and a set of agonists with different potencies allows us to explicitly test our model in vitro. First, when high concentration of C8-CAI-1 was applied, decreased CqsS and LuxU phosphorylation were observed, consistent with its weak agonist identity (Fig. 4A and B, right). Next, we combined C8-CAI-1, a weak agonist of CqsS, with CAI-1, a strong agonist of CqsS, and examined CqsS and LuxU phosphorylation. When C8-CAI-1 was present together with CAI-1, CAI-1-mediated inhibition of phosphorylation of CqsS and LuxU was reduced (Fig. 4, right). Therefore, C8-CAI-1 is a weak agonist of CqsS when it acts alone but it can also act as an antagonist in the presence of CAI-1, validating the predictions of the theoretical two-state model.
CAI-1 regulates CqsS His194 auto-phosphorylation
CAI-1 driven kinase inhibition of CqsS could be due to regulation of CqsS His194 auto-phosphorylation, phosphotransfer to Asp618, phosphotransfer from Asp618 to LuxU His58, or CAI-1 could control the CqsS phosphatase activity. To pinpoint which step(s) is controlled by CAI-1, we first tested if His194 auto-phosphorylation is regulated by CAI-1 using the CqsS Asp- receptor construct. Our rationale is that the CqsS Asp- construct can auto-phosphorylate but all subsequent phosphotransfer processes are eliminated. Thus, this construct allows us to exclusively examine the dependence of His194 auto-phosphorylation on CAI-1. Rapid auto-phosphorylation of the CqsS Asp- mutant construct occurred when it was incubated with [γ-32P] ATP, and maximum auto-phosphorylation was achieved at 1 min (Fig. 5A and B). Addition of CAI-1 decreased the initial phosphorylation rate sixfold (Fig. 5A and B). These results show that His194 auto-phosphorylation is indeed regulated by CAI-1. This result does not preclude CAI-1 regulation of other phosphotransfer events, and we address this in the subsequent sections.
Fig. 5.
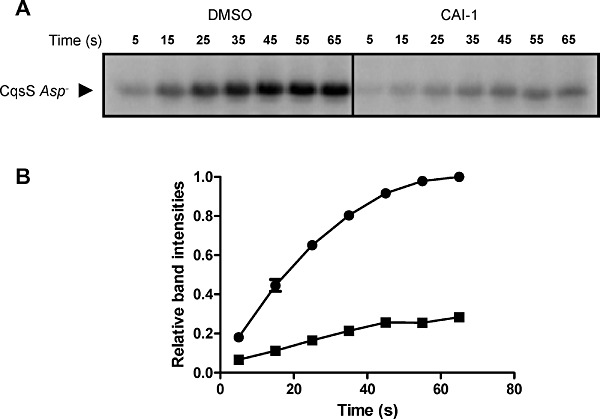
CAI-1 regulates CqsS auto-phosphorylation. A. CqsS auto-phosphorylation was examined using the CqsS Asp- mutant construct in the presence of DMSO (left) or 500 µM CAI-1 (right). Samples were taken at the indicated time points. B. Experiments in (A) were performed in duplicate. Band intensities from the DMSO control (circles) and CAI-1 treated (squares) samples were quantified and normalized to the 65 s time point band from the DMSO control in each experiment.
Phosphotransfer processes are not affected by CAI-1
In addition to this first phosphorylation step, downstream phosphotransfer processes (His194∼P to Asp618 within CqsS and/or CqsS Asp618∼P to LuxU His58) could also be inhibited by CAI-1 binding. We reason that if His194 auto-phosphorylation is the only step that is controlled by CAI-1, then CAI-1 inhibition of LuxU His58 phosphorylation should track with inhibition of CqsS His194 auto-phosphorylation. By contrast, if either or both of the subsequent phosphotransfer steps, His194∼P to Asp618 in CqsS or CqsS Asp618∼P to LuxU His58, is CAI-1 regulated, following CAI-1 addition, inhibition of LuxU His58 phosphorylation should be more severe than inhibition of CqsS His194 auto-phosphorylation. To assess this, we compared the extent of CqsS His194 auto-phosphorylation in the CqsS Asp- construct with that of LuxU phosphorylation by the wild-type CqsS receptor. When different concentrations of CAI-1 were added, both His194∼P on the CqsS Asp- construct and phosphotransfer to His58∼P on LuxU from wild-type CqsS were inhibited to the same extent (Fig. 6). These results suggest that CAI-1 does not affect phosphotransfer processes other than the initial His194 auto-phosphorylation.
Fig. 6.
