

Abstract
Sodium dodecyl sulfate (SDS) is one of the most popular laboratory reagents used for biological sample extraction; however, the presence of this reagent in samples challenges LC-MS-based proteomics analyses because it can interfer with reversed-phase LC separations and electrospray ionization. This study reports a simple SDS-assisted proteomics sample preparation method facilitated by a novel peptide-level SDS removal step. In an initial demonstration, SDS was effectively (>99.9%) removed from peptide samples through ion substitution-mediated DS- precipitation using potassium chloride (KCl), and excellent peptide recovery (>95%) was observed for <20 μg peptides. Further experiments demonstrated the compatibility of this protocol with LC-MS/MS analyses. The resulting proteome coverage obtained for both mammalian tissues and bacterial samples was comparable to or better than that obtained for the same sample types prepared using standard proteomics preparation methods and analyzed using LC-MS/MS. These results suggest the SDS-assisted protocol is a practical, simple, and broadly applicable proteomics sample processing method, which can be particularly useful when dealing with samples difficult to solubilize by other methods.
Keywords: SDS removal, KDS precipitation, proteomics, sample preparation, LC-MS
Introduction
During the past decades, sodium dodecyl sulfate (SDS) has been recognized as one of the most popular reagents for solubilizing biological materials. This ionic detergent binds to proteins through ionic and hydrophobic interactions, and solubilizes proteins with a wide range of physical properties by altering their secondary and tertiary structures1. It is particularly useful for studies on membrane proteins or aggregated proteins, which are usually hard to solubilize by other reagents. Additionally, SDS is routinely used in polyacrylamide gel electrophoresis (SDS-PAGE), a method commonly employed in biochemical experiments to separate, characterize, and/or quantify proteins of interest. However, application of SDS in liquid chromatography–mass spectrometry (LC-MS)-based proteomics has been largely limited due to its deleterious effects on reversed-phase LC and its surface activity that leads to large DS- related signals, as well as to ionization suppression of other species during electrospray ionization (ESI)2, 3. Moreover, it is well recognized that high concentrations of SDS will significantly interfere with enzymatic protein digestion4.
Given the advantages or even necessity of using SDS for effective protein extraction and denaturation, SDS removal prior to downstream LC-MS-based analysis is desirable. Several recent methods for removing SDS from samples destined for LC-MS are protein precipitation from SDS-containing solution, filter-aided sample preparation5, 6, in-gel protein digestion7, 8, ion-exchange chromatography9, detergent removal spin columns10, 11, dialysis12, and ethyl acetate extraction13. Moreover, SDS-assisted protein extraction can be utilized as a universal sample preparation method for proteome analysis when SDS is effectively removed by filter-aided sample preparation (FASP) prior to protein digestion6, 14. While these developments have made important contributions to the field of proteomics, among the drawbacks are incomplete SDS removal, labor or time intensive and undesired sample loss.
In this study, we report a simple SDS-assisted sample preparation method for proteomics facilitated by a novel peptide-level SDS removal step. With this method, proteins are extracted in a typical SDS containing buffer (>0.5% SDS), digested in ≤0.07% SDS, and then SDS is removed from the resulting peptide samples by potassium dodecyl sulfate (KDS) precipitation. This precipitate is removed by centrifugation. Initial evaluation of the protocol revealed the supernatant, i.e., the peptide sample, was extracted virtually free of detergent. After desalting using C-18 SPE, the peptide samples were analyzed using LC-MS/MS, and results were compared to those obtained using several standard sample preparation methods. The new SDS-assisted method provided proteome coverage similar to, and in some cases, even better than that provided by standard methods for both bacterial and mammalian samples.
Experimental Section
Materials
Urea, dithiothreitol (DTT), SDS, calcium chloride (CaCl2), potassium chloride (KCl), iodoacetamide, 2,2,2-trifluoroethanol (TFE), ammonium bicarbonate, methanol, acetonitrile, trifluoroacetic acid (TFA), and formic acid were obtained from Sigma-Aldrich (St. Louis, MO). BCA protein assay reagents and the Silver Stain Kit (Product # 24612) were obtained from Thermo Scientific Pierce (Rockford, IL). The SeeBlue Plus2 protein standard was from Invitrogen (Carlsbad, CA), and porcine trypsin, from Promega (Madison, WI).
