

Abstract
Disruption of ribosomal proteins is associated with hematopoietic phenotypes in cell culture and animal models. Mutations in ribosomal proteins are seen in patients with Diamond Blackfan anemia (DBA), a rare congenital disease characterized by red cell aplasia and distinctive craniofacial anomalies. A zebrafish screen uncovered decreased hematopoietic stem cells (HSCs) in embryos with mutations in ribosomal protein rps29. Here, we determined that rps29-/- embryos also have red blood cell defects and increased apoptosis in the head. As the p53 pathway has been shown to play a role in other ribosomal protein mutants, we studied the genetic relationship of rps29 and p53. Transcriptional profiling revealed that genes up-regulated in the rps29 mutant are enriched for genes up-regulated by p53 after irradiation. p53 mutation near completely rescues the rps29 morphological and hematopoietic phenotypes, demonstrating that p53 mediates the effects of rps29 knockdown. We also identified neuronal gene orthopedia protein a (otpa) as one whose expression correlates with rps29 expression, suggesting that levels of expression of some genes are dependent on rps29 levels. Together, our studies demonstrate a role of p53 in mediating the cellular defects associated with rps29 and establish a role for rps29 and p53 in HSCs and red blood cell development.
Keywords: Diamond Blackfan anemia, hematopoietic stem cells, ribosomal proteins
Introduction
Diamond Blackfan anemia (DBA) is a rare, congenital anemia often diagnosed by one year of age[1]. Patients with DBA have a block in erythropoiesis, and often have craniofacial anomalies. It is also thought that patients may have an HSC defect, as they may develop bone marrow failure and aplastic anemia. Treatment regimens include transfusions and steroids, but currently the only cure is hematopoietic stem cell transplant.
Over 50% of patients have a mutation in a ribosomal protein gene[2]. Pathogenic mutations in RPS19[3], RPS24[4], RPS17[5], RPL35A[6], RPL5, RPL11, RPS7[7], RPS10, and RPS26[8] have been identified in patients. Patients are always heterozygous for the mutations and maintain one copy of the unmutated ribosomal protein. In addition, some family members have been identified to have a mutation in a DBA gene but have only mild or no symptoms. The mechanism behind the incomplete penetrance seen in families is still unknown and suggests that other genetic modifiers exist.
Why ribosomal protein mutations specifically affect hematopoiesis is still unclear. To address the mechanism, several models of ribosomal protein mutation have been developed, including mouse models and shRNA knockdown in human cells. The first mouse model generated, an rps19 knockout, was homozygous lethal[9]. Heterozygous mice did not have a phenotype, but the wildtype rps19 locus was found to be upregulated, compensating for the mutant allele[10]. However, mice with a point mutation in rps19 do have a hematopoietic defect that is rescued by p53 knockdown[11]. p53 inactivation also rescues craniofacial abnormalities in mouse models of Treacher Collins Syndrome, where mutations affect ribosome biogenesis[12]. In a mouse model of conditional rps6 knockdown, p53 is activated[13]. Mechanistic studies in this system suggest that levels of some free ribosomal proteins are increased as a result of impaired ribosome biogenesis. These free ribosomal proteins, including rpL11 and rpL5, can enter the nucleus, bind mdm2, and sequester it from p53. Thus, rpL11 mediates the p53 activation seen in the rps6 knockdown mice, and this mechanism may hold true for other models of ribosomal protein knockdown.
Several groups have previously used zebrafish for modeling ribosomal protein knockdown, as zebrafish hematopoietic regulation is conserved with mammals and the model system is particularly amenable for high throughput in vivo genetics. Ribosomal protein mutants have been generated[14], or these genes can be knocked down in the embryo with morpholino technology. Zebrafish embryos homozygous for ribosomal protein mutations can survive for several days, likely the result of maternal RNA and protein contribution. Morpholinos against rps19 and rpL11[15-17], as well as an rpL11 mutant[18], recapitulate many aspects of the DBA phenotype, including hematopoietic specific defects and p53 activation.
Our lab previously identified a zebrafish ribosomal protein mutant with defects in hematopoietic stem cell (HSC) development. Rps29, a protein in the 40s small subunit of the ribosome important for rRNA processing and ribosome biogenesis[19], was found to be required for definitive hematopoiesis in the zebrafish embryo[20]. The rps29 mutant has decreased HSC markers runx and myb at 36 hours post fertilization (hpf) in the aorta gonad mesonephros (AGM) region. At this stage of development, definitive HSCs emerge from the hemogenic endothelium lining the aorta[21]. Thus, endothelial cell fate specification, vasculogenesis, and the establishment of artery identity are all required for HSC formation. In the rps29 mutant, vasculogenesis occurs normally, but artery identity is not established as shown by lack of flk1-expressing intersomitic vessels and ephrinB2 expression. Therefore, the HSC defect is likely the result of a defect in aorta specification.
