

SUMMARY
The mitochondrial sirtuin SIRT3 regulates metabolic homeostasis during fasting and calorie restriction. We identified mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 (HMGCS2) as an acetylated protein and a possible target of SIRT3 in a proteomics survey in hepatic mitochondria from Sirt3−/− (SIRT3KO) mice. HMGCS2 is the rate-limiting step in β-hydroxybutyrate synthesis and is hyperacetylated at lysines 310, 447, and 473 in the absence of SIRT3. HMGCS2 is deacetylated by SIRT3 in response to fasting in wild-type mice, but not in SIRT3KO mice. HMGCS2 is deacetylated in vitro when incubated with SIRT3 and in vivo by overexpression of SIRT3. Deacetylation of HMGCS2 lysines 310, 447, and 473 by incubation with wild-type SIRT3 or by mutation to arginine enhances its enzymatic activity. Molecular dynamics simulations show that in silico deacetylation of these three lysines causes conformational changes of HMGCS2 near the active site. Mice lacking SIRT3 show decreased β-hydroxybutyrate levels during fasting. Our findings show SIRT3 regulates ketone body production during fasting and provide molecular insight into how protein acetylation can regulate enzymatic activity.
INTRODUCTION
Organisms rely on adaptive metabolic mechanisms to maintain energy homeostasis under low nutrient conditions. In the transition from the fed to the fasted state, carbohydrate utilization and fatty acid synthesis cease in the liver, and fatty acid oxidation and ketogenesis are induced (McGarry and Foster, 1980). Acetyl-CoA generated from fatty acid oxidation is diverted away from the tricarboxylic acid (TCA) cycle and converted into acetoacetate, β-hydroxybutyrate, and acetone (ketone bodies) through ketogenesis in the mitochondria (Laffel, 1999). Specific tissues, such as the brain, consume acetoacetate and β-hydroxybutyrate to spare glucose when glucose levels are low.
β-hydroxybutyrate production is regulated by the enzyme 3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS2) in the mitochondria. HMGCS2 catalyzes the rate-limiting conversion of acetoacetyl-CoA and acetyl-CoA into 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA), which is further converted into acetoacetate by mitochondrial HMG-CoA lyase (Hegardt, 1999). Acetoacetate is finally converted into β-hydroxybutyrate by β-hydroxybutyrate dehydrogenase. Ketone body production is regulated by HMGCS2 via multiple signals from the overall metabolic state of the organism. Ketogenesis is activated during fasting, which upregulates HMGCS2 gene expression by increases in glucagon and cyclic-AMP (for a comprehensive review, see Hegardt, 1999). At the protein level, succinyl-CoA regulates HMGCS2 directly by binding to and competitively inhibiting the active site (Quant et al., 1990). Additionally, HMGCS2 is palmitoylated, a posttranslational modification predicted to regulate enzymatic activity (Kostiuk et al., 2008).
Lysine acetylation, another posttranslational modification, regulates multiple metabolic pathways in bacteria (Zhao et al., 2010) and in human liver cells (Wang et al., 2010). The mitochondrial sirtuin SIRT3 is a mitochondrial deacetylase and is emerging as an important regulator protein acetylation and metabolic regulation during fasting. SIRT3 expression is enhanced during fasting, deacetylates long-chain acyl-CoA dehydrogenase (LCAD), and increases fatty acid oxidation in the liver (Hirschey et al., 2010). In extrahepatic tissue, SIRT3 also deacetylates and activates mitochondrial acetylCoA synthase 2 (AceCS2) (Hallows et al., 2006; Schwer et al., 2006), an enzyme required in the fasting response (Sakakibara et al., 2009). Additionally, SIRT3 deacetylates a subunit of the electron transport chain and regulates ATP production (Ahn et al., 2008). In this study, we identify HMGCS2 as a substrate of SIRT3, further supporting a role for SIRT3 in adaptive response to fasting.
RESULTS
SIRT3 Regulates the Acetylation of HMGCS2
To identify possible SIRT3 targets, we purified hepatic mitochondria from Sirt3−/− (SIRT3KO) mice, subjected mitochondrial matrix lysates to trypsin proteolytic digestion and immunoprecipitated this peptide mix with an anti-acetyllysine antiserum. The acetyllysine-containing peptides were eluted with dilute acid and analyzed by nanoflow liquid chromatography-tandem mass spectrometry (LC-MS/MS) on a hybrid linear ion-trap Fourier transform mass spectrometer. HMGCS2 was identified as a possible substrate of SIRT3.
To confirm that HMGCS2 was acetylated in SIRT3KO mice and to assess the possibility that SIRT3 regulates the acetylation of HMGCS2, we probed the acetylation levels of endogenous HMGCS2 under fed and fasted conditions. HMGCS2 was immunoprecipitated with an antiserum specific for HMGCS2 from wild-type (WT) and SIRT3KO mouse liver mitochondria and analyzed by western blotting with an anti-acetyllysine antiserum. In the fed state, hepatic HMGCS2 was acetylated at a basal level (Figure 1A). However, after 24 hr of fasting, expression of SIRT3 protein is upregulated (Hirschey et al., 2010), and acetylation levels of HMGCS2 were reduced by 58% in WT mice (Figure 1A). In contrast, HMGCS2 was hyperacetylated under basal and fasting conditions in SIRT3KO mice (72% and 64% increase in acetylation compared to WT, respectively). These observations are consistent with a model in which increased SIRT3 expression during fasting leads to the deacetylation of HMGCS2.