Preparation of Shewanella oneidensis cell lysate and peptide mixture
Shewanella oneidensis MR-1 cell lysis was carried out in a Barocycler NEP 3229 PCT Sample Preparation System (Pressure Biosciences Inc., South Easton, MA). Briefly, cells were suspended in 100 mM NH4HCO3 (pH 8.0), transferred to FT500 PULSE™ tubes, and then lysed by 10 pressure cycles. Each cycle consisted of 20 s at 35,000 psi and 10 s at ambient pressure. The resulting lysate was transferred to a fresh 15 mL polypropylene tube and incubated in 7 M urea and 5 mM DTT for 30 min at 37 °C. The sample was then diluted 10-fold with 100 mM NH4HCO3 and incubated for 3 h at 37 °C in the presence of 1 mM CaCl2 and porcine trypsin at a 1:50 enzyme to protein ratio. The resulting peptides were centrifuged at 5,000×g for 5 min to remove undigested pellets and were then loaded onto a 1-mL SPE C18 column (Supelco, Bellefonte, PA) for clean-up. The sample was washed with 4 mL of 0.1% TFA/5% acetonitrile. Peptides were eluted from the SPE column using 1 mL of 0.1% TFA/80% acetonitrile and then lyophilized. Final peptide concentration was determined by BCA protein assay (Pierce). Peptide samples were stored at -80 °C until use as a standard peptide mixture.
In-solution protein digestion
Cell lysates from Shewanella and tissue homogenates of both mouse liver and mouse brain cortex were suspended in 50 mM NH4HCO3 (pH 8.0) buffer. Equal amounts of proteins (∼200 μg, ∼20 μg/μL) were used for in-solution protein digestions. For comparative purposes, four different digestion protocols were evaluated: urea, TFE, SDS, and FASP. Protein samples were initially denatured based on conditions specified by a specific protocol; that is: 1) 8M urea (at 37 °C for 1 h); 2) 50% TFE (at 60 °C for 2 h); 3) 1% SDS (at 95 °C for 5 min); and 4) 4% SDS (at 95 °C for 5 min) for the FASP protocol6. For the FASP protocol, details were the same as previously reported6, whereby SDS was removed by filtration prior to protein digestion. For the other protocols, samples were reduced using 10 mM DTT (urea protocol) or 2 mM DTT (TFE protocol) for 1 h at 37 °C, or 10 mM DTT for 5 min at 95 °C during protein denaturation (SDS protocol). Cysteine was alkylated by 40 mM iodoacetamide for 1 h at 37 °C in dark for all protocols except TFE, which was performed without alkylation. All samples were then diluted 5- to ∼20-fold with 50 mM NH4HCO3 buffer to ensure the protein concentrations were ∼ 1 μg/μL for digestion. All samples were digested using 20 ng/μL porcine trypsin (1:50 trypsin:protein ratio) for 3 h at 37 °C, and remaining SDS in the processed samples was removed using the KDS precipitation method described below. Peptides from the final urea and SDS-processed samples were cleaned using C-18 SPE columns prior to LC-MS/MS analysis. The TFE-processed samples were directly lyophilized using a Speed-Vac without cleaning prior to LC-MS/MS analysis15.
SDS removal by KDS precipitation
Each SDS containing peptide sample was mixed with an equal volume of 0.25 - 4 M KCl and incubated at room temperature for 5 min to form the KDS precipitate. The KDS precipitate was pelleted by centrifugation at 14,000×g for 5-10 min, and peptides in the supernatant were collected for Discovery C-18 SPE cleanup and LC-MS/MS analysis.