Here, we describe new aspects of the rps29 mutant phenotype that include defects in red blood cell development and an increase in apoptotic cells. To understand what pathways mediate this phenotype, we performed microarray analyses on both rps29 mutants and p53 mutants. Analysis of microarray data from the rps29 mutant embryos uncovered differential expression of genes associated with apoptotic pathways and a gene whose levels correlate with rps29 levels. Using gene set enrichment analysis (GSEA), we found that genes up-regulated in the rps29 mutant were enriched for genes activated by p53 upon irradiation. When p53 is mutated in homozygous rps29 mutant embryos, the apoptotic and hematopoietic phenotypes are rescued. Our microarray data also identified a neuronal gene, otpa, whose expression correlated with rps29 level. However, p53 mutation does not fully rescue expression of this gene, suggesting that other p53-independent mechanisms are affected by rps29 knockdown. Our data support a model where p53 activation results from ribosomal protein deficiency and mediates the hematopoietic phenotype.
Materials and Methods
Zebrafish maintenance and lines
Fish were maintained under approved laboratory conditions. The two fish lines used are: hi2903, an insertional mutant in the first intron of rps29[14], and p53M214K/M214K, a p53 point mutant in the DNA-binding domain[22].
In situ hybridization, stains, and immunohistochemistry
Whole-mount in situ hybridization was performed as described[23]. Antisense probes were synthesized from digested plasmid or PCR product template. O-dianisidine was performed as previously described[24]. Embryos treated with 1-phenyl 2-thiourea (PTU) to prevent pigmentation were stained with Sudan Black as previously described[25]. TUNEL staining was performed according to manufacturer's instructions using Millipore's ApopTag® Plus Peroxidase In Situ Apoptosis Kit. Globin antibody staining was performed as described[25] using a zebrafish antibody from Anaspec (Anti-Hbbe, Catalog Number 55608).
Quantitative PCR and microarrays
RNA was isolated from zebrafish embryos using Trizol and according to the manufacturer's directions. cDNA was made using the Invitrogen Superscript III First-Strand Synthesis system. Quantitative PCR was performed using Bio-Rad Ssofast EvaGreen Supermix and published primer sequences are available upon request. For the rps29 microarrays, 24 hpf mutant embryos were separated from wildtype and heterozygous siblings by morphology, and the wildtypes and heterozygotes were pooled together. 3 pairs of samples (6 total) were sent for analysis on a Nimblegen microarray. For the p53 microarray, wildtype (Tu) and p53M214K/M214K embryos received 16 Gy of gamma irradiation at 27 hpf. RNA isolation was performed on pooled embryos one hour post irradiation. A total of 12 samples were analyzed, 3 of each class (wildtype unirradiated, wildtype irradiated, p53M214K/M214K unirradiated, and p53M214K/M214K irradiated), and were hybridized to Affymetrix zebrafish arrays. For otpa and βe3 globin expression, tail DNA from 24 or 40 hpf embryos was used for genotyping. Five embryos of the same genotype were pooled for cDNA synthesis and qPCR.
Bioinformatics analysis
Differential gene sets were generated utilizing the Goldenspike workflow[26], identifying genes significantly different amongst groups with q values<0.05. To overcome annotation differences between the array platforms, we mapped the Nimblegen probe that corresponded to the appropriate Affymetrix probe. Rps29 microarray data was analyzed using Ingenuity Pathway Analysis (IPA) to identify pathways significantly affected in the rps29 mutant. For the GSEA analysis, we identified a gene signature of the rps29 mutants consisting of 524 up-regulated and 442 down-regulated genes. These were then used as a query set to identify enrichment within the p53 dataset using GSEA v2.