Figure 1. SIRT3 Regulates the Acetylation Level of HMGCS2.

(A) Liver extracts from fed or fasted (24 hr) WT and SIRT3KO mice were immunoprecipitated with an HMGCS2 antiserum and analyzed by western blotting with antisera specific for acetyllysine; integrated density values were calculated and are shown relative to WT mice; data are represented in arbitrary units (AU) ± SEM, n = 3/condition; *p < 0.05.
(B) Recombinant HMGCS2 expressed in E. coli was incubated in vitro with recombinant SIRT3 or catalytically inactive SIRT3-H248Y in the presence or absence of nicotinamide, and the HMGCS2 acetylation status was assessed.
(C) Expression vectors for WT SIRT3, catalytically inactive SIRT3-H248Y, SIRT4, or SIRT5 were cotransfected into HEK293 cells with expression vectors for FLAG-tagged HMGCS2, and the levels of HMGCS2 acetylation were assessed.
To measure the ability of SIRT3 to directly deacetylate HMGCS2, recombinant HMGCS2 was purified after overexpression in Escherichia coli and incubated with recombinant WT SIRT3 or a catalytically inactive SIRT3 mutant (H248Y). Changes in levels of acetylation of HMGCS2 were measured by western blotting with an anti-acetyllysine antibody. SIRT3, but not catalytically inactive SIRT3-H248Y, deacetylated HMGCS2 in vitro (Figure 1B).
To further confirm the role of SIRT3 in regulating the acetylation of HMGCS2, expression vectors encoding FLAG-tagged HMGCS2 were cotransfected with expression vectors for SIRT3, catalytically inactive SIRT3-H248Y mutant, SIRT4, or SIRT5 (HA-tagged) into HEK293 cells. HMGCS2 acetylation levels were measured after immunoprecipitation (anti-FLAG) and western blotting with an anti-acetyllysine antiserum. We found SIRT3, but not catalytically inactive SIRT3-H248Y, SIRT4, or SIRT5, deacetylated HMGCS2 (34% reduction compared to WT, Figure 1C). These data show SIRT3 mediates the deacetylation of HMGCS2 in vivo and in vitro.
Three Acetylated Lysines Regulate HMGCS2 Activity
To characterize the sites of acetylation regulated by SIRT3, HMGCS2 was further analyzed by MS. Acetylated peptides were immunoprecipitated from isolated mitochondria from WT and SIRT3KO mice, and semiquantitative MS data analysis was performed. Eleven acetylation sites were identified in hepatic murine HMGCS2 (see Figure S1 available online). Nine of those sites are conserved between mouse and human (Figure 2A and Figure S1). Three of the sites, K310, K447, and K473, were significantly hyperacetylated in SIRT3KO mice (Figure 2B).
Figure 2. HMGCS2 Acetylation at Lysines 310, 447, and 473 Regulate Activity.

(A) Sites of HMGCS2 acetylation identified by mass spectrometry of purified mitochondria from WT and SIRT3KO mice.
(B) Differential acetylation fold values for HMGCS2 acetylated lysines from WT and SIRT3KO hepatic mouse mitochondria compared to a separate standard mouse reference.
(C) Expression vectors for WT HMGCS2 were transfected into HEK293 cells with an empty vector control; SIRT3-HA; SIRT3-H248Y-HA; pSicoRMS2 shSIRT3; or HMGCS2-K310, −447, −473R (3KR), and the levels of acetylation were assessed.
(D and E) Steady-state kinetic analysis of HMGCS2 activity; rates of HMGCS2 activity were determined as a function of (acetyl-CoA), as measured by DTNB detection of CoASH; 1 unit = 1 μmol substrate converted to product per minute, n = 2–3 measurements/sample, ± SEM, curve is representative of two independent experiments.
To test the biological significance of HMGCS2 acetylation at these three sites, we mutated the three lysines targeted by SIRT3 (K310R, K447R, K473R [HMGCS2-3KR]) into arginines. HMGCS2-3KR was expressed in HEK293 cells and probed for acetylation by immunoblotting with anti-acetyllysine antiserum in comparison to WT. Since arginines cannot be acetylated like lysines and conserve the positive charge of the lysine residues, the 3KR mutant represents a constitutively unacetylated form of the protein. The triple point mutation of HMGCS2-3KR had significantly less acetylation than WT HMGCS2 (Figure 2C), demonstrating these three lysines are major sites of acetylation.