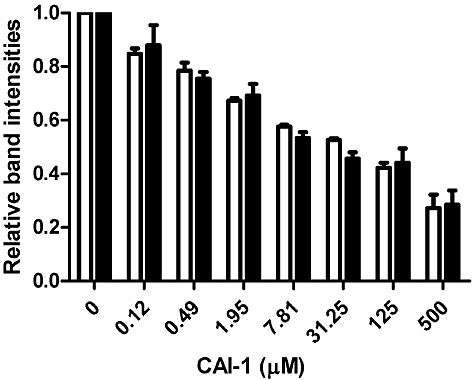
Phosphotransfer steps subsequent to CqsS auto-phosphorylation are not CAI-1 regulated. Auto-phosphorylation of His194 on CqsS Asp- and phosphotransfer from wild-type CqsS to LuxU were measured in the presence of various concentrations of CAI-1. Reactions were carried out for 30 s. Experiments were performed in duplicate and band intensities of phosphorylated CqsS Asp- (white bars) and LuxU (black bars) were quantified and normalized to their respective DMSO controls.
CqsS phosphatase activity is independent of CAI-1
In addition to kinase activity, CqsS possesses a phosphatase activity that dephosphorylates LuxU∼P. We tested if this activity is regulated by CAI-1. To obtain LuxU∼P, LuxU was first phosphorylated by CqsS. Subsequently, CqsS was removed by ultracentrifugation and ATP was removed by gel filtration. To determine which region of CqsS contains the phosphatase activity, we assayed LuxU∼P dephosphorylation by CqsS His-, CqsS Cat- and CqsS Asp-. Both CqsS His- and CqsS Cat- could dephosphorylate LuxU∼P, indicating that His194 or the CqsS catalytic site is not required for CqsS phosphatase activity (Fig. S3). By contrast, CqsS Asp- was incapable of dephosphorylation of LuxU∼P, suggesting that Asp618 in the CqsS receiver domain is essential for the CqsS phosphatase activity (Fig. S3). To test for CAI-1 regulation of phosphatase activity, the wild-type CqsS was used to dephosphorylate LuxU∼P in the presence and absence of CAI-1. There was no difference between dephosphorylation of LuxU∼P with and without CAI-1 (Fig. 7), demonstrating that CAI-1 does not regulate the CqsS phosphatase activity.
Fig. 7.
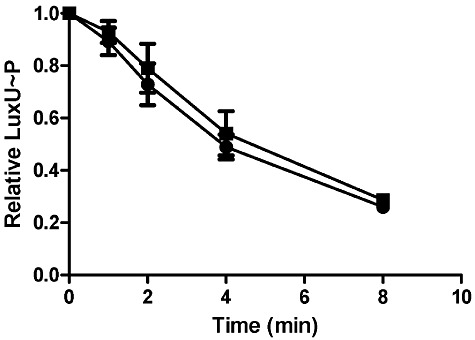
The CqsS phosphatase activity is not regulated by CAI-1. Phosphorylated LuxU was purified and divided in half. DMSO (circles) or 500 µM CAI-1 (squares) was added to one half of the sample. CqsS was added to both reaction mixtures at time zero and aliquots were subsequently removed at the indicated time points. Experiments were performed in duplicate and band intensities were normalized to the time zero band from each experiment.
CAI-1 binding causes a conformational change
To explore the mechanism by which CAI-1 binding inhibits His194 auto-phosphorylation, we relied on a phenomenon discovered in studies of the chemotaxis TCS system showing that the phosphorylated histidine residue of the CheA sensor kinase could be dephosphorylated by ADP (Tawa and Stewart, 1994). Thus, in CheA, the His∼P is accessible to ADP. We investigated whether CqsS His194∼P is also capable of being dephosphorylated by ADP and whether CAI-1 binding alters this reaction. To do this, we used the CqsS Asp- construct because it is functional for the initial His194 auto-phosphorylation step but further phosphotransfer processes do not occur (Fig. 2A). Following auto-phosphorylation, the 32P labelled His194∼P rapidly decreased when ADP was added, showing that CqsS His194 is dephosphorylated by ADP (Fig. 8). However, 32P labelled His194∼P decreased more slowly when CAI-1 was added together with ADP, indicating that dephosphorylation of His∼P is inhibited by CAI-1 (Fig. 8). In the presence of EDTA, the His∼P remained stable when either CAI-1 or DMSO was included (Fig. 8), indicating the importance of Mg2+ and presumably the ATP binding domain, for the dephosphorylation process to occur. We conclude that CAI-1 binding to the CqsS transmembrane domain results in a conformational change that protects the His194∼P from being accessed by the catalytic ATP binding domain.
Fig. 8.
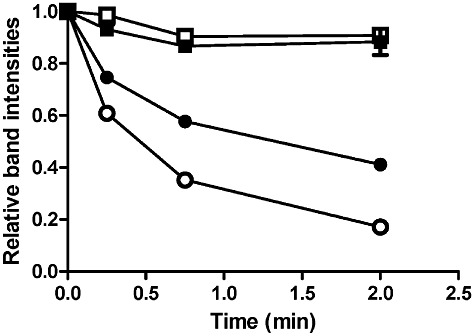
CAI-1 affects dephosphorylation of CqsS His∼P by ADP. 100 µM ADP was added to phosphorylated CqsS Asp- together with DMSO (open circles) or 500 µM CAI-1 (closed circles). At that time of ADP and ligand addition, 1 mM ATP was also added to terminate new labelling. Aliquots were removed at the indicated time points. The same experiment was performed with 100 µM EDTA present together with DMSO (open squares) or CAI-1 (closed squares). Experiments were performed in duplicate and band intensities were normalized to the time zero band from each experiment.