Results
RPS29 is required for definitive HSCs and primitive RBCs
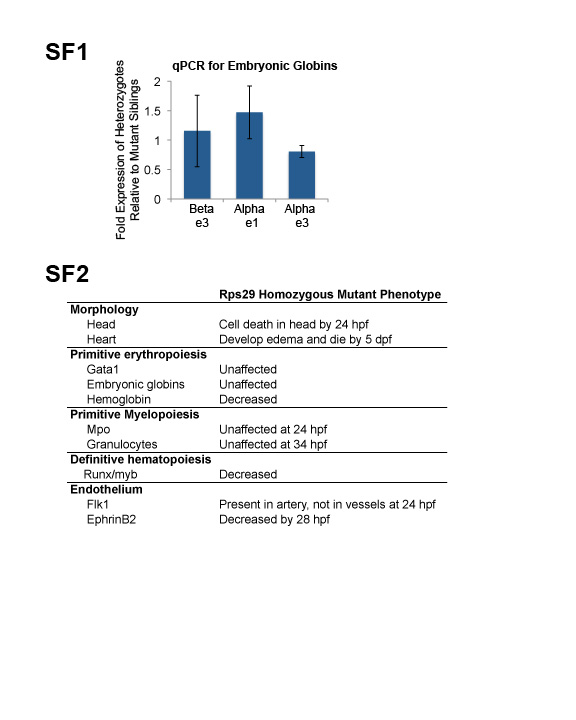
Zebrafish embryos undergo both primitive and definitive waves of hematopoiesis. We wanted to determine the effects of an rps29 insertional mutation[14] on these waves in the zebrafish. Rps29-/- embryos survive for five days[27], probably due to maternal rps29 protein deposited in the embryo. We previously showed that rps29-/- embryos have decreased definitive HSC formation, likely the result of a defect in aorta specification[20]. Because ribosomal proteins are mutated in patients with erythroid specific deficiencies, we hypothesized that the zebrafish rps29 mutation might also cause a defect in primitive red blood cell development independent of the definitive HSC defect. In the zebrafish embryo, primitive erythropoiesis begins in the posterior lateral plate mesoderm, and blood cells can be seen in circulation soon after 24 hpf[28]. Using whole mount in situ hybridization [29], we analyzed rps29 mutants for gata1, a marker of erythroid precursors, and βe3 globin, which also marks more mature erythrocytes. Gata1 expression in primitive erythroid cells and precursors appears normal at the 18 somite stage (ss) in the rps29 mutant, and βe3 globin levels in the erythroid lineage at 40-48 hpf are unaffected (Figure 1A and 1B). RNA levels of αe1 and αe3 are comparable between heterozygous and mutant embryos, as determined by qPCR (Supplemental Figure 1). βe3 globin protein levels were also unchanged in the rps29 mutant (Figure 1A). In contrast, at the same stage, o-dianisidine (benzidine) staining is decreased (15 out of 21 embryos affected, Fisher's exact p=4.15×10-13), demonstrating a decrease in hemoglobin levels (Figure 1A). These data indicate a late stage defect in red cell differentiation or hemoglobinization and are suggestive of a hypochromic anemia that is typically due to a lack of iron, heme, or globin expression[30]. In contrast, primitive granulocytes remain unaffected, as determined by mpo RNA expression at 24 hpf and Sudan Black staining for mature granulocytes at 34 hpf (Figure 1C), suggesting an erythroid specific defect in primitive hematopoiesis. A summary of the mutant phenotypes, both primitive and definitive, is shown in Supplemental Figure 2.
Figure 1. Zebrafish rps29 mutant has a late-stage erythropoietic defect.

(A) Whole embryo ISH for blood markers - gata1 at 18 somites; βe3 globin at 40-48 hpf. Fluorescence antibody staining for βe3 globin at 48 hpf. O-dianisidine (benzidine) stain at 40 hpf.
(B) RT-PCR for βe3 globin. Data shown are fold changes of expression in rps29+/+ and rps29+/- embryos compared to rps29-/- siblings at 40 hpf.
(C) Whole embryo ISH for mpo at 24 hpf; Sudan Black stain at 34 hpf.
Microarray identifies hematopoietic, skeletal, and neural pathways affected in the rps29 mutant
To identify pathways and gene networks affected by the rps29 mutation, we performed a Nimblegen microarray comparing 24 hpf rps29 homozygous mutants vs. their heterozygous and wildtype siblings (Figure 2A). Because wildtype and heterozygous embryos cannot be separated by morphology, they were pooled for the microarray analysis. Rps29 was the most down-regulated gene, and rps29 levels are decreased in both heterozygous and homozygous embryos as early as 8ss (Figure 2B). Approximately 1000 genes were differentially regulated (Supplemental Table 1). To highlight statistically significant pathways from this list, we used Ingenuity Pathway Analysis (IPA). Tissue-specific pathways, including blood, skeleton, and neuron development, were among the pathways altered by rps29 mutation (Figure 2C). We assayed representative genes from these pathways by ISH, allowing analysis of individual wildtype, heterozygous, and mutant embryos. Genes involved in hematopoiesis, including bcl11a and Notch, were decreased in homozygous mutants but not in heterozygous embryos (data not shown). However, orthopedia protein a (otpa), a homeobox containing gene expressed in the brain by 24 hpf[31], was affected in both heterozygous and homozygous mutants. Otpa levels were substantially decreased in the homozygous mutant embryo, but ISH analysis revealed that over one-third of heterozygous embryos had an intermediate phenotype (9 embryos out of 24, Fisher's exact p=0.0013) (Figure 2D). This was confirmed by qPCR (Figure 2E). Amongst the genes we examined by ISH, otpa is the only one that showed a distinct heterozygous vs. homozygous phenotype. The existence of a gene whose levels directly correlate to the expression of a ribosomal protein suggests a dose-dependent mechanism.