SDS-PAGE
SDS-PAGE was used to evaluate digestion efficiency. Samples for SDS-PAGE were neutralized using 1.0 M Tris-HCl (pH 6.8) before mixing with an equal volume of Laemmli Sample Buffer (Bio-Rad, #161-0737EDU) that contained 1% SDS and 10 mM DTT. After heating at 95 °C for 5 min, all samples were separated on a 12% SDS-PAGE gel prepared in house. Following electrophoresis, gels were fixed in a fixing solution (50% methanol, 10% acetic acid) for 10 min and subsequently washed with water for at least 40 min. Protein and peptide bands were visualized using a Silver Stain Kit (Thermo Scientific Pierce, Product # 24612) or Coomassie brilliant blue G-250 staining.
LC-MS/MS analysis
LC-MS/MS was performed as previously described16. Briefly, 2.5 μg peptides were loaded onto a 65-cm-long, 75-μm-i.d. reversed-phase capillary column packed in house with 3 μm Jupiter C18 particles (Phenomenex, Torrance, CA). The mobile phase was held at 100% A (0.1% formic acid) for 20 min, followed by a linear gradient from 0 to 70% mobile phase B (0.1% formic acid in 90% acetonitrile) over 85 min. Eluted peptides were ionized via a nanoelectrospray ionization interface manufactured in house and analyzed on a linear ion trap mass spectrometer (Thermo Scientific, San Jose, CA). The instrument was operated in data-dependent mode with m/z ranging from 400–2000, in which a full MS scan was followed by 10 MS/MS scans. The normalized collision energy of collision-induced dissociation was 35%, and the dynamic exclusion duration was 1 min. The heated capillary was maintained at 200 °C while the ESI voltage was maintained at 2.2 kV.
Data analysis
LC-MS/MS raw data were converted into .dta files using Extract_MSn (version 3.0) in Bioworks Cluster 3.2 (Thermo Fisher Scientific, Cambridge, MA). The SEQUEST algorithm17 (version 27, revision 12) was used to search all MS/MS spectra against either a mouse protein FASTA file (Uniprot, released on April 20, 2010) or a Shewanella oneidensis MR-1 protein FASTA file created in house as previously described18. Searching parameters were: 3 Da tolerance for precursor ion masses and 1 Da for fragment ion masses with no enzyme restraint and a maximum of three missed tryptic cleavages. Static carboxamidomethylation of cysteine was used during the database search except for the TFE processed samples. MS Generating-Function (MSGF) scores were generated for each identified spectrum by computing rigorous p-values (spectral probabilities)19. Fully tryptic peptides with an MSGF score <1E-9 and partially tryptic peptides with an MSGF score <1E-10 were accepted as identifications. A decoy-database searching methodology20, 21 was used to control the FDR at the unique peptide level to <0.5%.
Results and Discussion
SDS removal by KDS precipitation
While KDS precipitation has been applied to visualize protein bands in SDS-PAGE22 and remove SDS from protein samples23, 24, the precipitation method has not previously been explored for removing SDS during MS-based proteomic sample processing. Therefore, in this study, we investigated KDS precipitation as a method for removing SDS from peptide mixtures prior to LC-MS/MS analyses. To evaluate SDS removal efficiency from proteomic buffer systems, we initially mixed 1 mL of 50 mM NH4HCO3 buffer containing various concentrations of SDS with 1 mL of 0.25-4 M KCl, and then visually observed KDS precipitation (Table 1). Note, the minimum SDS concentration observed following KDS precipitation was 0.001%. Similar results were obtained from buffers containing SDS in 0.1% TFA, or in 50 mM NH4HCO3 that contained 0.1 μg/μL standard Shewanella peptides. These results suggest that for samples containing 1% SDS, over 99.9% of SDS can be removed by KDS precipitation. As the precipitation process is dependent on both SDS and KCl concentrations, more complete SDS removal can be achieved by concentrating samples to increase SDS concentrations prior to KDS precipitation.