Discussion
Bacterial TCSs play crucial roles in environmental adaptation. Although many model TCSs are well established, how their activities are controlled by stimuli has not, in general, been rigorously investigated, primarily due to the profound lack of identified ligands and the difficulties encountered in biochemical and structural studies of membrane spanning proteins (Lee et al., 1999; Timmen et al., 2006; Cheung and Hendrickson, 2010). In addition, TCSs containing many components are difficult to reconstitute in vitro. Even the paradigmatic bacterial chemotaxis TCS has not been examined in a completely reconstituted system in vitro. This is due to the fact that the membrane bound chemotaxis receptors (e.g. Tar) are not histidine kinases; rather, these receptors regulate the activity of the soluble histidine kinase CheA (Hazelbauer and Lai, 2010). Second, the chemotaxis system relies on the specific organization of the receptors in arrays (Webre et al., 2004). Third, chemotaxis signalling is subject to multiple types of regulation such as methylation of the receptor by CheR, demethylation by CheB, and dephosphorylation of the response regulator CheY by the phosphatase CheZ (Ninfa et al., 1991). In V. cholerae, by contrast, QS relies on a three protein phosphorelay system to detect the major autoinducer CAI-1 (Miller et al., 2002). Moreover, the structure of the ligand CAI-1 is known, and CAI-1 and analogues are readily available (Kelly et al., 2009; Ng et al., 2010; Bolitho et al., 2011; Wei et al., 2011). Finally, a panel of CqsS ‘gatekeeper’ mutants, with altered ligand specificities, has been engineered (Ng et al., 2010). Thus, the CqsS system is tractable to mechanistic dissection in vitro. Using inverted E. coli membranes containing the full-length CqsS receptor together with purified downstream protein components, we were able to reconstitute the V. cholerae QS phosphorylation pathway in vitro. The complete phosphorylation cascade was established from CqsS, the hybrid sensor histidine kinase, through LuxU, the HPt protein, to LuxO, the response regulator. Our set of sensing mutants and ligand analogues allowed us to discover that ligand-receptor specificities are maintained in vitro, and that CAI-1 binding regulates His194 auto-phosphorylation, whereas phosphotransfer and phosphatase activity are not affected by ligand binding.
It is interesting that QS relies on a phosphorelay system rather than a simple TCS system. We speculate that having the HPt protein LuxU provides a hub for signal integration. Indeed, V. cholerae QS relies on two parallel sensory pathways. In addition to the CAI-1/CqsS pathway, input from the AI-2/LuxPQ sensory pathway converges with that from CAI-1/CqsS at LuxU (Miller et al., 2002). Likewise, in the related bacterium V. harveyi, information from three hybrid sensor histidine kinases, CqsS, LuxPQ and LuxN all converge at V. harveyi LuxU (Henke and Bassler, 2004). The particular domain architecture of the Vibrio QS pathway ensures that the enzymatic activities required for phosphotransfer to and dephosphorylation of LuxU are colocalized within the same protein (CqsS, LuxQ and LuxN). Therefore, modulation of receptor levels potentially regulates phospho-flow in both directions. Feed-back regulation of QS receptor levels has been demonstrated in V. harveyi (Teng et al., 2011). We speculate that V. cholerae CqsS could also be subject to similar feed-back regulation.
Proteins like CqsS that contain both the H1 and D1 modules are common in bacteria. Some other bacterial hybrid histidine kinases have domain arrangements similar to ArcB and BvgS, which contain the H1, D1 and the H2 domains (Iuchi et al., 1990; Uhl and Miller, 1996). ArcB responds to the redox state of the membrane and anaerobic metabolites, while BvgS responds to temperature, SO42–, and nicotinic acid. CqsS, interestingly, resembles the domain architectures of sensory proteins present in eukaryotic organisms. In Arabidopsis thaliana, over 10 sensor histidine kinases have been identified, all of which contain the histidine kinase domain (H1) and an attached receiver domain (D1) (Mizuno, 2005). While most of these sensor histidine kinases have unknown functions, interestingly, in A. thaliana, detection of ethylene, a plant hormone that controls growth and development, relies on the hybrid sensor histidine kinase ETR1 that resembles CqsS (Voet-van-Vormizeele and Groth, 2008; Kim et al., 2011). Response to another A. thaliana master growth hormone, cytokinin, requires a phosphorelay system consisting of several hybrid sensor histidine kinases CRE1, AHK2 and AHK3 (H1, D1), multiple HPt proteins called APHs (H2), and multiple response regulators called ARRs (D2) (Inoue et al., 2001; Sheen, 2002). It is striking to us that ‘hormone-like’ signalling molecules in both bacteria and plants are detected by similarly arranged sensory systems. We wonder if a fundamental advantage of these systems is in signal integration. HPt proteins linking sensor histidine kinases and response regulators might serve as ideal merging points for inputs from multiple receptors.
