

Abstract
Modern drug discovery relies on the continual development of synthetic methodology to address the many challenges associated with the design of new pharmaceutical agents1. One such challenge arises from the enzymatic metabolism of drugs in vivo by cytochrome P450 oxidases, which use single-electron oxidative mechanisms to rapidly modify small molecules to facilitate their excretion2. A commonly used synthetic strategy to protect against in vivo metabolism involves the incorporation of electron-withdrawing functionality, such as the trifluoromethyl (CF3) group, into drug candidates3. The CF3 group enjoys a privileged role in the realm of medicinal chemistry because its incorporation into small molecules often enhances efficacy by promoting electrostatic interactions with targets, improving cellular membrane permeability, and increasing robustness towards oxidative metabolism of the drug4–6. Although common pharmacophores often bear CF3 motifs in an aromatic system, access to such analogues typically requires the incorporation of the CF3 group, or a surrogate moiety, at the start of a multi-step synthetic sequence. Here we report a mild, operationally simple strategy for the direct trifluoromethylation of unactivated arenes and heteroarenes through a radical-mediated mechanism using commercial photocatalysts and a household light bulb. We demonstrate the broad utility of this transformation through addition of CF3 to a number of heteroaromatic and aromatic systems. The benefit to medicinal chemistry and applicability to late-stage drug development is also shown through examples of the direct trifluoromethylation of widely prescribed pharmaceutical agents.
The CF3 moiety is typically installed by means of cross-coupling technologies catalysed by transition metals. These methods historically required the stoichiometric use of metal salts or organometallic complexes, and have shown limited generality7. Recently, however, copper and palladium complexes have been successfully shown to enable this transformation in a catalytic fashion8. Using either a nucleophilic or electrophilic source of CF3 (Ruppert–Prakash or Umemoto's reagent, respectively), it is now possible to install an aryl trifluoromethyl moiety in place of halides and boronic acids, or adjacent to tailored directing groups (Fig. 1b)9–12. These cross-coupling reactions have facilitated the synthesis of a range of CF3 analogues without the reliance on prefluorinated building blocks. However, the ideal position of CF3 substitution on a drug candidate must still be determined through parallel, multi-step syntheses employing aryl precursors bearing activating groups at various positions around an aromatic ring. Therefore, a remaining challenge in the synthesis of organofluorine drugs is this reliance on substituted precursors and their varying amenability to cross-coupling reactions.
Figure 1. Direct trifluoromethylation of aryl and heteroaryl C–H bonds.
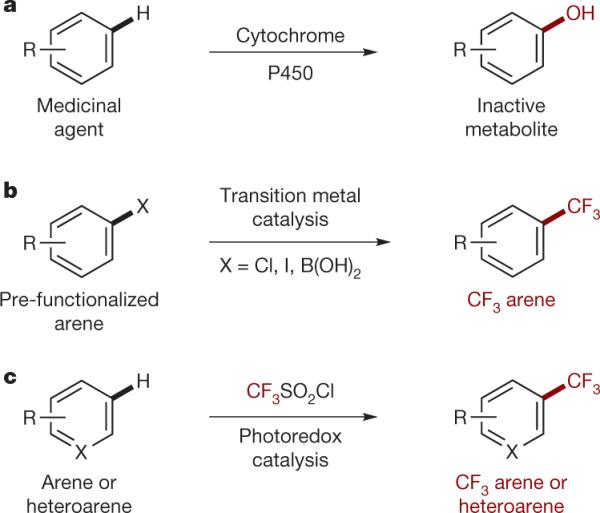
The excretion of medicinal agents is facilitated by remote functionalization of aromatic moieties (a). A common medicinal chemistry approach to block this catabolism involves cross-coupling of CF3 reagents to pre-functionalized arenes (b). In analogy to enzymatic processes, our photoredox strategy (c) enables the direct C–H functionalization of unfunctionalized arenes and heteroarenes with the CF3 pharmacophore using a cheap and easy to handle •NCF3 source.