Table 1. Efficient removal of SDS by KDS precipitation.
| SDS (w/v) | KCl (Molar) | ||||
|---|---|---|---|---|---|
| 4 | 2 | 1 | 0.5 | 0.25 | |
| 1.000% | +++++ | +++++ | +++++ | +++++ | +++++ |
| 0.100% | ++++ | ++++ | ++++ | ++++ | ++++ |
| 0.010% | +++ | +++ | ++ | + | + |
| 0.005% | ++ | ++ | + | - | - |
| 0.001% | + | + | - | - | - |
Note: 1 mL SDS and 1 mL KCl of the indicated concentrations were mixed to induce KDS precipitation. “+++++”: precipitation with an observable pellet, “+”: barely visible precipitation. “-”: no precipitation visually observed.
Peptide recovery after KDS precipitation
We assessed peptide recovery following KDS precipitation based on BCA assay measurements. Peptide recovery of 96 ± 1% after KDS precipitation was obtained for 20 μg of standard Shewanella peptides treated with 2 M KCl and 1% SDS, i.e., demonstrating minimal peptide loss from the precipitation process (Figure 1A). High peptide recovery was also achieved from 5 μg standard peptide samples (data not shown), which indicates that the KDS precipitation procedure works well for samples containing very small amounts of peptides.
Figure 1. Peptide recovery after SDS removal.

A Shewanella whole cell lysate was digested by the urea digestion protocol and peptides were cleaned up by C-18 SPE column before use. 20 μg of peptides were used in each experiment. A, peptide recovery after treatment with 2M KCl, 1% SDS, or 1% SDS followed by SDS removal via KDS precipitation. B, peptide recovery after SDS removal and C-18 SPE clean up. C, peptide recovery from different available SDS removal protocols. All data were summarized from triplicates. #, below the detect limitation.
To make samples compatible for LC-MS/MS analysis, excess KCl salt has to be removed by C-18 SPE cleanup. Figure 1B shows peptide recovery was ∼80% after KDS precipitation and C-18 SPE cleanup of samples containing 20 μg standard peptides both with (2 M KCl alone, 0.5% SDS plus 2M KCl, 0.5% SDS plus 1M KCl, or 0.5% SDS plus 0.5M KCl) and without (Ctl) treatment (Figure 1B). Again, the results suggest that KDS precipitation has a minimal effect on overall peptide recovery and the SPE clean-up process.
Comparison of the KDS method with other available SDS removal methods (Figure 1C) revealed KDS precipitation provided superior peptide recovery. Nearly zero peptide recovery was observed for acetone precipitation and chloroform/methanol precipitation methods in spite of the fact they work well for removing SDS from protein samples3. Application of an SDS removal spin column10 resulted in peptide recovery of ∼20% for a 20 μg standard peptide sample with 1% SDS.
Sample compatibility with LC-MS/MS analysis after KDS precipitation
To evaluate the compatibility of peptide samples after KDS precipitation with LC-MS/MS, we spiked 0.3% SDS in 30 μg standard Shewanella peptides and then applied the KDS precipitation methodology to remove SDS and C-18 SPE cleanup to remove salts. A comparison between LC-MS chromatograms obtained for control and KDS precipitated samples (Figure 2A) showed high similarity, which indicates that no significant peak loss or peak contamination was introduced after the precipitation step. Furthermore, peptide and protein identification data confirmed that the SDS-depleted sample was fully compatible with LC-MS/MS analyses based on the fact that comparable numbers of peptides and proteins were identified from both the untreated control and the peptide sample after KDS precipitation (Figure 2B-2E). These results are consistent with a previous study that reported reliable LC-MS characterization of peptides could be obtained from samples containing up to 0.01% SDS3 since the SDS concentration in the sample after KDS precipitation was <0.001% (Table 1).
Figure 2. Compatibility of the SDS removed samples with LC-MS/MS analyses.