Our work shows that while histidine auto-phosphorylation is inhibited by CAI-1, the CqsS phosphatase activity is not regulated by the ligand. Having a constant CqsS phosphatase activity could be crucial for properly timed QS transitions. During the low cell density to high cell density transition, although CAI-1 inhibition of the CqsS kinase ensures that little new LuxO∼P is generated, existing LuxO∼P could still activate transcription, due to its high stability (Fig. S2). However, because CqsS possesses a constant phosphatase activity, it ensures rapid dephosphorylation of any existing LuxO∼P. During the high cell density to low cell density transition, the constant CqsS phosphatase activity would not significantly affect LuxO∼P generation due to the much stronger CqsS kinase activity. Thus, rapid phosphorylation of LuxU and LuxO occurs when CAI-1 disappears.
His194 auto-phosphorylation is inhibited by CAI-1. This result is consistent with previously proposed models suggesting that TCS binding of ligands triggers downstream conformational changes in sensor histidine kinases that affect interactions between the catalytic domains and the histidines in the DHp domains (Borkovich and Simon, 1990; Neiditch et al., 2006). We verified this idea by showing that dephosphorylation of His194∼P by ADP is slower in the presence of CAI-1, suggesting that binding of CAI-1 in the transmembrane domain causes a conformational change that repositions the His194 on the DHp domain away from the catalytic domain. Therefore, in the presence of CAI-1, new rounds of phosphorylation are inhibited. Additionally, already phosphorylated His194 also becomes less accessible to the catalytic domain, resulting in its higher stability compared with in the absence of the ligand.
Our previous structural study of an analogous QS receptor, LuxPQ, suggests that upon ligand binding, a symmetry-breaking conformational change occurs in the periplasm that is transduced across the membrane to alter the relative positions of the histidine in the DHp domain and the catalytic domain active site (Neiditch et al., 2006). However, in the LuxPQ case, two proteins are required for transducing the AI-2 autoinducer signal. LuxP, the periplasmic autoinducer binding protein, binds AI-2 and induces the conformational change in the partner hybrid sensor kinase LuxQ through protein–protein interactions (Neiditch et al., 2005; 2006). Extensive studies with CheA have led to a similar model (Borkovich and Simon, 1990). In the CheA case, the chemotaxis receptor Tar is membrane associated and it regulates the cytoplasmic kinase CheA. When the ligand binds to the Tar receptor, it is proposed to induce a ‘closed’ confirmation of the CheA kinase. It is curious to us that three distinct TCSs with three different modular domain architectures use a related mechanism for regulating kinase activity. Evolution has produced a variety of domain arrangements for histidine kinases in TCSs, yet; at least in those cases examined, the underlying regulation mechanism seems to be conserved.
In our current study, P-CAI-1, a CqsS antagonist, also antagonizes CqsS in the presence of CAI-1 in vitro. In addition, C8-CAI-1, a weak agonist of CqsS, also acts as an antagonist of CqsS in the presence of CAI-1 in vitro. This result is consistent with our two-state model (see Results). Competition between a weak agonist and a strong agonist for the ligand binding pocket results in an intermediate effect rather than a synergistic effect. Antagonism can also be replicated in vitro with the CqsSC170Y mutant receptor, which possesses altered specificity for ligands. In vitro, C8-CAI-1 is a strong agonist and CAI-1, which does not affect CqsSC170Y when acting alone, is an antagonist of the CqsSC170Y receptor. Therefore, CAI-1 likely binds and stabilizes the ‘kinase on’ and ‘kinase off’ states of CqsSC170Y receptor with equal preference, and thus appears neutral in the absence of other molecules. Alternatively, to function as an antagonist, CAI-1 could prefer to bind and stabilize the ‘kinase on’ state of CqsSC170Y. In either case, CAI-1 certainly competes for the binding site with C8-CAI-1 and therefore acts functionally as an antagonist. Thus, predictions of the two-state theoretical model are consistent with the results of the experiments using our in vitro phosphorylation system.
Bacteria exist in niches containing complex microbial-species compositions. Thus, the chemical environments they encounter contain numerous signalling cues, including many classes of autoinducers, the concentrations of which change in time and in space. In a simplified system, we have examined receptor histidine kinase regulation by a set of agonists and antagonists. To the best of our knowledge, this study shows, for the first time, that histidine auto-phosphorylation of the sensor is the only step that is regulated by the ligands. Structural studies of apo-CqsS and the CqsS receptor bound to agonists and antagonists should reveal the conformational changes induced by these ligands. Additionally, future work with reconstituted circuits that contain both the CqsS and LuxPQ sensors will be useful to understand QS signal integration.