To address this challenge, we reasoned that the direct installation of a CF3 group to a drug candidate—ideally at the position(s) of metabolic susceptibility—would obviate the need for pre-functionalization. Thus, this direct trifluoromethylation of aryl and heteroaryl compoundswould, in a single chemical operation, preclude the need for redundant synthetic efforts and simultaneously protect against metabolic oxidation in vivo. In the first part of our design plan, we proposed that a single-electron pathway might provide such an approach to address the unsolved challenge of trifluoromethylation of non-activated arenes (Fig. 1c). Specifically, we sought to take advantage of photoredox catalysis, which provides a mild and efficient method for accessing electrophilic radicals, such as •CF3, by means of photosynthesis-inspired redox chemistry13–15. This mode of reactivity employs polypyridyl organometallic complexes, such as Ru(phen)3Cl2 (1 in Fig. 2, phen=phenanthroline), whose excitation by visible light (for example, a household light bulb) at room temperature provides a strongly oxidizing or reducing catalyst that can rapidly engage a variety of substrates to generate high energy, reactive species16.
Figure 2. Proposed mechanism for the direct trifluoromethylation of aryl C–H bonds via photoredox catalysis.
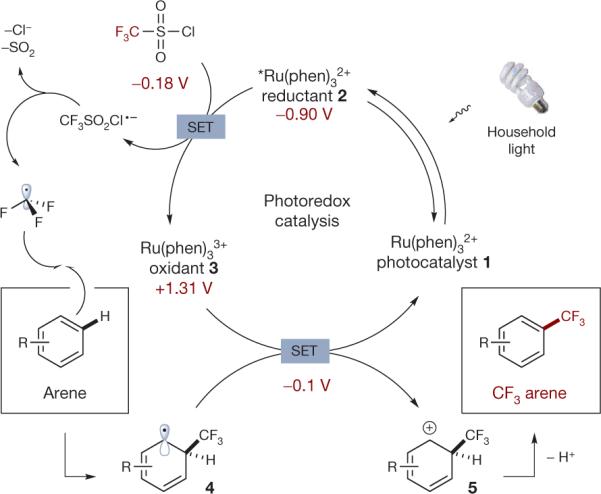
The photoredox catalytic cycle is initiated via excitation of photocatalyst 1 to excited state 2 with a household light bulb. Subsequent reduction of triflyl chloride by reductant 2 (via single electron transfer, that is, SET) provides oxidant 3 along with the radical anion of triflyl chloride. This high energy species spontaneously collapses to form CF3 radical (top left), which selectively combines with aromatic systems enabling direct CF3 substitution. Catalyst 3-promoted oxidation of the ensuing radical 4 completes the catalytic cycle and provides cyclohexadienyl cation 5, whose facile deprotonation provides the desired CF3 arene. This mechanism is consistent with measured redox values (shown below respective compounds, in maroon).
As a second part of our design plan, we recognized that the moderate to low nucleophilicity of many classes of unfunctionalized arenes has been typically underemployed in substitution reactions, presumably owing to the harsh conditions required to manipulate such aromatic rings via two-electron pathways (for example, nucleophilic or electrophilic aromatic substitution). As an alternative, we considered that a strategy based on radical addition could exploit this large area of aromatic structural space, thereby rendering a more general approach to CF3 incorporation. Our strategy, outlined in Fig. 1, involves site-specific incorporation of electrophilic radicals at metabolically susceptible positions of arenes and heteroarenes—in analogy to the single-electron aryl modification processes employed by enzymes. This approach precludes the need for pre-functionalization of arenes, and offers a complementary method of accessing aryl trifluoromethyl functionality from late-stage synthetic intermediates.