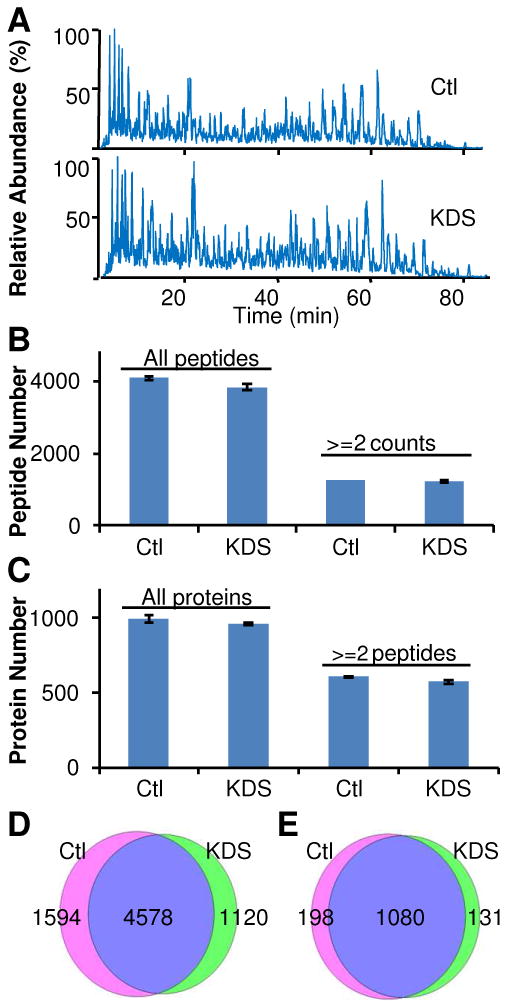
Standard peptides digested from the Shewanella whole cell lysate were used in these experiments. All samples were analyzed on an LTQ instrument. A, LC-MS/MS base peak chromatograms; B, peptide identifications from LC-MS/MS analyses (n=3); C, protein identifications from LC-MS/MS analyses (n=3); D-E, overlaps of peptide and protein identifications between control samples and SDS-depleted samples. Data for each condition were summarized from triplicates. D, peptide overlap; E, protein overlap. “Ctl” denotes the control sample. KDS indicates the peptide sample treated with 0.5% SDS followed by SDS removal.
SDS facilitated in-solution tryptic digestion
While well recognized that high concentrations of SDS inhibit trypsin activity, the effect of low concentration SDS (<0.1%) on protein digestion was unclear and not fully characterized. To evaluate the effect of SDS on tryptic digestion efficiency, aliquots of mouse liver homogenates (∼100 μg proteins per aliquot) were denatured and digested for 3 h in 50 mM NH4HCO3 containing various concentrations of SDS. The digested samples were then loaded onto 12% SDS-PAGE and visualized using Coomassie brilliant blue G250 staining. Digestion efficiency was assessed based on the absence of high molecular weight protein bands and the presence of low molecular weight peptide bands visualized in the gel. Figure 3A shows optimal digestion efficiency was achieved for samples containing 0.07% SDS or less.
Figure 3. SDS facilitates protein digestion by trypsin.
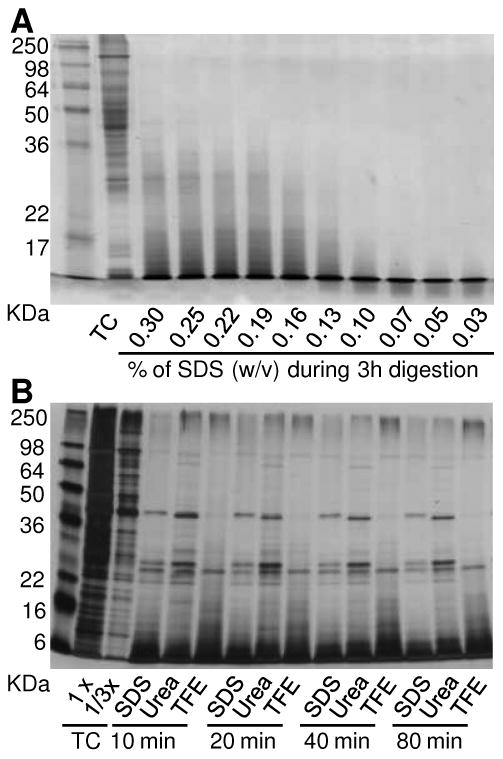
A, titration of SDS concentration during trypsin digestion of mouse liver total cell lysates. B, digestion efficiency comparison of different protocols on Shewanella total cell lysates. TC: total cell lysate.