To measure the effect of HMGCS2 acetylation on enzymatic activity, we purified HMGCS2. An expression vector for HMGCS2 was cotransfected in HEK293 with SIRT3 expression vectors (WT or catalytically inactive) or with an shRNA targeting SIRT3. An expression vector for HMGCS2-3KR was also transfected separately. WT and mutated HMGCS2 proteins were immunoprecipitated (anti-FLAG), and their enzymatic activity was measured. A steady-state kinetic analysis of HMGCS2 activity was performed, and the initial rates of CoA formation were measured as a function of acetyl-CoA concentration. The resulting saturation curves were fitted to the Michaelis-Menten equation and the parameters compared among the various HMGCS2 preparations. When HMGCS2 was coexpressed with WT SIRT3, we observed a reduction in acetylation and a 125%–165% increase in Vmax, compared to control HMGCS2 coexpressed with an empty vector or coexpressed with catalytically inactive SIRT3, respectively (Figures 2D and 2E). However, no significant changes were observed in Km values (Figures 2D and 2E). Additionally, when HMGCS2 was cotransfected with an shRNA targeting SIRT3, HMGCS2 became hyperacetylated (Figure 2C) and displayed reduced activity (43% reduction in Vmax compared to HMGCS2 cotransfected with an empty vector control; Figures 2D and 2E). Furthermore, constitutively unacetylated HMGCS2-3KR had reduced acetylation and increased enzymatic activity (109% increase in Vmax compared to HMGCS2 cotransfected with an empty vector control), similar to HMGCS2 cotransfected with WT SIRT3 (Figures 2D and 2E). These results indicate that the acetylation state of HMGCS2 directly correlates with its enzymatic activity. Since K310, K447, and K473 represent the primary acetylation sites of HMGCS2 that are regulated by SIRT3, these observations are consistent with the model that SIRT3 regulates HMGCS2 enzymatic activity via the deacetylation of these residues in vivo.
Lysine Deacetylation Induces HMGCS2 Conformational Changes
To further investigate how changes in the acetylation level of HMGCS2 modulate enzymatic activity, we performed molecular dynamics simulations on the unacetylated and triply acetylated on K310, K447, and K473 mouse HMGCS2 (based on homology to human HMGCS2 [Protein Data Bank (PDB) 2WYA]). The overall conformation of HMGCS2 was minimally perturbed by lysine acetylation: approximately 80% of Cα atoms in acetylated HMGCS2 deviated less than 2 Å from the unacetylated form (Figure 3A). However, significant differences in protein conformation were observed in two distinct regions in triply acetylated HMGCS2 compared to the unacetylated model.
Figure 3. Lysine Deacetylation Affects HMGCS2 Conformation and Dynamics.

The tubes represent the average backbone conformation of the protein over the last 5 ns of five independent simulations, and the widths of the tubes represent the fluctuations from the average conformation (i.e., thicker lines indicate increased conformational flexibility).
(A) Overview of the entire structure for the unacetylated (yellow) and triply acetylated (cyan) mouse HMGCS2. Acetyl-CoA in the model is shown using spheres, whereas K310, K447, K473, and their acetylated forms are shown as balls and sticks.
(B) Closer view of the region around a distal portion of the active site (the green box in A). The two loops showing significant conformational and dynamical changes are labeled L1 (residues 242–251) and L2 (residues 131–140).
(C) Closer view of the region around K310 (the red box in A).
First, acetylation affects HMGCS2 conformation around the acetyl-CoA binding site (Figure 3C). When HMGCS2 was unacetylated, the 3-amino group of K310 formed ion pairs with both the 3′ phosphate of the acetyl-CoA (>90% of the time, during the last 5 ns of five independent simulations [Figure S2]) and aspartate residues (D351, D353) on helix 350–367 (Figures 4A and 4B). However, these interactions were abrogated by acetylation, which eliminates the positive charge on the lysine side chain (Figures 4A and 4C). As a result, conformational changes occur in the vicinity of K310. In particular, large changes were observed in the conformation of helix 350–367 (Figure 3C), while the helix containing the K310 acetylation site (residues 305–316) was minimally perturbed. Specifically, the helix 350–367 moved further away from the acetyl-CoA in the acetylated state compared to the unacetylated state (Figures 3C and 4C). Similar conformational changes occurred in simulations with only K310 acetylated (Figure S3).
Figure 4. Acetylation Shifts Catalytic Residues in the Acetyl-CoA Binding Region.

(A) The percentage of time the distance between the 3′ phosphate atom of acetyl-CoA P(3′) and the nitrogen atom (N) in the sidechain of K310, K306, K350, and K354 is smaller than 4.2 Å, during which the phosphate group of acetyl-CoA and lysine sidechain form salt bridge (Mandell et al., 2007); comparing the unacetylated (yellow) and triply acetylated (cyan) HMGCS2; data extracted from the last 5 ns of each simulation and averaged over five independent simulations for either state of HMGCS2.
(B) Representative snapshot image during a simulation of the electrostatic network formed by K310, K306, K350, K354, and the 3′ phosphate group of acetyl-CoA in unacetylated HMGCS2.
(C) Same as (B) for the triply acetylated (K310, K447, K473) HMGCS2.
(D) The percentage of time the distance between the catalytic cysteine (C166) and nearby histidine (H301) (or acetyl-CoA) is smaller than 4.0 Å; comparing the unacetylated (yellow) and triply acetylated (cyan) HMGCS2; data extracted from the last 5 ns of each simulation and averaged over five independent simulations for either state of HMGCS2.
(E) Representative snapshot image during a simulation of the catalytic amino acids and the acetyl-CoA in unacetylated HMGCS2.
(F) Same as (E) for the triply acetylated (K310, K447, K473) HMGCS2.