Experimental procedures
Cloning of CqsS, LuxU and LuxO
DNA manipulations were performed using standard methods (Sambrook et al., 1989). The gene encoding CqsS was PCR amplified from V. cholerae genomic DNA and cloned into the pET21b vector. Plasmids encoding CqsS H194Q (CqsS His-), CqsS D618N (CqsS Asp-), CqsS G379A/G381A (CqsS Cat-), CqsSC170Y and CqsSF162A were constructed from the plasmid containing wild-type CqsS using the Quikchange II XL Site-Directed Mutagenesis Kit (Stratagene). The genes encoding LuxU and LuxO were PCR amplified from V. cholerae genomic DNA and cloned into the pET28b vector. We call the LuxO site of phosphorylation D47 to be consistent with earlier publications and to match reported nomenclature of mutants. As part of the present work, we discovered the in vivo LuxO translational start site and found it is 14 codons 5′ to the previously reported start site. Thus, the phosphorylated Asp residue is amino acid 61 in the LuxO protein.
Preparation of inverted membranes
Escherichia coli BL21 (DE3) harbouring plasmids encoding wild type or mutant CqsS constructs were grown with shaking in Luria–Bertani (LB) with 100 µg ml−1 kanamycin at 37°C. Overnight bacterial cultures were diluted 1:100 into fresh LB medium with kanamycin. After growth with aeration for 3 h at 37°C, protein production was induced with 1 mM IPTG. The cultures were shifted to room temperature and grown for an additional 5 h with shaking. Cells were harvested by centrifugation, resuspended in lysis buffer (50 mM Tris pH 8.0, 200 mM NaCl, 5 mM β-mercaptoethanol, 5 mM MgCl2 and 20 mM imidazole), and lysed under 15 000 psi. Cell lysates were centrifuged at 9300 g for 30 min and the cleared fluids were harvested and subjected to ultra-centrifugation at 180 000 g for 1 h. Membrane pellets were resuspended in kinase buffer [50 mM Tris pH 8.0, 100 mM KCl, 5 mM MgCl2, and 10% (v/v) glycerol] and quantified by SDS-PAGE and Western blot using CqsS purified in detergent as the standard.
Purification of LuxU and LuxO
Escherichia coli BL21 (DE3) containing plasmids encoding LuxU or LuxO were grown and lysed using the above conditions. Cell lysates were centrifuged at 21 000 g for 30 min and the cleared fluids were harvested and passed through 5 ml Hi-Trap Chelating columns (GE healthcare) pre-charged with Ni2+ ion. Following a wash with 30 ml lysis buffer, proteins were eluted with lysis buffer except that imidazole was included at 500 mM. Proteins were examined by SDS-PAGE followed by Coomassie Brilliant Blue staining and found to be ∼ 95% pure. Purified proteins were dialysed against lysis buffer lacking imidazole and concentrated. Protein concentrations were determined using the Bio-rad protein assay.
Phosphorylation assays
Phosphorylation assays were performed with inverted membranes containing 2 µM wild-type CqsS or CqsS mutant proteins. In assays containing LuxU and/or LuxO, LuxU and LuxO were supplied at 10 µM. Reactions were carried out in phosphorylation buffer [50 mM Tris pH 8.0, 100 mM KCl, 5 mM MgCl2, and 10% (v/v) glycerol], and were initiated with the addition of 100 µM ATP and 2 µCi [γ-32P]-ATP (from a stock of 3000 Ci mmol−1: Perkin Elmer). For experiments with CAI-1 and analogues, compounds were added 10 min before the initiation of the reactions. For the reconstitution of the complete CAI-1/CqsS, LuxU, LuxO circuit, CAI-1 was supplied at 500 µM. Regulation of histidine phosphorylation by CAI-1 was assayed with 500 µM CAI-1. Agonism was tested at 100 µM. Antagonism was tested with 10 µM agonist and 100 µM (for P-CAI-1 antagonism of CqsSWT and CAI-1 antagonism of CqsSC170Y) or 500 µM (for C8-CAI-1 antagonism of CqsSWT) antagonist as specified. Reactions were incubated at room temperature and terminated with SDS-PAGE loading buffer. Reaction products were separated using SDS-PAGE. Gels were dried at 80°C on filter paper under vacuum, exposed to a phosphoscreen overnight, and subsequently analysed using a Typhoon 9400 scanner and ImageQuant software.
Dephosphorylation of LuxU∼P
LuxU was phosphorylated for 5 min in reactions with membrane vesicles containing 4 µM CqsS, 125 µM LuxU, 100 µM ATP and 10 µCi [γ-32P]-ATP. Subsequently, the membrane vesicles were removed by ultracentrifugation at 180 000 g for 40 min. The supernatants containing LuxU∼P were applied to gel filtration spin columns (Probe Quant G-50, GE healthcare) to remove ATP. Dephosphorylation reactions were initiated by adding inverted membranes containing 2 µM CqsS receptor and either 500 µM CAI-1 or DMSO. Aliquots were taken at the indicated time points and analysed as described above.