To further evaluate digestion efficiency, 400 μg of insoluble fractions from Shewanella lysate were subjected to three in-solution digestion protocols, i.e., urea, SDS-assisted, and TFE (see Experimental Section). Aliquots taken at different times after the addition of trypsin were quenched by adding 0.1% TFA and then separated using SDS-PAGE gels. Digestion buffer containing 0.05% SDS provided the fastest and most complete protein digestion compared to urea and TFE digestion protocols, as evidenced by the more intense undigested or partially digested protein bands for the latter two methods in Figure 3B. Similar results were observed for both mouse liver and mouse plasma samples (Supplementary Figure 1). These results are consistent with a previous study that reported an SDS-assisted sample preparation method (0.11% during denaturing and 0.025% during digestion) provided high tryptic peptide production and signal intensities in MRM assays4, indicating that a low concentration of SDS results in more complete in-solution tryptic digestion of complex biological samples.
Application of the SDS-assisted sample preparation methods for proteome profiling
To evaluate the applicability of SDS-assisted proteomic sample preparation, we compared proteome profiling results of different sample preparation methods for several sample types. Aliquots of tissue homogenates from mouse brain cortex, mouse liver and the insoluble fractions of Shewanella lysates were solubilized in 1% SDS, 4% SDS, 8 M urea or 50% TFE, and processed using the KDS precipitation, FASP, TFE and urea-based protocols, respectively (as described in the Experimental Section). The proteome coverage obtained from triplicate LC-MS/MS sample analyses are shown in Figure 4 for mouse brain cortex (A), mouse liver (B), and Shewanella insoluble fractions (C) samples. In all cases, the coverage obtained from using KDS precipitation (i.e., the SDS-assisted protocol) was comparable to those obtained using any of the standard protocols tested. Further comparison of the four protocols in terms of numbers of proteins identified from mouse brain samples (Figure 4D) reveals that the majority of identifications were common among the different methods. Collectively, these results confirm the compatibility of the SDS-removed samples with LC-MS/MS analyses. These results also suggest that the four protocols are comparable in terms of numbers of unique proteins identified. The TFE method provided fewer identifications in the mouse brain cortex samples, presumably due to endogenous contaminations (e.g., soluble lipids and metabolites that can potentially interfer with LC-MS/MS analysis) since no clean-up was performed after protein digestion15.
Figure 4. Comparison of protein identification between different in-solution digestion protocols.

A, mouse brain cortex; B, mouse liver; C, insoluble fraction of Shewanella oneidensis lysate; D, mouse brain cortex protein identification overlap between different methods. Only proteins identified by at least two unique peptides or at least in 2 different replicate samples were used. Data were summarized from triplicate experiments.
Pros and cons of the SDS-assisted sample preparation
Our results demonstrate a simple SDS-assisted sample preparation method for LC-MS/MS analysis that, given the ability of SDS to solubilize biological materials, is universally applicable to a diversity of sample types. After extracting proteins with high concentrations of SDS, protein samples can be diluted (SDS <0.07%) and digested, after which the SDS can be effectively removed from the digested peptide samples via a precipitation step to render samples fully compatible with LC-MS/MS analysis.