A second region more distant from the active site also showed significant changes in conformation and dynamics between the acetylated and unacetylated proteins (Figure 3B). Two loops (L1 [residues 242–251] and L2 [residues 131–140]) are in close proximity to each other but not to K310, K447, or K473. Acetylated HMGCS2 showed larger fluctuations in loop 1 and a shift in the average position of loop 2 compared to the unacetylated protein (Figure 3B). The changes in the conformation and dynamics of these two loops propagated to the end of the active site where the acetyl-CoA resides. Simulations with the enzyme singly acetylated on K473, and to a lesser extent on K310 or K447, showed similar changes in both loops, as well as helix 350–3677 (Figures S4 and S5).
Finally, the acetylation-induced changes around the acetyl-CoA binding site discussed above affected the positioning of critical catalytic residues. Histidine 301 (H301) and cysteine 166 (C166) are required for catalysis (Lowe and Tubbs, 1985). The molecular dynamics simulations showed acetylation of K310, K447, and K473 on HMGCS2 shifted the relative positioning of C166 to H301 and to acetyl-CoA (Figures 4E and 4F). Misalignment of the catalytic residues may reduce the probability of H301 acting as a hydrogen bond acceptor for the nucleophilic C166 and the probability of C166 attacking the acetyl group of acetyl-CoA, because the average distances between C166-H301 and C166-(acetyl-CoA) increased (Figure 4D).
Taken together, these simulations suggest that deacetylation of K310, K447, and K473 causes changes in HMGCS2 that propagate far beyond the sites of acetylation, and significantly affect protein conformation, dynamics, and electrostatics near the active site while minimally affecting the remainder of the protein.
β-Hydroxybutyrate Production Is Decreased in SIRT3KO Mice
Because we observed lower levels of HMGCS2 enzymatic activity in SIRT3KO mice and since HMGCS2 is the rate-limiting step in β-hydroxybutyrate synthesis, we measured β-hydroxybutyrate production in WT and SIRT3KO mice. No differences between WT and SIRT3KO mice in terms of plasma β-hydroxybutyrate were observed in the fed state (Hirschey et al., 2010). However, under fasting conditions, β-hydroxybutyrate levels were lower in 1-, 3-, and 12-month-old SIRT3KO mice than in WT mice (Figure 5A). We also measured the level of plasma β-hydroxybutyrylcarnitine, a mitochondrial intermediate derived from β-hydroxybutyryl-CoA that reflects the pool of β-hydroxybutyrate within cells (An et al., 2004). β- hydroxybutyrylcarnitine levels were 40% lower in 3-month-old SIRT3KO mice than in WT mice (Figure 5B), further supporting lower levels of β-hydroxybutyrate production during fasting in SIRT3KO mice.
Figure 5. β-Hydroxybutyrate Production Is Decreased in SIRT3KO Mice.
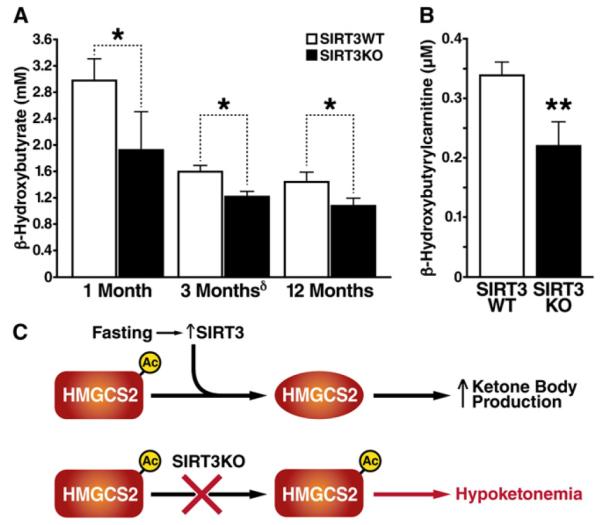
(A) β-Hydroxybutyrate (β-OHB) levels in SIRT3KO and WT mice at 1, 3, and 12 months of age after a 24 hr fast; n = 5–10 for each genotype ± SEM †(Hirschey et al., 2010), *p < 0.05.
(B) Serum β-hydroxybutyrylcarnitine levels in SIRT3KO mice compared to WT at 3 months of age after a 24 hr fast; n = 5 for each genotype ± SEM, **p < 0.01. (C) Proposed model for regulation of HMGCS2 by acetylation and deacetylation by SIRT3.
DISCUSSION
Mitochondrial HMGCS2 catalyzes the conversion of acetoacetyl-CoA to HMG-CoA, the rate-limiting step in β-hydroxybutyrate production. Here we show that mitochondrial HMGCS2 is acetylated in the fed state and deacetylated by SIRT3 during fasting. When HMGCS2 is hyperacetylated, such as in the SIRT3KO mouse, the enzymatic activity is reduced, resulting in decreased β-hydroxybutyrate synthesis. Deacetylation of HMGCS2 by SIRT3 during fasting elevates HMGCS2 enzymatic activity and β-hydroxybutyrate production (Figure 5C).