Dephosphorylation of His194∼P by ADP
Phosphorylation of the CqsS Asp- mutant construct was carried out under the conditions described above for 2 min. His∼P dephosphorylation reactions were initiated by the addition of ADP at 100 µM final concentration and either 500 µM CAI-1 or DMSO. ATP was also added at 1 mM together with DMSO or CAI-1 to terminate new labelling of CqsS Asp-. In experiments with EDTA, 100 µM EDTA was added together with ligand and nucleotides. Aliquots were taken at the indicated time points and analysed as described above.
Chemical synthesis
Chemical syntheses of CAI-1, C8-CAI-1, P-CAI-1, HAI-1 and AI-2 have been described (Semmelhack et al., 2005; Higgins et al., 2007; Swem et al., 2008; Ng et al., 2010). Decanoic acid was purchased from Sigma-Aldrich.
Acknowledgments
We thank Dr. Guozhou Chen for assistance in CqsS cloning and for providing purified CqsS for concentration analyses. We thank Dr. Frederick Hughson, Dr. Jeffry Stock, the entire Bassler group, and especially Dr. Devin Stauff for insightful discussions. This work was supported by the Howard Hughes Medical Institute, National Institutes of Health (NIH) Grant 5R01GM065859, NIH Grant 5R01AI054442, National Science Foundation (NSF) Grant MCB-0343821 to B.L.B.
Supporting information
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Bassler BL, Wright M, Silverman MR. Multiple signalling systems controlling expression of luminescence in Vibrio harveyi: sequence and function of genes encoding a second sensory pathway. Mol Microbiol. 1994;13:273–286. doi: 10.1111/j.1365-2958.1994.tb00422.x. [DOI] [PubMed] [Google Scholar]
- Bolitho ME, Perez LJ, Koch MJ, Ng WL, Bassler BL, Semmelhack MF. Small molecule probes of the receptor binding site in the Vibrio cholerae CAI-1 quorum sensing circuit. Bioorg Med Chem. 2011;19:6906–6918. doi: 10.1016/j.bmc.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkovich KA, Simon MI. The dynamics of protein phosphorylation in bacterial chemotaxis. Cell. 1990;63:1339–1348. doi: 10.1016/0092-8674(90)90429-i. [DOI] [PubMed] [Google Scholar]
- Casino P, Rubio V, Marina A. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell. 2009;139:325–336. doi: 10.1016/j.cell.2009.08.032. [DOI] [PubMed] [Google Scholar]
- Casino P, Rubio V, Marina A. The mechanism of signal transduction by two-component systems. Curr Opin Struct Biol. 2010;20:763–771. doi: 10.1016/j.sbi.2010.09.010. [DOI] [PubMed] [Google Scholar]
- Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, Hughson FM. Structural identification of a bacterial quorum-sensing signal containing boron. Nature. 2002;415:545–549. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]
- Cheung J, Hendrickson WA. Sensor domains of two-component regulatory systems. Curr Opin Microbiol. 2010;13:116–123. doi: 10.1016/j.mib.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Qin L, Inouye M. Histidine kinases: diversity of domain organization. Mol Microbiol. 1999;34:633–640. doi: 10.1046/j.1365-2958.1999.01646.x. [DOI] [PubMed] [Google Scholar]
- Freeman JA, Bassler BL. Sequence and function of LuxU: a two-component phosphorelay protein that regulates quorum sensing in Vibrio harveyi. J Bacteriol. 1999a;181:899–906. doi: 10.1128/jb.181.3.899-906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JA, Bassler BL. A genetic analysis of the function of LuxO, a two-component response regulator involved in quorum sensing in Vibrio harveyi. Mol Microbiol. 1999b;31:665–677. doi: 10.1046/j.1365-2958.1999.01208.x. [DOI] [PubMed] [Google Scholar]
- Freeman JA, Lilley BN, Bassler BL. A genetic analysis of the functions of LuxN: a two-component hybrid sensor kinase that regulates quorum sensing in Vibrio harveyi. Mol Microbiol. 2000;35:139–149. doi: 10.1046/j.1365-2958.2000.01684.x. [DOI] [PubMed] [Google Scholar]
- Greenberg EP. Bacterial communication and group behavior. J Clin Invest. 2003;112:1288–1290. doi: 10.1172/JCI20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer BK, Bassler BL. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol Microbiol. 2003;50:101–104. doi: 10.1046/j.1365-2958.2003.03688.x. [DOI] [PubMed] [Google Scholar]
- Hazelbauer GL, Lai WC. Bacterial chemoreceptors: providing enhanced features to two-component signaling. Curr Opin Microbiol. 2010;13:124–132. doi: 10.1016/j.mib.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke JM, Bassler BL. Three parallel quorum-sensing systems regulate gene expression in Vibrio harveyi. J Bacteriol. 2004;186:6902–6914. doi: 10.1128/JB.186.20.6902-6914.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins DA, Pomianek ME, Kraml CM, Taylor RK, Semmelhack MF, Bassler BL. The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature. 2007;450:883–886. doi: 10.1038/nature06284. [DOI] [PubMed] [Google Scholar]