Figure 2. Rps29 mutant microarray reveals dose-dependent genes.

(A) Microarray schematic. Rps29+/- fish were incrossed, and mutant embryos were sorted at 24 hpf. RNA was isolated from sibling embryos and mutant embryos for microarray.
(B) Rps29 expression at the 8 somite stage by whole mount ISH.
(C) Pathways significantly affected in the rps29 mutant, identified by Ingenuity Pathway Analysis.
(D) Dose-dependent expression of otpa. Orthopedia protein a (otpa) levels were assayed by ISH at 24 hpf. Expression can be seen in the telencephalon and hindbrain.
(E) RT-PCR for otpa. Data shown are fold changes of expression in rps29+/- and rps29-/- embryos compared to wildtype siblings at 24 hpf.
Cell cycle and apoptotic effects of the rps29 mutation
As genes involved in cell cycle, cell death, and metabolism were also identified by gene expression studies with microarrays, we determined if proliferation was affected or apoptosis was aberrantly induced at 24 hpf. By flow activated cell sorting (FACS) analysis on whole embryos treated with propidium iodide, we generated cell cycle profiles. We determined that the profile of homozygous mutant embryos was not different from their wildtype and heterozygous siblings, suggesting that cell cycle progression is not affected (Figure 3A). In contrast, there is an increase in TUNEL-positive cells in the rps29 mutant compared to wildtype and heterozygous siblings, particularly in the head (7/8 mutant embryos, Fisher's exact p=0.001), demonstrating increased apoptosis (Figure 3B). We looked at p53 and its target p21 by quantitative PCR, and this assay confirmed p53 and p21 RNA levels were up-regulated over 3-fold in the 24 hpf rps29 mutant embryos (Figure 3C). These data indicate that a p53 apoptotic pathway is being activated in the mutant, mediated by p21 activation.
Figure 3. Apoptosis is induced in rps29-/- embryos.

(A) Cell cycle analysis. Propidium iodide was used to generate a cell cycle profile of rps29 homozygous mutants and their heterozygous and wildtype siblings at 24 hpf.
(B) TUNEL staining. TUNEL staining was performed on 24 hpf embryos from a rps29+/- incross, as well as embryos from a p53; rps29+/- incross.
(C) RT-PCR for p53 and p21. Data shown are fold changes of expression in rps29-/- mutant embryos compared to their heterozygous and wildtype siblings at 24 hpf.
p53-dependent irradiation signature activated in the rps29 mutant
Based on these data and data from the literature[32], we hypothesized that rps29 loss acts similarly to p53 activation. To assess this idea at the gene expression level, we compared the rps29 deficiency transcriptional profile to that of a p53 activation signature. To generate a p53 activation signature by microarray, we analyzed the transcriptome of both wildtype and p53M214K/M214K mutant embryos. (The p53M214K mutation is in the DNA binding domain.) We used gamma-radiation to activate p53, as radiation is an insult that is known to activate the p53 pathway[33]. We have previously shown that the p53 mutant no longer up-regulates p21 in response to irradiation[22]. We performed an Affymetrix microarray on the following four groups of 27 hpf embryos (in triplicate): wildtype unirradiated, wildtype irradiated, p53M214K/M214K unirradiated, and p53M214K/M214K irradiated (Supplemental Table 2). Comparing wildtype embryos to p53 mutants produces a list of genes affected by basal levels of p53, but does not identify genes up-regulated upon p53 activation. We compared wildtype unirradiated and irradiated lists to generate a p53 activation signature, and we excluded genes that failed to be up-regulated in p53M214K/M214K mutant embryos to ensure that the irradiation signature was specific to p53.