HMGCS2 proceeds via a “bi bi ping-pong” kinetic mechanism in which acetyl-CoA binds to and forms a covalent acetylenzyme intermediate (Lowe and Tubbs, 1985). The intermediate undergoes a condensation reaction with the second substrate, acetoacetyl-CoA, to form HMG-CoA that is ultimately hydrolyzed from the enzyme. We observed a marked increase in HMGCS2 Vmax in the unacetylated state, after deacetylation by WT SIRT3 or in the constitutively unacetylated HMGCS2-3KR mutant. Conversely, the Vmax of HMGCS2 was reduced after SIRT3 knockdown, demonstrating that the acetylation state of HMGCS2 directly influences catalytic Vmax. The formation of the acetyl-HMGCS2 substrate-enzyme intermediate at C166 is proposed to be the rate-limiting step of the enzyme (Miziorko and Lane, 1977). Thus we interpret the effect of acetylation on Vmax as reflecting this chemical step. We observed no significant effect of acetylation on the Km value for acetyl-CoA, suggesting that deacetylation does not directly affect acetyl-CoA binding affinity to HMGCS2. Notably, the Km of HMGCS2-3KR was significantly reduced, demonstrating that the 3KR mutant does not perfectly mimic the unacetylated HMGCS2.
Molecular dynamics simulations performed on the triply acetylated and unacetylated HMGCS2 structures revealed a possible mechanism of how acetylation regulates the Vmax of HMGCS2. Acetylation of K310, K447, and K473 leads to substantial changes in conformation and dynamics of the enzyme primarily in two regions close to the active site, leaving the remainder of the protein largely unchanged. Moreover, the conformational changes were qualitatively similar when acetylating each lysine individually (Figures S3–S5) or all nine acetylated lysines (Figure S6).
Due to the complexity of the reaction mechanism, determining how these conformational changes affect enzyme kinetics with certainty is not possible. However, we predict that acetylation regulates HMGCS2 enzymatic activity by altering the local protein conformation near the acetyl-CoA catalytic residues, consistent with our interpretation that the observed reduction in Vmax represents a modulation of this chemical step. Acetyl transfer from acetyl-CoA to C166 is the first step in HMG-CoA formation. For this reaction to occur, H301 must act as the H+-bond acceptor from C166, and thus an optimally close distance between H301 and C166 is required. Similarly, acetyl transfer from coenzyme A to the enzyme at C166 requires that acetyl-CoA be positioned nearby C166. Acetylation of HMGCS2 at K310, K447, and K473 increases the average distance between H301-C166 and C166-(acetyl-CoA), which could reduce the efficiency with which the active site cysteine attacks the acetyl group of acetyl-CoA and initiates the reaction. We hypothesize that deacetylation by SIRT3 reverses this impairment and induces a change in protein conformation to place these residues into a more favorable position for catalysis.
The biological roles and structural mechanisms of protein regulation by lysine acetylation remain less well understood than those of posttranslational phosphorylation. However, lysine acetylation changes the net charge of the lysine side chain and can thereby induce local changes in protein conformation or affinity. Here, we present evidence for a mechanism whereby lysine acetylation at distant sites can induce changes in the conformation of the enzyme near the active site and changes in enzymatic activity. Phosphorylation at amino acids distant from the active site can effect conformational changes that propagate between the sites (Johnson and Lewis, 2001; Narayanan and Jacobson, 2009). Similarly, protonation changes on a histidine side chain in talin cause alterations in the conformation and dynamics of the actin binding site approximately 40 Å away, thereby modulating binding affinity (Srivastava et al., 2008). Molecular characterization of additional acetylated enzymes will determine if deacetylation-induced conformational change represents a general regulatory mechanism.
Recent reports implicate SIRT3 in the regulation of energy homeostasis during nutrient deprivation. SIRT3 regulates fatty acid catabolism (Hirschey et al., 2010) and acetate metabolism (Hallows et al., 2006; Schwer et al., 2006), two metabolic pathways that are activated during fasting. Here, we report that ketone body synthesis is regulated by SIRT3 during fasting. SIRT3 has also been implicated in regulating metabolism during calorie restriction (CR) (Shi et al., 2005). Since both fatty acid oxidation and ketone body production also increase during CR, we speculate that SIRT3 could regulate metabolism during CR. Future studies will determine if SIRT3 plays a role or is required for the increase in ketone bodies during CR or ketogenic diet feeding conditions, and possibly the associated beneficial metabolic effects.
EXPERIMENTAL PROCEDURES
Animal Measurements
Animal measurements were performed on IACUC-approved protocols. Studies used WT and SIRT3KO 129Sv (as described in Lombard et al., 2007) male 1-, 3-, or 12-month-old mice, maintained on a standard chow diet (5053 PicoLab diet, Ralston Purina Company, St. Louis, MO). Mice were sacrificed at 7:00 a.m. for fed mouse studies or transferred to a new cage without food for 24 hr from 7:00 a.m. to 7:00 a.m. and then sacrificed for fasted mouse studies. Concentration of β-hydroxybutyrate was determined using a β-hydroxybutyrate detection kit (Stanbio Laboratory, Boerne, TX). For β-hydroxylbutrylcarnitine measurements, hepatic proteins were precipitated with methanol, and supernatants were dried, esterified with hot, acidic methanol, and then analyzed by tandem MS (Quattro Micro, Waters Corporation, Milford, MA). All acylcarnitines were assayed by adapting described methods for analysis of amino acids in dried blood spots (Wu et al., 2004).