- Hoch JA. Two-component and phosphorelay signal transduction. Curr Opin Microbiol. 2000;3:165–170. doi: 10.1016/s1369-5274(00)00070-9. [DOI] [PubMed] [Google Scholar]
- Huynh TN, Noriega CE, Stewart V. Conserved mechanism for sensor phosphatase control of two-component signaling revealed in the nitrate sensor NarX. Proc Natl Acad Sci USA. 2010;107:21140–21145. doi: 10.1073/pnas.1013081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igo MM, Ninfa AJ, Stock JB, Silhavy TJ. Phosphorylation and dephosphorylation of a bacterial transcriptional activator by a transmembrane receptor. Genes Dev. 1989;3:1725–1734. doi: 10.1101/gad.3.11.1725. [DOI] [PubMed] [Google Scholar]
- Inoue T, Higuchi M, Hashimoto Y, Seki M, Kobayashi M, Kato T, et al. Identification of CRE1 as a cytokinin receptor from Arabidopsis. Nature. 2001;409:1060–1063. doi: 10.1038/35059117. [DOI] [PubMed] [Google Scholar]
- Iuchi S, Matsuda Z, Fujiwara T, Lin EC. The arcB gene of Escherichia coli encodes a sensor-regulator protein for anaerobic repression of the arc modulon. Mol Microbiol. 1990;4:715–727. doi: 10.1111/j.1365-2958.1990.tb00642.x. [DOI] [PubMed] [Google Scholar]
- Kelly RC, Bolitho ME, Higgins DA, Lu W, Ng WL, Jeffrey PD, et al. The Vibrio cholerae quorum-sensing autoinducer CAI-1: analysis of the biosynthetic enzyme CqsA. Nat Chem Biol. 2009;5:891–895. doi: 10.1038/nchembio.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Helmbrecht EE, Stalans MB, Schmitt C, Patel N, Wen CK, et al. Ethylene receptor ETHYLENE RECEPTOR1 domain requirements for ethylene responses in Arabidopsis seedlings. Plant Physiol. 2011;156:417–429. doi: 10.1104/pp.110.170621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krell T, Lacal J, Busch A, Silva-Jimenez H, Guazzaroni ME, Ramos JL. Bacterial sensor kinases: diversity in the recognition of environmental signals. Annu Rev Microbiol. 2010;64:539–559. doi: 10.1146/annurev.micro.112408.134054. [DOI] [PubMed] [Google Scholar]
- Lee AI, Delgado A, Gunsalus RP. Signal-dependent phosphorylation of the membrane-bound NarX two-component sensor-transmitter protein of Escherichia coli: nitrate elicits a superior anion ligand response compared to nitrite. J Bacteriol. 1999;181:5309–5316. doi: 10.1128/jb.181.17.5309-5316.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell. 2004;118:69–82. doi: 10.1016/j.cell.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Lupas A, Stock J. Phosphorylation of an N-terminal regulatory domain activates the CheB methylesterase in bacterial chemotaxis. J Biol Chem. 1989;264:17337–17342. [PubMed] [Google Scholar]
- Martin M, Albanesi D, Alzari PM, de Mendoza D. Functional in vitro assembly of the integral membrane bacterial thermosensor DesK. Protein Expr Purif. 2009;66:39–45. doi: 10.1016/j.pep.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Miller MB, Skorupski K, Lenz DH, Taylor RK, Bassler BL. Parallel quorum sensing systems converge to regulate virulence in Vibrio cholerae. Cell. 2002;110:303–314. doi: 10.1016/s0092-8674(02)00829-2. [DOI] [PubMed] [Google Scholar]
- Mizuno T. Two-component phosphorelay signal transduction systems in plants: from hormone responses to circadian rhythms. Biosci Biotechnol Biochem. 2005;69:2263–2276. doi: 10.1271/bbb.69.2263. [DOI] [PubMed] [Google Scholar]
- Neiditch MB, Federle MJ, Miller ST, Bassler BL, Hughson FM. Regulation of LuxPQ receptor activity by the quorum-sensing signal autoinducer-2. Mol Cell. 2005;18:507–518. doi: 10.1016/j.molcel.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Neiditch MB, Federle MJ, Pompeani AJ, Kelly RC, Swem DL, Jeffrey PD, et al. Ligand-induced asymmetry in histidine sensor kinase complex regulates quorum sensing. Cell. 2006;126:1095–1108. doi: 10.1016/j.cell.2006.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng WL, Bassler BL. Bacterial quorum-sensing network architectures. Annu Rev Genet. 2009;43:197–222. doi: 10.1146/annurev-genet-102108-134304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng WL, Wei Y, Perez LJ, Cong J, Long T, Koch M, et al. Probing bacterial transmembrane histidine kinase receptor-ligand interactions with natural and synthetic molecules. Proc Natl Acad Sci USA. 2010;107:5575–5580. doi: 10.1073/pnas.1001392107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng WL, Perez LJ, Wei Y, Kraml C, Semmelhack MF, Bassler BL. Signal production and detection specificity in Vibrio CqsA/CqsS quorum-sensing systems. Mol Microbiol. 2011;79:1407–1417. doi: 10.1111/j.1365-2958.2011.07548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninfa EG, Stock A, Mowbray S, Stock J. Reconstitution of the bacterial chemotaxis signal transduction system from purified components. J Biol Chem. 1991;266:9764–9770. [PubMed] [Google Scholar]