We then used gene set enrichment analysis (GSEA) to compare the genes up-regulated in the rps29 mutant to the p53 dataset. The genes up-regulated in the rps29 mutant were enriched for genes up-regulated in response to irradiation (Figure 4A). Of the 31 genes up-regulated in response to irradiation, 22 are also up-regulated in the rps29 mutant (Figure 4B). None of these 22 genes are up-regulated in the p53 mutant post irradiation. This result confirms that the gene expression signature of the rps29 mutant is similar to p53-dependent radiation responsive genes. The enriched gene list includes genes involved in DNA damage response and apoptosis, such as GADD45AL and BAX (Figure 4C). Our data demonstrate that the p53 pathway is being activated in the rps29 mutant, and suggest that mutations in p53 may rescue the rps29 mutant phenotype.
Figure 4. GSEA identifies a p53-dependent irradiation signature in rps29 mutants.

(A) GSEA enrichment plot comparing genes up-regulated in the rps29-/- embryo to genes up-regulated in wildtype embryos in response to irradiation.
(B) Comparison of gene lists. Of the 31 genes up-regulated in the wildtype embryo post-irradiation, 22 are also up-regulated in the rps29 mutant. There are no genes that overlap between all 3 lists: rps29 mutant, wildtype irradiated, and p53 irradiated.
(C) Genes up-regulated in both rps29-/- embryos and wildtype irradiated embryos.
p53 mutation rescues the rps29 mutant phenotype
To test our hypothesis that the rps29 mutant phenotype was also p53 dependent in vivo, we generated rps29-/-;p53M214K/M214K embryos. The following phenotypes were restored to wildtype levels in double mutants compared to the rps29 homozygous mutant: head morphology (18/18 mutant embryos rescued), TUNEL staining (2/2), o-dianisidine staining (7/7), HSC markers (5/5), and flk1 expression in the intersomitic vessels (11/11) (Figure 5A-D, Figure 3B). The ability of the p53 mutation to rescue these phenotypes demonstrates that p53 activation is involved in the rps29 mutant phenotype. However, p53 mutation could not rescue the embryonic lethality of rps29 mutants. The inability to rescue embryonic lethality is likely because once maternal rps29 is completely lost, embryos can no longer make new ribosomes, as has been demonstrated in mammalian cell culture[19]. Even if p53 is also removed, the embryos still cannot make new proteins and therefore will die without rps29. We also looked at otpa levels in p53M214K/M214K;rps29-/- embryos; although otpa was not restored to WT levels, it had higher expression than rps29-/- alone (4/7 mutant embryos, Figure 5F). In the case of the otpa phenotype, other mechanisms may be involved in addition to p53 activation. Our studies of the rps29 mutant were able to identify p53 as a modifier of the mutant phenotype, and this system can be used to identify novel modifiers of ribosomal protein mutation.
Figure 5. p53 mutation rescues the rps29 mutant.

(A) Morphology of embryos at 24 hpf.
(B) Hemoglobin at 40-48 hpf measured by staining with o-dianisidine (benzidine).
(C) HSCs at 36 hpf measured by ISH for myb.
(D) Vasculature at 24 hpf assayed by ISH for flk1.
(E) Otpa expression at 24 hpf assayed by ISH.
Discussion
Ribosomal protein mutations in genes such as rps19 and rpL11 cause blood specific defects, and this is seen in patients with DBA. We described erythroid and HSC defects in the zebrafish embryo resulting from homozygous mutation of rps29, a protein in the 40s small subunit of the ribosome. We identified a gene expression signature of p53 activation in the rps29 mutant and tested the role of p53 activation genetically by generating rps29-/-;p53-/- embryos. Loss of p53 rescues all of the hematopoietic and morphological phenotypes in the rps29 mutant that we tested. Skeletal and neuronal tissues are also affected, including a gene, otpa, whose expression is dependent on rps29 expression level.
Our work builds upon the previously characterized HSC and artery defects of the rps29 mutant[20]. We went on to characterize red blood cells in the mutant, as ribosomal protein mutations cause erythroid specific defects in patients with DBA. (N.B. Mutations in rps29 have not been found in DBA patients.) Red blood cells in the homozygous rps29 mutant are indeed affected as hemoglobin levels are decreased. Since embryonic globin RNA is normal, this defect may be at a terminal stage of differentiation or specific to hemoglobinization. This phenotype is in contrast with the erythroid progenitor block seen in DBA patients, but mirrors what is seen in the zebrafish rps19 morphant and rpL11 mutant. Although the specific stage of the erythropoietic defect differs in zebrafish ribosomal protein mutants and patients, the trends of the zebrafish mutants phenocopy DBA in both skeletal and hematopoietic tissues. Erythroid cells are affected, but myeloid tissues remain largely unaffected in both cases. Mechanistically, the role of p53 activation in the rps29 mutant is consistent with other ribosomal protein mutants and DBA models.