Cell Culture and Plasmid Construction
HEK293 cells were cultured in DMEM supplemented with 10% FCS. All expression constructs were generated by standard PCR-based cloning strategies, and all expression constructs were verified by DNA sequencing. The human HMGCS2 coding sequence was PCR amplified from human full-length Mammalian Gene Collection cDNA (GenBank accession number NM_005518; obtained through Open Biosystems, Huntsville, AL) and cloned into the pcDNA3.1+-derived vectors pcDNA-Flag or pcDNA-HA to yield HMGCS2 with a C-terminal Flag or hemagglutinin tag (Invitrogen, Carlsbad, CA). Human SIRT3, SIRT4, and SIRT5 cDNA were also cloned into pcDNA-Flag or pcDNA-HA. Recombinant expression vectors encoding mature human SIRT3 (amino acids 102–399) were as described (Schwer et al., 2006). For recombinant HMGCS2 expression plasmid, full-length human HMGCS2 was cloned into pTrcHis2 vector. Expression and purification of recombinant proteins were as described (Schwer et al., 2006). For shSIRT3 generation, a short 19-mer hairpin corresponding to human SIRT3 (482-GGAGTGGCCTGTACAGCAA-500) was cloned into pSicoRMS2 vector, a modified pSicoR (EF1alphamCherry-T2A-Puro) (Ventura et al., 2004).
Computational Methods
System Preparation
The crystal structure of human HMGCS2 (PDB 2WYA) was used as a template to build the homology model of mouse HMGCS2 with in-house software (Protein Localization Optimization Program [PLOP; distributed as Prime by Schrodinger LLC], Portland, OR). To study the role of lysine acetylation in regulating the enzymatic activity of mouse HMGCS2, simulations on six states were performed: (1) the unacetylated state with all lysine residues unmodified; (2) a triply acetylated state with only K310, K447, and K473 acetylated; (3) a singly acetylated state with only K310 acetylated; (4) a singly acetylated state with only K473 acetylated; (5) a singly acetylated state with only K447 acetylated; and (6) a state with acetylation on nine lysine residues (K243, K306, K310, K333, K350, K354, K437, K447, and K473). Each state initially was energy minimized using PLOP (Zhu et al., 2007). The force field parameters for the substrate (acetyl-coenzyme A) were obtained from R.E.DD.B. (Dupradeau et al., 2008) using the version for Amber force field ff03 (Duan et al., 2003; Lee and Duan, 2004). The partial charges for acetylated lysine were the same as those in Table 1 of Liu and Duan (2008). The backbone torsion parameters of unmodified lysine in the Amber ff03 force field (Duan et al., 2003; Lee and Duan, 2004) were used for the acetylated lysines.
Molecular Dynamics Simulations
Each of the six states of mouse HMGCS2 was placed in a water box with a minimum 8 Å from any protein heavy atom to the edge of the box. Na+ counterions were added to make the system electrically neutral. All of the molecular dynamics simulations were performed with the Amber10 simulation package (Case et al., 2008). The ff03 force field was used for protein and substrate (Duan et al., 2003; Lee and Duan, 2004), and the TIP3P force field was used for water (Jorgensen et al., 1983). A two-stage approach was used to energy minimize the simulation systems, with a steepest descent algorithm followed by a conjugate gradient method. In the first stage, the protein complex was fixed, and only the water and ions were energy minimized. In the second stage, the entire system was energy minimized. A 20 ps MD simulation at constant volume with weak restraints on the protein complex was performed to heat the system to 300 K, and Langevin dynamics was used to control the temperature at a collision frequency of 1.0 ps−1. Then each system was simulated with a time step of 2 fs at a constant temperature of 300K and a constant pressure of 1 atm with a time constant of 1 ps. A cutoff of 12 Å was used for both van der Waals and short-range electrostatic interactions. Long-range electrostatic forces were treated with the particle mesh Ewald (PME) method (Darden et al., 1993). For all simulations, the SHAKE algorithm (Ryckaert et al., 1977) was used to constrain bond lengths. For each of the six states of HMGCS2, four other independent simulations (at 300K and 1 atm) were also performed with different initial velocities. Structures were taken every 100 ps of the first 400 ps of the simulations after the 20ps position restrained simulations. Each simulation has a length of 11–20 ns. Statistical analyses were performed using the last 5 ns of each simulation, during which the backbone root-meansquare deviation (rmsd) was stable (Figure S2), and then averaged over the five independent simulations.
Immunoblotting
Antibodies used were anti-mtHsp70 (Affinity Bioreagents, Neshanic Station, NJ), anti-HMGCS2 (GenWay Biotech, San Diego, CA), anti-Flag M2 or rabbit polyclonal anti-Flag (Sigma-Aldrich, St. Louis, MO), anti-HA (12CA5 and 3F10; Roche Diagnostics, Indianapolis, IN), acetylated-lysine polyclonal antibody (Cell Signaling Technology, Danvers, MA), and anti-c-myc (Santa Cruz Biotechnology, Santa Cruz, CA). SIRT3 antiserum was raised as described (Lombard et al., 2007). Immunoblots were developed with enhanced chemiluminescence (Amersham Pharmacia Biosciences, Piscataway, NJ) or West SuperSignal reagent (Pierce, Rockford, IL).
Immunoprecipitation
Cells were lysed on ice in NP1 buffer (PBS with 0.5% Nonidet P-40 and 0.2 mM PMSF) with protease inhibitor cocktail (Roche). Flag-tagged proteins were immunoprecipitated and washed in NP1 buffer four times. For HMGCS2 activity assays, immunoprecipitated Flag-HMGCS2 preparations were eluted with 0.5 mg/ml Flag peptide and dialyzed with PBS/10% glycerol/0.1 mM DTT. Immunoprecipitated HA-tagged sirtuins for deacetylation assays were washed in NP1 buffer three times containing 500 mM NaCl and twice in sirtuin deacetylase buffer (SDAC) (50 mM Tris$HCl [pH 9.0], 4 mM MgCl2, 50 mM NaCl, 0.5 mM DTT).