This SDS-assisted preparation method has several potentially unique benefits. First, the simple SDS removal step enables the broad use of SDS in proteomic sample preparation, especially for difficult-to-solubilize samples. SDS is believed to be a superior reagent for protein solubilization and denaturation when proteins of interest, such as membrane proteins25, are resistant to other reagents. Additionally, SDS is useful for extracting proteins from challenging samples, such as bacteria, fungi, and plant samples, which usually contain materials that are resistant to dissolution in an aqueous buffer without the assistance of detergents. Previous work by Proc, et al.4 avoided complications by using very low concentrations of SDS for in-solution digestions; however, the starting concentration of SDS during protein denaturation (0.11%) was too low to solubilize many hydrophobic proteins. To utilize the full power of SDS for solubilizing proteins, a high concentration is necessary for protein denaturation, after which the remaining SDS in the digested samples needs to be removed prior to LC-MS/MS analysis. We note that while TFE-based sample preparation is much simpler (no SPE cleanup required) than the SDS-assisted method that utilizes KDS precipitation, when dealing with samples that are difficult to solubilize, the KDS-based method should offer a significant advantage. Second, KDS precipitation provides high peptide recovery for general applications and is much less time-consuming and laborious than the popular FASP method6. Third, as SDS removal occurs at the peptide level, the same SDS-containing buffer can be used to extract proteins from biological materials for both proteomic profiling and concurrent Western blotting validation studies. The ability to use the same SDS-extracted protein samples for orthogonal experimental procedures may significantly improve consistency between proteomics data and biochemically validated data.
The SDS-assisted sample preparation method also has several potential limitations that should be considered. First, we note the necessity of diluting the sample to make the SDS concentration ≤0.07% during digestion. This requirement needs to be considered when determining the sample starting volume and the concentration of SDS required for protein extraction in order to achieve optimal results for proteomic processing. Second, in some experiments, we observed signal loss for some very hydrophobic peaks at the end of the LC elution profile in the SDS sample (Supplementary Figure 2). There are two possible reasons for this observation. One possibility is that these peaks are from large peptides that were not effectively digested in methods other than the SDS-assisted digestion. This possibility suggests a greater number of smaller peptides are being generated, as evidenced by the enhanced peak intensities of some early elution peaks ions in the SDS sample and the enhanced trypsin digestion efficiency (Figure 3B). The second possibility is that hydrophobic molecules are lost during the KDS precipitation. Regardless, our data confirmed that the majority of proteins identified from late elution peaks in other methods were also identified by other peptides in the SDS samples.
Conclusions
A simple SDS-assisted sample preparation method for LC-MS/MS-based proteomics applications takes advantage of SDS for protein extraction and denaturation at SDS concentrations >0.5% w/v, and facilitates more complete tryptic protein digestion at SDS concentrations ≤0.07%. The method is primarily enabled by a novel peptide-level SDS removal step involving KDS precipitation and centrifugation. The resulting proteome coverage from this method is comparable to those observed from several other standard protocols. This simple sample preparation method is universally applicable to a diversity of sample types that require processing for LC-MS/MS analyses.
Supplementary Material
Acknowledgments
Portions of this research were supported by National Institutes of Health grants R01 DK074795 and RR018522, and a U. S. Department of Energy (DOE) Early Career Research Award. Experimental work was performed in the Environmental Molecular Sciences Laboratory, a DOE/BER national scientific user facility at Pacific Northwest National Laboratory, is multi-program national laboratory operated by Battelle for the DOE under Contract No. DE-AC05-76RLO 1830.
Footnotes
Supporting Information: Supplementary material includes the following figures: Supplemental Figure 1 displays the time course of different digestion protocol on different biological samples. Supplementary Figure 2 displays the chromatographic comparison between different digestion protocols. Supplemental Table 1-6 list the spectral counts of all proteins and peptides identified in different methods.