Our data demonstrate that p53 is a mediator of the rps29 mutant phenotype. Previous studies have suggested a critical role for p53 in ribosomal protein mutant phenotypes, and our findings extend this observation to an additional ribosomal protein, rps29. GSEA shows strong enrichment for a p53 activation gene signature, as 22 out of 31 genes in a p53-dependent irradiation signature are also up in the rps29 mutant. Studies from others suggest that the mechanism of p53 activation is nucleolar/ribosomal stress[13]. In this model, a mutated ribosomal protein prevents ribosomal subunit formation, leading to an increase in free ribosomal proteins. Some of these, including rpL11 and rpL5, can bind mdm2 and sequester it from p53, leading to p53 activation. Our data suggest that the mechanism of p53 activation in the mutant mirrors that of irradiation, the source of p53 activation in our analysis. Interestingly, there is evidence that some DNA damage agents can initiate ribosomal stress[34], and a common pathway could explain the similar gene signatures. However, the downstream targets of p53 in the rps29 mutant are not yet fully elucidated.
It is interesting that p53 knockdown also rescues the hemoglobin defect, as this suggests that p53 activation is causing the hemoglobin defect. Other zebrafish studies have tested the role of p53 activation with conflicting results; a p53 morpholino rescues the hemoglobin defect in the rpL11 mutant, but does not rescue the defect induced by rps19 morpholino knockdown[16, 18]. Interestingly, morpholino knockdown of telomerase reverse transcriptase (TERT) in zebrafish embryos also causes apoptosis and hematopoietic specific defects[29]. Like the rps29 mutant, many of the zTERT morphant phenotypes are p53-dependent; in contrast, the zTERT hemoglobin defect cannot be rescued by p53 knockdown. The reason for these discrepancies is unclear, but could be the result of differences between mutants and morpholino knockdown. Homozygous embryos can get maternally deposited RNA from a heterozygous parent, whereas an ATG morpholino would inhibit translation of maternal RNAs and cause a more severe knockdown, making rescue more difficult. We still do not know how p53 activation inhibits heme biosynthesis. One hypothesis is based on evidence that p53 in the mitochondria is involved in heme biosysthesis and iron-sulfur (Fe-S) cluster formation. Heme biosynthesis requires synthesis of ALAS2, which our lab has previously shown to be inhibited when Fe-S cluster biogenesis is impacted[30]. Assembly of Fe-S clusters requires intact mitochondria[35], but p53 activation causes mitochondrial membrane permeabilization[36]. In this model, p53 activation would affect Fe-S cluster biogenesis and subsequent heme biosynthesis, and the effect would be rescued by p53 knockdown.
An advantage of using a zebrafish mutant for these studies is the ability to compare both heterozygous and homozygous mutants, and subsequently identify gene expression patterns that correlate with rps29 level. Although we could not separate wildtype and heterozygous embryos for the microarray, we could still identify dosage specific defects by whole mount ISH. Orthopedia protein a (otpa) is a homeobox containing gene expressed in the brain and important for neuron development[31], and the levels of otpa depend on rps29 levels. The existence of dose-dependent genes with respect to a ribosomal protein suggests a dosage sensitivity model. In wildtype embryos, the ribosome can function normally, translation occurs normally, and genes such as myb, flk1, and otpa are expressed as expected. In the homozygous rps29 mutant, there is a downstream effect on myb, flk1, and otpa expression. When only one copy of rps29 is mutated, only otpa is down-regulated and many other genes are unaffected. An alternate hypothesis is that otpa expression is not differentially regulated, but stability of otpa mRNA is affected by rps29 mutation. Regulation of RNA expression or stability could still be dose-dependent on rps29 mRNA levels. Interestingly, there are cases where relatives of DBA patients have the same mutation but milder symptoms of the disease, and this may be the result of different expression levels of the functional ribosomal protein. Our data are in agreement with the hypothesis that differences of a ribosomal protein level could cause a change in disease presentation.
p53 mutation partially rescues otpa in the homozygous mutant to heterozygous levels, suggesting that p53 activation may play a role in both the heterozygous and homozygous rps29 phenotype. The fact that p53 mutation only partially rescues otpa mRNA levels further suggests that otpa is affected by rps29 knockdown by both p53-dependent and p53-independent mechanisms. It is possible that a protein not efficiently translated in the rps29 heterozygote (or homozygote) directly regulates otp transcription, leading to a dose-dependent decrease in otpa mRNA expression in the rps29 mutant. Another possibility is that otpa directly interacts with the ribosome. There may also be a p53-independent stress signal in the rps29 heterozygote and homozygote, causing some neuronal tissues to die without p53 activation. Further testing is required to determine the exact p53-independent mechanism causing a decrease of otpa mRNA in the rps29 mutant.