In Vitro Assays
SIRT3 Deacetylation
Equimolar purified recombinant HMGCS2 and purified recombinant sirtuins were incubated in SDAC buffer in the presence or absence of NAD+ (1 mM) with trichostatin A (500 nM) for 3 hr at 30°C (Hirschey et al., 2009; Schwer et al., 2006).
HMGCS2 Activity
HMGCS2 enzymatic activity was measured by monitoring the conversion of acetyl-CoA and acetoacetyl-CoA to 3-hydroxy-3-methylglutaryl-CoA, as measured by DTNB detection of CoASH (Andrew Skaff and Miziorko, 2010). Assay mixtures contained 100 mM Tris-Cl (pH 8.0), 130 μM DTNB (A412 nm = 13.6 mM−1), 10–1400 μM acetyl-CoA, 10 μM acetoacetyl-CoA, and 1 μg of enzyme in a total volume of 200 μL. Absorbance at 412 nm was recorded over 4 min, reflecting linear rates for all enzymes. Data were fitted using nonlinear regression to the Michaelis-Menten equation (R2 > 0.95) to determine Km and Vmax. Each point represents the average of two or three measurements at that concentration, with error bars reflecting the standard deviation of these measurements. Curves are representative of two independent experiments. A unit of enzyme activity is defined as the amount of enzyme that causes 1 μmol of substrate to be transformed into product per minute.
Mass Spectrometry
MS analysis was performed on a 7 Tesla LTQ-FT (Thermo Fisher Scientific, Waltham, MA) connected to an Agilent 1100 nanoflow HPLC system (Agilent, Santa Clara, CA) with a nanoelectrospray ion source (Proxeon Biosystems, Cambridge, MA). Peptides were separated by reversed phase chromatography with an in-house fused silica emitter (75 μm ID) packed with Reprosil-Pur C18-AQ 3 μm reverse-phase material (Dr. Maisch GmbH). Data were searched using Mascot (Matrix-Science, Boston, MA) against the mouse IPI database and analyzed with MSQuant (Mortensen et al., 2010) and MaxQuant (Cox and Mann, 2008). Semiquantitative MS analyses were as described (Rikova et al., 2007; Rush et al., 2005). Briefly, peptides containing acetyllysines were isolated directly from protease-digested (trypsin) mitochondrial extracts from WT and SIRT3KO mouse livers with an anti-acetyllysine-specific antibody and were identified by tandem MS.
Statistical Analyses
Results are given as the mean ± standard error. Statistical analyses represent a nonparametric Student’s t test, and null hypotheses were rejected at 0.05.
Supplementary Material
ACKNOWLEDGMENTS
Special thanks to A. Wilson and J. Carroll for figure preparation and to G. Howard and S. Ordway for editorial review. This work was supported in part by a National Science Foundation (NSF) predoctoral fellowship (K.E.D.-R.), a K08 award (AG022325) from the National Institute on Aging/National Institutes of Health (NIA/NIH) (D.B.L.), an Ellison Medical Foundation/AFAR Senior Postdoctoral Research Grant (B.S.), NIH grant GM-081710 (M.P.J.), NIH grant GM-065386 (J.M.D.), a Senior Scholarship in Aging from the Ellison Medical Foundation (F.W.A. and E.V.), and by institutional support from the J. David Gladstone Institutes. F.W.A. is an Investigator of the Howard Hughes Medical Institute. The J. David Gladstone Institutes received support from a National Center for Research Resources Grant RR18928-01. E.V. and F.W.A. are members of the scientific advisory board for Sirtris, a GSK company. M.P.J. is a member of the scientific advisory board for Schrodinger LLC.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and can be found with this article online at doi:10.1016/j.cmet.2010.11.003.