Building on our recent investigations of the α-trifluoromethylation of carbonyls with CF3I as a •CF3, source17, our initial exploration of a photoredox strategy towards the development of this transformation was successful within the limited context of π-rich arenes. We quickly observed, however, that reaction efficiency was diminished by competitive aryl iodination, presumably as a result of CF3–I homolysis. Our mechanistic understanding of this undesired pathway—wherein initiation of the photocatalytic cycle relies on sacrificial oxidation of an electron-rich arene—prompted us to seek an alternative method of catalytic initiation. Because direct reduction of CF3I (at a potential of −1.52 V versus the saturated calomel electrode, SCE, in cyclic voltammetry)18 by visible-light-excited photocatalyst *Ru(phen)32+ 2 (Fig. 2; −0.90 V versus SCE)19 is thermodynamically challenging, we questioned whether the incorporation of an electron-deficient system such as SO2 within CF3X might enable more facile reduction20. Our investigations led us to the use of trifluoromethanesulphonyl chloride (known as triflyl chloride, TfCl), which has been employed in a non-general sense to effect aryl trifluoromethylation of benzene and other simple arenes via atom transfer catalysis21–23. In particular, the relative cost and ease of handling of TfCl in comparison to other •CF3 sources rendered this a highly attractive reagent.
For the proposed photoredox catalytic cycle, we assumed that single-electron transfer (SET) reduction of triflyl chloride should be concurrent with oxidation of *Ru(phen)32+ 2 to Ru(phen)331 3 (Fig. 2). The ensuing CF3SO2Cl radical anion should then rapidly collapse to generate the stabilized •CF3, a process that should be entropically driven by the release of SO2 and chloride24. This electron deficient trifluoromethyl radical is then well-suited to add—in a selective fashion—to the most electron-rich position of any arene or heteroarene25,26. In the case of benzene, the resultant cyclohexadienyl radical 4 (Fig. 2; −0.1 V versus SCE)27 should then undergo a second SET event with the now strongly oxidizing Ru3+ photocatalyst 3 (Fig. 2; +1.31 V versus SCE) to regenerate ground-state photocatalyst 1. Finally, facile deprotonation of the cyclohexadienyl cation 5 (Fig. 2) with a suitable base would provide the desired trifluoromethyl arene in a redox, catalytic fashion without theneed for arene pre-functionalization.
As illustrated in Fig. 3, we found that this new photoredox protocol allows the direct incorporation of CF3 into a broad range of arene and heteroarene rings using only TfCl and a household light bulb. In contrast to atom transfer pathways for trifluoromethylation that require high temperature (120 °C), our photoredox strategy is successful at room temperature, allowing efficient production of the trifluoromethyl radical. Our initial studies revealed that exposure of five-atom heterocycles to a household 26-W fluorescent light source in the presence of triflyl chloride, Ru(phen)3Cl2 (1) photocatalyst, and base, enabled trifluoromethylation with excellent levels of efficiency (Fig. 3a). More specifically, the fluoroalkylation of pyrroles, furans and thiophenes occurred with excellent selectivity and yield (compounds 6–12, 78–94% yield). Our rationale for the observed selectivity at the C2 position of these heteroaromatics relies on the formation of the conjugated (rather than cross-conjugated) radical and cationic intermediates. We also found that both mono- and bis-trifluoromethylation could be obtained by varying the amount of CF3 source used (8 and 9, 88 and 91% yield, respectively). Additionally, a wide range of ring substitution and heteroatom protecting groups are well tolerated (10–17, 70–87% yield). Electron-deficient five-atom heterocycles such as thiazoles are also amenable to this trifluoromethylation protocol, and useful levels of efficiency are obtained by increasing equivalents of the CF3 source (14, 70% yield).
Figure 3. Radical trifluoromethylation of five- and six-membered heteroarenes and C–H arenes via photoredox catalysis.

Aromatic and heteroaromatic systems with varying stereoelectronics are efficiently trifluoromethylated under these standard conditions (top, generalized reaction). Substrate scope includes electron-rich five-atom heteroarenes (a), electron-deficient six-atom heteroarenes (b), and unactivated arenes (c). Isolated yields are indicated below each entry (19F NMR yields for volatile compounds). See Supplementary Information for experimental details. Abbreviations: X, Y, A, B, C represent either C, N, O, or S; MeCN, acetonitrile; Me, methyl; Boc, tert-butoxycarbonyl; Ac, acyl; tBu, tert-butyl. *The minor regioisomeric position is labelled with the respective carbon atom number.