There are still lingering questions about the exact mechanism downstream of ribosomal protein mutation, and modifier screens in the zebrafish could address these questions. The zebrafish embryo is amenable to in vivo chemical and genetic screens. For example, a genetic screen for rescue of the rps29 mutant would identify novel pathways, p53 dependent or independent, mediating the mutant phenotype. These pathways can provide insight into exactly how erythropoiesis and HSC formation are affected by ribosomal protein mutation and p53 activation. An in vivo chemical screen for compounds that rescue the hematopoietic defect would identify therapeutics that may be more likely to translate to patients than hits from an in vitro screen. The unique advantages of a zebrafish DBA model and ribosomal deficiencies hold promise for the field of ribosomal protein biology.
Supplementary Material
{kind=link}
Supplemental Figure 1. Expression of embryonic globins by RT-PCR.
RT-PCR for αe1, αe3, and βe3 globins in 48 hpf heterozygous embryos compared to mutant siblings.
Supplemental Figure 2. Summary of Hematopoietic Defects in the Rps29 Mutant.
Acknowledgements
We would like to thank Yi Zhou and Anthony DiBiase for help with analysis of the microarray data, Ronald Mathieu for flow cytometry assistance, and Benjamin Ebert for helpful feedback on this manuscript.
Support and Financial Disclosure Declaration
A.M.T. was supported in part by NIH Cell and Developmental Biology Training Grant GM07226. L.I.Z. is supported by HHMI, and this work was funded by NHLBI 5U01 HL10001-02. L.I.Z. is a founder and stock holder of Fate, Inc. and a scientific advisor for Stemgent.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142:859–876. doi: 10.1111/j.1365-2141.2008.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vlachos A, Muir E. How I treat Diamond Blackfan anemia. Blood. 2010;116:3715–3723. doi: 10.1182/blood-2010-02-251090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Draptchinskaia N, Gustavsson P, Andersson B, et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet. 1999;21:169–175. doi: 10.1038/5951. [DOI] [PubMed] [Google Scholar]
- 4.Choesmel V, Fribourg S, Aguissa-Toure AH, et al. Mutation of ribosomal protein RPS24 in Diamond-Blackfan anemia results in a ribosome biogenesis disorder. Hum Mol Genet. 2008;17:1253–1263. doi: 10.1093/hmg/ddn015. [DOI] [PubMed] [Google Scholar]
- 5.Cmejla R, Cmejlova J, Handrkova H, Petrak J, Pospisilova D. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum Mutat. 2007;28:1178–1182. doi: 10.1002/humu.20608. [DOI] [PubMed] [Google Scholar]
- 6.Farrar JE, Nater M, Caywood E, et al. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood. 2008;112:1582–1592. doi: 10.1182/blood-2008-02-140012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gazda HT, Sheen MR, Vlachos A, et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am J Hum Genet. 2008;83:769–780. doi: 10.1016/j.ajhg.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doherty L, Sheen MR, Vlachos A, et al. Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2010;86:222–228. doi: 10.1016/j.ajhg.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsson H, Davey EJ, Draptchinskaia N, et al. Targeted disruption of the ribosomal protein S19 gene is lethal prior to implantation. Mol Cell Biol. 2004;24:4032–4037. doi: 10.1128/MCB.24.9.4032-4037.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsson H, Davey EJ, Frojmark AS, et al. Erythropoiesis in the Rps19 disrupted mouse: Analysis of erythropoietin response and biochemical markers for Diamond-Blackfan anemia. Blood Cells Mol Dis. 2006;36:259–264. doi: 10.1016/j.bcmd.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 11.McGowan KA, Li JZ, Park CY, et al. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet. 2008;40:963–970. doi: 10.1038/ng.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones NC, Lynn ML, Gaudenz K, et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med. 2008;14:125–133. doi: 10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fumagalli S, Di Cara A, Neb-Gulati A, et al. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol. 2009;11:501–508. doi: 10.1038/ncb1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amsterdam A, Sadler KC, Lai K, et al. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004;2:E139. doi: 10.1371/journal.pbio.0020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danilova N, Sakamoto KM, Lin S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 2008;112:5228–5237. doi: 10.1182/blood-2008-01-132290. [DOI] [PubMed] [Google Scholar]