REFERENCES
- Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Muoio DM, Shiota M, Fujimoto Y, Cline GW, Shulman GI, Koves TR, Stevens R, Millington D, Newgard CB. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat. Med. 2004;10:268–274. doi: 10.1038/nm995. [DOI] [PubMed] [Google Scholar]
- Andrew Skaff D, Miziorko HM. A visible wavelength spectropho-tometric assay suitable for high-throughput screening of 3-hydroxy-3-methylglutaryl-CoA synthase. Anal. Biochem. 2010;396:96–102. doi: 10.1016/j.ab.2009.08.030. [DOI] [PubMed] [Google Scholar]
- Case D, Darden T, Cheatham T, III, Simmerling C, Wang J, Duke R, Luo R, Crowley M, Walker RC, Zhang W, et al. AMBER. University of California; San Francisco: 2008. p. 10. [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N [center-dot] log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- Dupradeau F-Y, Cézard C, Lelong R, Stanislawiak E, Pêcher J, Delepine JC, Cieplak P. R.E.DD.B.: a database for RESP and ESP atomic charges, and force field libraries. Nucleic Acids Res. 2008;36:D360–D367. doi: 10.1093/nar/gkm887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. USA. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegardt FG. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis. Biochem. J. 1999;338:569–582. [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Huang J, Verdin E. Acetylation of mitochondrial proteins. Methods Enzymol. 2009;457:137–147. doi: 10.1016/S0076-6879(09)05008-3. [DOI] [PubMed] [Google Scholar]
- Hirschey M, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard D, Grueter C, Harris C, Biddinger S, Ilkayeva O, et al. SIRT3 regulates mitochondrial fatty acid oxidation via reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LN, Lewis RJ. Structural basis for control by phosphorylation. Chem. Rev. 2001;101:2209–2242. doi: 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- Jorgensen W, Chandrasekhar J, Madura J, Impey R, Klein M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- Kostiuk MA, Corvi MM, Keller BO, Plummer G, Prescher JA, Hangauer MJ, Bertozzi CR, Rajaiah G, Falck JR, Berthiaume LG. Identification of palmitoylated mitochondrial proteins using a bio-orthogonal azido-palmitate analogue. FASEB J. 2008;22:721–732. doi: 10.1096/fj.07-9199com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffel L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999;15:412–426. doi: 10.1002/(sici)1520-7560(199911/12)15:6<412::aid-dmrr72>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Lee MC, Duan Y. Distinguish protein decoys by using a scoring function based on a new AMBER force field, short molecular dynamics simulations, and the generalized born solvent model. Proteins. 2004;55:620–634. doi: 10.1002/prot.10470. [DOI] [PubMed] [Google Scholar]
- Liu H, Duan Y. Effects of posttranslational modifications on the structure and dynamics of histone H3 N-terminal peptide. Biophys. J. 2008;94:4579–4585. doi: 10.1529/biophysj.107.115824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe DM, Tubbs PK. 3-hydroxy-3-methylglutaryl-coenzyme A synthase from ox liver. Purification, molecular and catalytic properties. Biochem. J. 1985;227:591–599. doi: 10.1042/bj2270591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell DJ, Chorny I, Groban ES, Wong SE, Levine E, Rapp CS, Jacobson MP. Strengths of hydrogen bonds involving phosphorylated amino acid side chains. J. Am. Chem. Soc. 2007;129:820–827. doi: 10.1021/ja063019w. [DOI] [PubMed] [Google Scholar]
- McGarry JD, Foster DW. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 1980;49:395–420. doi: 10.1146/annurev.bi.49.070180.002143. [DOI] [PubMed] [Google Scholar]
- Miziorko HM, Lane MD. 3-Hydroxy-3-methylgutaryl-CoA synthase. Participation of acetyl-S-enzyme and enzyme-S-hydroxymethylgutaryl-SCoA intermediates in the reaction. J. Biol. Chem. 1977;252:1414–1420. [PubMed] [Google Scholar]
- Mortensen P, Gouw JW, Olsen JV, Ong S-E, Rigbolt KTG, Bunkenborg J, Cox J, Foster LJ, Heck AJR, Blagoev B, et al. MSQuant, an open source platform for mass spectrometry-based quantitative proteomics. J. Proteome Res. 2010;9:393–403. doi: 10.1021/pr900721e. [DOI] [PubMed] [Google Scholar]
- Narayanan A, Jacobson MP. Computational studies of protein regulation by post-translational phosphorylation. Curr. Opin. Struct. Biol. 2009;19:156–163. doi: 10.1016/j.sbi.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Quant PA, Tubbs PK, Brand MD. Glucagon activates mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in vivo by decreasing the extent of succinylation of the enzyme. Eur. J. Biochem. 1990;187:169–174. doi: 10.1111/j.1432-1033.1990.tb15291.x. [DOI] [PubMed] [Google Scholar]
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- Rush J, Moritz A, Lee K, Guo A, Goss V, Spek E, Zhang H, Zha X, Polakiewicz R, Comb M. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 2005;23:94–101. doi: 10.1038/nbt1046. [DOI] [PubMed] [Google Scholar]
- Ryckaert J-P, Ciccotti G, Berendsen H. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- Sakakibara I, Fujino T, Ishii M, Tanaka T, Shimosawa T, Miura S, Zhang W, Tokutake Y, Yamamoto J, Awano M, et al. Fasting-induced hypothermia and reduced energy production in mice lacking acetyl-CoA synthetase 2. Cell Metab. 2009;9:191–202. doi: 10.1016/j.cmet.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. USA. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- Srivastava J, Barreiro G, Groscurth S, Gingras AR, Goult BT, Critchley DR, Kelly MJS, Jacobson MP, Barber DL. Structural model and functional significance of pH-dependent talin-actin binding for focal adhesion remodeling. Proc. Natl. Acad. Sci. USA. 2008;105:14436–14441. doi: 10.1073/pnas.0805163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, Jacks T. Cre-lox-regulated conditional RNA interference from transgenes. Proc. Natl. Acad. Sci. USA. 2004;101:10380–10385. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY, Kao HJ, Li SC, Stevens R, Hillman S, Millington D, Chen YT. ENU mutagenesis identifies mice with mitochondrial branched-chain aminotransferase deficiency resembling human maple syrup urine disease. J. Clin. Invest. 2004;113:434–440. doi: 10.1172/JCI19574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Shirts M, Friesner R, Jacobson M. Multiscale optimization of a truncated Newton minimization algorithm and application to proteins and protein-ligand complexes. J. Chem. Theory Comput. 2007;3:640–648. doi: 10.1021/ct600129f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.