We next turned our attention to six-atom heterocycles with the knowledge that there is an unmet need for CF3 cross-coupling technologies for these valuable pharmacophores. As shown in Fig. 3b, we found that an array of pyrazine, pyrimidine, pyridine and pyrone substrates are compatible with this new trifluoromethylation approach (18–32, 70–94% yield). We further observed that radical addition and the ensuing oxidation of electronically deficient halide-substituted heteroaromatics (such as 2,6-dichloropyrazine) were also operative. Unsubstituted analogues of each of these heterocycles can also be trifluoromethylated, albeit with somewhat diminished efficiency. Notably, the commercial photocatalyst Ir(Fppy)3 (Fppy 5 2-(2,4-difluorophenyl)pyridine, see Supplementary Information for commercial sources, synthesis and photophysical properties) is capable of providing higher levels of reactivity (presumably due to a longer-lived excited state), although lower levels of regiocontrol are observed with this system (the role of catalyst reduction potentials in these outcomes is being explored). Finally, the trifluoromethylation of arenes (shown in Fig. 3c) demonstrates the complementary reactivity offered by this radical process. Ortho-trifluoromethyl containing anilines, anisoles, thioanisoles and xylenes—typically challenging motifs to access via cross-coupling strategies—are also readily prepared using this methodology (34–40, 70–84% yield). Tolerance of halides, multiple substitution and large steric bulk are additional benefits of this radical-based process (38–47, 72–92% yield).
Our mechanistic hypothesis for the photoredox process described above is supported by emission quenching experiments as well as cyclic voltammetry. In the first case, no quenching interactions are observed between excited state 2 (Fig. 2) and a range of electronically diverse arenes and heteroarenes that arecompetent partners in this transformation; whereas triflyl chloride exhib its significant fluorescence quenching (Stern–Volmer constant, 22 M−1; Supplementary Fig. 1). Furthermore, we have determined the oxidation potential of triflyl chloride (−0.18 V versus SCE; Supplementary Fig. 2) to be in accord with the proposed reduction step using excited state 2, as shown in Fig. 2. It is also noteworthy that decreased reactivity is observed in the absence of base, presumably due to HCl formation in the reaction. Inorganic bases are preferred to organic bases, as pyridines are trifluoromethylated and alkyl amines are prone to oxidative decomposition.
Ultimately, a major benefit of our mild, visible-light-induced trifluoromethylation procedure is its amenability to late-stage synthetic applications. To demonstrate this potential, biologically active molecules (Fig. 4) were subjected to our photoredox trifluoromethylation protocol to obtain their CF3 analogues. In some cases, highly regioselective addition is observed, presumably corresponding to positions of high electron density and potential metabolic susceptibility (Fig. 4a). In medicinal chemistry, the simultaneous identification and blocking of such positions with a CF3 group may prove quite useful. For instance, a DNA base (uracil) analogue, an Alzheimer's drug (Aricept) precursor, and a vitamin (flavone) were each selectively trifluoromethylated at a single position with excellent efficiency (85–94% yield). The first example represents a facile, one-step synthesis of a known CF3-uracil derivative from its abundant non-fluoro analogue. These uridines have been extensively studied owing to their anti-viral and cancer-treating properties28.
Figure 4. Direct trifluoromethylation of biologically active molecules.

Subjecting common medicinal agents and other biologically active molecules to our standard photoredox protocol enables direct CF3 installation selectively at metabolically susceptible positions of some molecules (a). Alternatively, C–H functionalization of more metabolically stable medicines occurs non-selectively, allowing for rapid access to drug analogues (b). The promiscuous modification of equally reactive π-systems, such as the arenes in Lipitor, may be followed by separation of isomers (for example, via supercritical fluid chromatography, SFC) for rapid screening of biological activity (c).
Alternatively, a successful drug discovery programme is likely to eliminate potential sites of metabolic reactivity from a medicinal agent during the course of its development29. Without an especially electron-rich π-system present, competitive trifluoromethylation of the remaining aromatic sites should be observed. Not surprisingly, radical trifluoromethylation of lidocaine and ibuprofen results in alkylation at both of their available aromatic positions (Fig. 4b). Subjection of the cholesterol-lowering statin, Lipitor, to our protocol similarly results in promiscuous functionalization at three positions (Fig. 4c; 74% yield, 1:1:1). In the context of drug discovery, the isolation of these separate isomers offers the potential for rapid testing of fluorinated analogues for biological activity from a single late-stage synthetic product. This streamlined process represents an attractive alternative to multi-step, parallel syntheses. Perhaps most importantly, this technology provides a strategically new approach to the introduction of metabolism-blocking fluoroalkyl groups at the end of a medicinal chemistry development effort.