- 16.Torihara H, Uechi T, Chakraborty A, Shinya M, Sakai N, Kenmochi N. Erythropoiesis failure due to RPS19 deficiency is independent of an activated Tp53 response in a zebrafish model of Diamond-Blackfan anaemia. Br J Haematol. 2011;152:648–654. doi: 10.1111/j.1365-2141.2010.08535.x. [DOI] [PubMed] [Google Scholar]
- 17.Chakraborty A, Uechi T, Higa S, Torihara H, Kenmochi N. Loss of ribosomal protein L11 affects zebrafish embryonic development through a p53-dependent apoptotic response. PLoS One. 2009;4:e4152. doi: 10.1371/journal.pone.0004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Danilova N, Sakamoto KM, Lin S. Ribosomal protein L11 mutation in zebrafish leads to haematopoietic and metabolic defects. Br J Haematol. 2011;152:217–228. doi: 10.1111/j.1365-2141.2010.08396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Donohue MF, Choesmel V, Faubladier M, Fichant G, Gleizes PE. Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J Cell Biol. 2010;190:853–866. doi: 10.1083/jcb.201005117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burns CE, Galloway JL, Smith AC, et al. A genetic screen in zebrafish defines a hierarchical network of pathways required for hematopoietic stem cell emergence. Blood. 2009;113:5776–5782. doi: 10.1182/blood-2008-12-193607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertrand JY, Chi NC, Santoso B, Teng S, Stainier DY, Traver D. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature. 2010;464:108–111. doi: 10.1038/nature08738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berghmans S, Murphey RD, Wienholds E, et al. tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc Natl Acad Sci U S A. 2005;102(2):407–12. doi: 10.1073/pnas.0406252102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 24.Paffett-Lugassy NN, Zon LI. Analysis of hematopoietic development in the zebrafish. Methods Mol Med. 2005;105:171–198. doi: 10.1385/1-59259-826-9:171. [DOI] [PubMed] [Google Scholar]
- 25.Le Guyader D, Redd MJ, Colucci-Guyon E, et al. Origins and unconventional behavior of neutrophils in developing zebrafish. Blood. 2008;111:132–141. doi: 10.1182/blood-2007-06-095398. [DOI] [PubMed] [Google Scholar]
- 26.Choe SE, Boutros M, Michelson AM, Church GM, Halfon MS. Preferred analysis methods for Affymetrix GeneChips revealed by a wholly defined control dataset. Genome Biol. 2005;6:R16. doi: 10.1186/gb-2005-6-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci U S A. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Jong JL, Zon LI. Use of the zebrafish system to study primitive and definitive hematopoiesis. Annu Rev Genet. 2005;39:481–501. doi: 10.1146/annurev.genet.39.073003.095931. [DOI] [PubMed] [Google Scholar]
- 29.Imamura S, Uchiyama J, Koshimizu E, et al. A non-canonical function of zebrafish telomerase reverse transcriptase is required for developmental hematopoiesis. PLoS One. 2008;3:e3364. doi: 10.1371/journal.pone.0003364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wingert RA, Galloway JL, Barut B, et al. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature. 2005;436:1035–1039. doi: 10.1038/nature03887. [DOI] [PubMed] [Google Scholar]
- 31.Ryu S, Mahler J, Acampora D, et al. Orthopedia homeodomain protein is essential for diencephalic dopaminergic neuron development. Curr Biol. 2007;17:873–880. doi: 10.1016/j.cub.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009;16:369–377. doi: 10.1016/j.ccr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langheinrich U, Hennen E, Stott G, Vacun G. Zebrafish as a model organism for the identification and characterization of drugs and genes affecting p53 signaling. Curr Biol. 2002;12:2023–2028. doi: 10.1016/s0960-9822(02)01319-2. [DOI] [PubMed] [Google Scholar]
- 34.Llanos S, Serrano M. Depletion of ribosomal protein L37 occurs in response to DNA damage and activates p53 through the L11/MDM2 pathway. Cell Cycle. 2010;9:4005–4012. doi: 10.4161/cc.9.19.13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veatch JR, McMurray MA, Nelson ZW, Gottschling DE. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell. 2009;137:1247–1258. doi: 10.1016/j.cell.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta. 2009;1787:414–420. doi: 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Expression of embryonic globins by RT-PCR.
RT-PCR for αe1, αe3, and βe3 globins in 48 hpf heterozygous embryos compared to mutant siblings.
Supplemental Figure 2. Summary of Hematopoietic Defects in the Rps29 Mutant.