- Paul K, Brunstetter D, Titen S, Blair DF. A molecular mechanism of direction switching in the flagellar motor of Escherichia coli. Proc Natl Acad Sci USA. 2011;108:17171–17176. doi: 10.1073/pnas.1110111108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova OE, Sauer K. A novel signaling network essential for regulating Pseudomonas aeruginosa biofilm development. PLoS Pathog. 2009;5:e1000668. doi: 10.1371/journal.ppat.1000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford ST, van Kessel JC, Shao Y, Bassler BL. AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes Dev. 2011;25:397–408. doi: 10.1101/gad.2015011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Semmelhack MF, Campagna SR, Federle MJ, Bassler BL. An expeditious synthesis of DPD and boron binding studies. Org Lett. 2005;7:569–572. doi: 10.1021/ol047695j. [DOI] [PubMed] [Google Scholar]
- Sheen J. Phosphorelay and transcription control in cytokinin signal transduction. Science. 2002;296:1650–1652. doi: 10.1126/science.1071883. [DOI] [PubMed] [Google Scholar]
- Swem LR, Swem DL, Wingreen NS, Bassler BL. Deducing receptor signaling parameters from in vivo analysis: LuxN/AI-1 quorum sensing in Vibrio harrveyi. Cell. 2008;134:461–473. doi: 10.1016/j.cell.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima H, Imada K, Sakuma M, Hattori F, Nara T, Kamo N, et al. Ligand specificity determined by differentially arranged common ligand-binding residues in the bacterial amino acid chemoreceptors Tsr and Tar. J Biol Chem. 2011;286:42200–42210. doi: 10.1074/jbc.M111.221887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai N, Nakajima M, Oyama T, Kito R, Sugita C, Sugita M, et al. A KaiC-associating SasA-RpaA two-component regulatory system as a major circadian timing mediator in cyanobacteria. Proc Natl Acad Sci USA. 2006;103:12109–12114. doi: 10.1073/pnas.0602955103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawa P, Stewart RC. Kinetics of CheA autophosphorylation and dephosphorylation reactions. Biochemistry. 1994;33:7917–7924. doi: 10.1021/bi00191a019. [DOI] [PubMed] [Google Scholar]
- Teng SW, Schaffer JN, Tu KC, Mehta P, Lu W, Ong NP, et al. Active regulation of receptor ratios controls integration of quorum-sensing signals in Vibrio harveyi. Mol Syst Biol. 2011;7:491. doi: 10.1038/msb.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmen M, Bassler BL, Jung K. AI-1 influences the kinase activity but not the phosphatase activity of LuxN of Vibrio harveyi. J Biol Chem. 2006;281:24398–24404. doi: 10.1074/jbc.M604108200. [DOI] [PubMed] [Google Scholar]
- Uhl MA, Miller JF. Central role of the BvgS receiver as a phosphorylated intermediate in a complex two-component phosphorelay. J Biol Chem. 1996;271:33176–33180. doi: 10.1074/jbc.271.52.33176. [DOI] [PubMed] [Google Scholar]
- Voet-van-Vormizeele J, Groth G. Ethylene controls autophosphorylation of the histidine kinase domain in ethylene receptor ETR1. Mol Plant. 2008;1:380–387. doi: 10.1093/mp/ssn004. [DOI] [PubMed] [Google Scholar]
- Webre DJ, Wolanin PM, Stock JB. Modulated receptor interactions in bacterial transmembrane signaling. Trends Cell Biol. 2004;14:478–482. doi: 10.1016/j.tcb.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Wei Y, Perez LJ, Ng WL, Semmelhack MF, Bassler BL. Mechanism of Vibrio cholerae autoinducer-1 biosynthesis. ACS Chem Biol. 2011;6:356–365. doi: 10.1021/cb1003652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss V, Claverie-Martin F, Magasanik B. Phosphorylation of nitrogen regulator I of Escherichia coli induces strong cooperative binding to DNA essential for activation of transcription. Proc Natl Acad Sci USA. 1992;89:5088–5092. doi: 10.1073/pnas.89.11.5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Cai S, Inouye M. Interaction of EnvZ, a sensory histidine kinase, with phosphorylated OmpR, the cognate response regulator. Mol Microbiol. 2002;46:1283–1294. doi: 10.1046/j.1365-2958.2002.03240.x. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Qin L, Egger LA, Inouye M. Transcription regulation of ompF and ompC by a single transcription factor, OmpR. J Biol Chem. 2006;281:17114–17123. doi: 10.1074/jbc.M602112200. [DOI] [PubMed] [Google Scholar]
- Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci USA. 2002;99:3129–3134. doi: 10.1073/pnas.052694299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.