In this work, we have introduced a photoredox-based method that allows for facile trifluoromethylation of aromatic and heteroaromatic systems without the need for aryl ring pre-activation. The value of this transformation has been highlighted via the trifluoromethylation of biologically active molecules with commercially available photocatalysts and a household light bulb. While this manuscript was under review, a complementary method for heterocyclic trifluoromethylation was reported by others30. Their approach also employs radical intermediates to harness innate chemical reactivity of unactivated substrates, although their use of peroxides as a radical initiator affords divergent substrate tolerance and reaction efficiency. We expect these two simple protocols to be of broad utility for the synthesis and development of new medicinal agents.
METHODS SUMMARY
General procedure
An oven-dried vial was equipped with photocatalyst (1–2 mol%), dry K2HPO4 (3 equiv.), arene or heteroarene (0.5 mmol, 1 equiv.) and MeCN (0.125 M), and degassed by alternating vacuum evacuation and argon backfill at −78 °C. The triflyl chloride (1–4 equiv.) was then added and the solution was stirred at room temperature adjacent to a 26-W compact fluorescent light bulb. After 24 h, the reaction mixture was purified by column chromatography to furnish the desired CF3 product. Full experimental details and characterization data (1H, 13C, 19F NMR, infrared spectroscopy, high-resolution mass spectrometry) for all new compounds are included in Supplementary Information.
Supplementary Material
Acknowledgements
Financial support was provided by the NIH General Medical Sciences (R01 01 GM093213-01) and gifts from Merck, Amgen, Abbott and Bristol-Myers Squibb. We thank C. Kraml and N. Byrne of Lotus Separations LLC for their development of preparatory supercritical fluid chromatography (SFC) methods and for the separation of all three CF3-Lipitor analogues.
Footnotes
Author Contributions D.A.N. performed and analysed experiments. D.A.N. and D.W.C.M. designed experiments to develop this reaction and probe its utility, and also prepared this manuscript.
Author Information Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of this article at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Li JJ, Johnson DS, editors. Modern Drug Synthesis. Wiley; 2010. [Google Scholar]
- 2.Montellano PRO, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. Springer; 2005. [Google Scholar]
- 3.Filler R, Kobayashi Y, Yagupolskii LM, editors. Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications. Elsevier; 1993. [Google Scholar]
- 4.Muller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 5.Purser S, Moore PR, Swallow S, Gouverneur V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 6.Hagmann WK. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 7.Tomashenko OA, Grushin VV. Aromatic trifluoromethylation with metal complexes. Chem. Rev. 2011;111:4475–4521. doi: 10.1021/cr1004293. [DOI] [PubMed] [Google Scholar]
- 8.Furuya T, Kamlet AS, Ritter T. Catalysis for fluorination and trifluoromethylation. Nature. 2011;473:470–477. doi: 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oishi M, Kondo H, Amii H. Aromatic trifluoromethylation catalytic in copper. Chem. Commun. 2009:1909–1911. doi: 10.1039/b823249k. [DOI] [PubMed] [Google Scholar]
- 10.Cho EJ, et al. The palladium-catalyzed trifluoromethylation of aryl chlorides. Science. 2010;328:1679–1681. doi: 10.1126/science.1190524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Truesdale L, Yu JQ. Pd (II)-catalyzed ortho-trifluoromethylation of arenes using TFA as a promoter. J. Am. Chem. Soc. 2010;132:3648–3649. doi: 10.1021/ja909522s. [DOI] [PubMed] [Google Scholar]