References
- 1.Andersen KK, Oliveira CL, Larsen KL, Poulsen FM, Callisen TH, Westh P, Pedersen JS, Otzen D. J Mol Biol. 2009;391:207–226. doi: 10.1016/j.jmb.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 2.Rundlett KL, Armstrong DW. Anal Chem. 1996;68:3493–3497. doi: 10.1021/ac960472p. [DOI] [PubMed] [Google Scholar]
- 3.Botelho D, Wall MJ, Vieira DB, Fitzsimmons S, Liu F, Doucette A. J Proteome Res. 2010;9:2863–2870. doi: 10.1021/pr900949p. [DOI] [PubMed] [Google Scholar]
- 4.Proc JL, Kuzyk MA, Hardie DB, Yang J, Smith DS, Jackson AM, Parker CE, Borchers CH. J Proteome Res. 2010;9:5422–5437. doi: 10.1021/pr100656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manza LL, Stamer SL, Ham AJ, Codreanu SG, Liebler DC. Proteomics. 2005;5:1742–1745. doi: 10.1002/pmic.200401063. [DOI] [PubMed] [Google Scholar]
- 6.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 7.Rosenfeld J, Capdevielle J, Guillemot JC, Ferrara P. Anal Biochem. 1992;203:173–179. doi: 10.1016/0003-2697(92)90061-b. [DOI] [PubMed] [Google Scholar]
- 8.Lapierre LA, Avant KM, Caldwell CM, Ham AJ, Hill S, Williams JA, Smolka AJ, Goldenring JR. American journal of physiology Gastrointestinal and liver physiology. 2007;292:G1249–1262. doi: 10.1152/ajpgi.00505.2006. [DOI] [PubMed] [Google Scholar]
- 9.Weber K, Kuter DJ. Journal of Biological Chemistry. 1971;246:4504–4509. [PubMed] [Google Scholar]
- 10.Antharavally BS, Mallia KA, Rosenblatt MM, Salunkhe AM, Rogers JC, Haney P, Haghdoost N. Anal Biochem. 2011;416:39–44. doi: 10.1016/j.ab.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 11.Bereman MS, Egertson JD, MacCoss MJ. Proteomics. 2011;11:2931–2935. doi: 10.1002/pmic.201100045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hjelmeland LM. Methods in enzymology. 1990;182:277–282. doi: 10.1016/0076-6879(90)82023-u. [DOI] [PubMed] [Google Scholar]
- 13.Yeung YG, Nieves E, Angeletti RH, Stanley ER. Analytical biochemistry. 2008;382:135–137. doi: 10.1016/j.ab.2008.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wisniewski JR, Nagaraj N, Zougman A, Gnad F, Mann M. Journal of proteome research. 2010;9:3280–3289. doi: 10.1021/pr1002214. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Qian WJ, Mottaz HM, Clauss TR, Anderson DJ, Moore RJ, Camp DG, 2nd, Khan AH, Sforza DM, Pallavicini M, Smith DJ, Smith RD. Journal of proteome research. 2005;4:2397–2403. doi: 10.1021/pr050160f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou JY, Schepmoes AA, Zhang X, Moore RJ, Monroe ME, Lee JH, Camp DG, Smith RD, Qian WJ. J Proteome Res. 2010;9:5698–5704. doi: 10.1021/pr100508p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eng JK, McCormack AL, Yates JR. Journal of The American Society for Mass Spectrometry. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 18.Romine MF, Carlson TS, Norbeck AD, McCue LA, Lipton MS. Applied and environmental microbiology. 2008;74:3257–3265. doi: 10.1128/AEM.02720-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim S, Gupta N, Pevzner PA. J Proteome Res. 2008;7:3354–3363. doi: 10.1021/pr8001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qian WJ, Liu T, Monroe ME, Strittmatter EF, Jacobs JM, Kangas LJ, Petritis K, Camp DG, 2nd, Smith RD. Journal of proteome research. 2005;4:53–62. doi: 10.1021/pr0498638. [DOI] [PubMed] [Google Scholar]
- 21.Elias JE, Gygi SP. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 22.Higgins RC, Dahmus ME. Anal Biochem. 1979;93:257–260. doi: 10.1016/s0003-2697(79)80148-7. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki H, Terada T. Anal Biochem. 1988;172:259–263. doi: 10.1016/0003-2697(88)90440-x. [DOI] [PubMed] [Google Scholar]
- 24.Carraro U, Doria D, Rizzi C, Sandri M. Biochem Biophys Res Commun. 1994;200:916–924. doi: 10.1006/bbrc.1994.1537. [DOI] [PubMed] [Google Scholar]
- 25.Wisniewski JR, Zougman A, Mann M. J Proteome Res. 2009;8:5674–5678. doi: 10.1021/pr900748n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.