- 12.Xu J, et al. Copper-catalyzed trifluoromethylation of aryl boronic acids using a CF31 reagent. Chem. Commun. 2011;47:4300–4302. doi: 10.1039/c1cc10359h. [DOI] [PubMed] [Google Scholar]
- 13.Nicewicz DA, MacMillan DWC. Merging photoredox catalysis with organocatalysis: the direct asymmetric alkylation of aldehydes. Science. 2008;322:77–80. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoon TP, Ischay MA, Du J. Visible light photocatalysis as a greener approach to photochemical synthesis. Nature Chem. 2010;2:527–532. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]
- 15.Narayanam JMR, Stephenson CRJ. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 2011;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]
- 16.Juris A, et al. Ru(II) polypyridine complexes: photophysics, photochemistry, electrochemistry, and chemiluminescence. Coord. Chem. Rev. 1988;84:85–277. [Google Scholar]
- 17.Nagib DA, Scott ME, MacMillan DWC. Enantioselective α-trifluoromethylation of aldehydes via photoredox organocatalysis. J. Am. Chem. Soc. 2009;131:10875–10877. doi: 10.1021/ja9053338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrieux CP, Gelis L, Medebielle M, Pinson J, Saveant J-M. Outer-sphere dissociative electron transfer to organic molecules: a source of radicals or carbanions? Direct and indirect electrochemistry of perfluoroalkyl bromides and iodides. J. Am. Chem. Soc. 1990;112:3509–3520. [Google Scholar]
- 19.Skarda V, et al. Luminescent metal complexes. Part 3. Electrochemical potentials of ground and excited states of ring-substituted 2,2'-bipyridyl and 1,10-phenanthroline tris-complexes of ruthenium. J. Chem. Soc. Perkin Trans. 1984;2:1309–1311. [Google Scholar]
- 20.Heaton CA, Miller AK, Powell RL. Predicting the reactivity of fluorinated compounds with copper using semi-empirical calculations. J. Fluor. Chem. 2001;107:1–3. [Google Scholar]
- 21.Heaton CA, Powell RL. Introduction of perfluoroalkyl groups — a new approach. J. Fluor. Chem. 1989;45:86. [Google Scholar]
- 22.Kamigata N, Fukushima T, Yoshida M. Reactions of perfluoroalkanesulfonyl chlorides with aromatic compounds catalyzed by a ruthenium (II) complex. Chem. Lett. 1990;19:649–650. [Google Scholar]
- 23.Kamigata N, Ohtsuka T, Fukushima T, Yoshida M, Shimizu T. Direct perfluoroalkylation of aromatic and heteroaromatic compounds with perfluoroalkanesulfonyl chlorides catalysed by a ruthenium(II) phosphine complex. J. Chem. Soc. Perkin Trans. 1994;1:1339–1346. [Google Scholar]
- 24.Dolbier W. Fluorinated free radicals. Top. Curr. Chem. 1997;192:97–163. [Google Scholar]
- 25.Langlois BR, Laurent E, Roidot N. Trifluoromethylation of aromatic compounds with sodium trifluoromethanesulfinate under oxidative conditions. Tetrahedron Lett. 1991;32:7525–7528. [Google Scholar]
- 26.Wiehn MS, Vinogradova EV, Togni A. Electrophilic trifluoromethylation of arenes and N-heteroarenes using hypervalent iodine reagents. J. Fluor. Chem. 2010;131:951–957. [Google Scholar]
- 27.Bahtia K, Schuler RH. Oxidation of hydroxycyclohexadienyl radical by metal ions. J. Phys. Chem. 1974;78:2335–2338. [Google Scholar]
- 28.Dexter DL, Wolberg WH, Ansfield FJ, Helson L, Heidelberger C. The clinical pharmacology of 5-trifluoromethyl-2'-deoxyuridine. Cancer Res. 1972;32:247–253. [PubMed] [Google Scholar]
- 29.Roth BD. The discovery and development of atorvastatin, a potent novel hypolipidemic agent. Prog. Med. Chem. 2002;40:1–22. doi: 10.1016/s0079-6468(08)70080-8. [DOI] [PubMed] [Google Scholar]
- 30.Ji Y, et al. Innate C–H trifluoromethylation of heterocycles. Proc. Natl Acad. Sci. USA. 2011;108:14411–14415. doi: 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.