

Abstract
Infectious complications are a serious cause of morbidity and mortality following hematopoietic stem cell transplantation (HSCT), and the lung is a particular target organ post-transplant. Our laboratory has used a murine bone marrow transplant model to study alterations in immunity that occur as a result of transplantation. Our studies focus on immune responses that occur following immune cell reconstitution in the absence of immunosuppressive drug therapy or graft-versus-host disease. We have found that impaired clearance of both bacterial and viral pulmonary infections is related to specific alterations in immune cell function and cytokine production. Our data offer insight into mechanisms that contribute to opportunistic infections in HSCT recipients.
Keywords: Hematopoietic stem cell transplant, Pneumonia, Pseudomonas aeruginosa, Murine gammaherpesvirus-68, Opportunistic infections, Innate immunity, Adaptive immunity
Overview of HSCT
Since the first transplants in human patients were performed in the late 1950s [1, 2], hematopoietic stem cell transplantation (HSCT) has become an important therapy in the treatment of several malignant, inherited, and autoimmune disorders [3]. During HSCT, patients are treated with a conditioning regimen and are then infused intravenously with their own hematopoietic stem cells (autologous) or HSCs from a donor (allogeneic). Autologous transplants are performed more frequently than allogeneic [4]. Various preparative regimens for HSCT have been used, but conditioning generally includes chemotherapy, such as cyclophosphamide and/or total body irradiation [5]. These regimens have several consequences, including eradication of malignant or self-reactive cells and providing an immunosuppressed environment for the transplantation of allogeneic cells [5, 6]. Stem cells for transplantation are derived from a variety of sources. Cells may be collected by aspiration of bone marrow, harvested from umbilical cord blood [7], or donors may be treated with growth factors such as granulocyte colony-stimulating factor (G-CSF) to mobilize stem cells to peripheral blood [8]. Additionally, cell infusions may or may not include specific subsets of cells. For instance, the inclusion of donor T cells in allogeneic grafts can be useful for promoting an immune response of grafted cells against malignant cells [9].
Post-transplant complications
Despite promising success in the treatment of many diseases, the efficacy of HSCT is limited due to significant transplant-related morbidity and mortality. There are a wide variety of complications that can occur following HSCT, both non-infectious and infectious. Specifically, pulmonary complications comprise a large group of post-transplant problems and are reported to occur in up to 60% of HSCT recipients [10]; one study of deceased HSCT recipients found pulmonary complications in 89% of the cohort [11].
Noninfectious complications may include graft-versus-host disease (GVHD) in the allogeneic setting [12], second malignancy [13], and late non-infectious lung complications, such as bronchiolitis obliterans and idiopathic pneumonia syndrome [14].
Infectious complications can occur in both allogeneic [15] and autologous recipients [16, 17], though they are more common in allogeneic transplants [10], presumably due to GVHD and immunosuppressive drug therapy. Infections tend to occur in the lung, skin, and genitourinary tract and are associated with the presence of indwelling catheters [18]. Historically, cytomegalovirus (CMV) was the most significant viral complication of HSCT; mortality rates of CMV pneumonia in HSCT recipients were near 85% prior to the development of anti-viral drug therapy [19]. Despite advances in prophylaxis and antiviral therapy, CMV disease remains a significant complication of HSCT [20]. HSCT recipients are additionally at-risk for infections by other herpesviruses, such as human herpes-virus-6, herpes simplex virus, and varicella zoster [10, 21, 22], as well as community-acquired respiratory viruses, such as influenza [21]. Pseuodomonas aeruginosa and Klebsiella pneumonia represent important gram-negative bacterial infections in HSCT recipients [23], and common gram-positive bacterial infections in HSCT populations are Staphylococcus epidermidis, Staphylococcus aureus, and viridians streptococci [18]. Fungal infections caused by Candida and Aspergillus species are also significant threats post-HSCT [24].
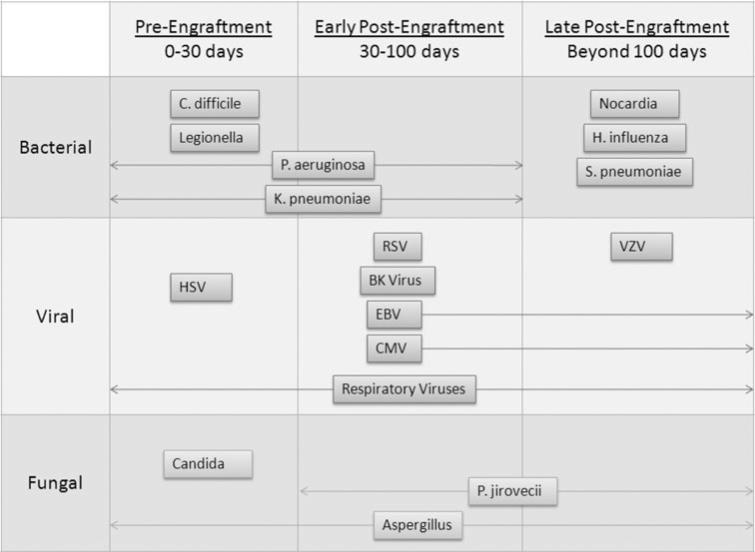
Infectious complications can be characterized by the time point during which they occur post-HSCT, including pre-engraftment (during neutropenia, 1 month post-HSCT), early post-engraftment, and late post-engraftment (approximately 3 months post-transplant) [18, 23, 25]. Table 1 summarizes infections reported to occur during these post-transplant time periods. Opportunistic infections, though rare, have been reported to occur late post-transplant in autologous patients [26, 27]. This fact suggests that transplantation, even in the absence of immunosuppressive therapy and GVHD, can lead to long-term immune dysfunction.
Table 1.
|
Immune defects post-HSCT
Beginning with the emergence of the innate immune system, reconstitution of donor-derived immune cells spans over the course of several months to a year following HSCT [28–30]. However, reconstitution of various immune cell compartments does not typically coincide with restoration of immune function. Within the first few months of transplant, marked reduction in neutrophil chemotaxis, phagocytosis, and bacterial killing is observed, contributing to patient susceptibility to a number of infections [31]. Similar defects are seen in tissue macrophage function [32]. Impaired mitogen proliferation and cytokine production also remain a common feature among both B- and T-cell subsets [28, 29, 33]. Although these defects have been broadly reported, mechanisms behind reduced cellular-mediated immunity following immune reconstitution are poorly understood. Our work has employed a murine model of HSCT to determine potential causes for impaired immune function following donor-cell reconstitution, with particular focus on the impact of reduced immunity on host defense in the lung.
Animal modeling
Our laboratory has developed two murine models of bone marrow transplantation (BMT) to examine the effect of HSCT on pulmonary immune function and host defense. This includes syngeneic and allogeneic BMT, where bone marrow is harvested from C57BL/6 or Balb/c mice, respectively, and infused by tail vein injection into lethally irradiated C57BL/6 recipients. Ablation of host-derived HSC in our mice has involved either TBI or cyclophosphamide/busulfan chemotherapy preparative regimens. However, we chose TBI as the primary means to ablate host-derived HSC, given that TBI eradicates host-derived HSC more efficiently than chemotherapy regimens and maximizes reconstitution of donor-derived cells [34]. Using this method, recipient mice are given a fractionated dose of 13 Gy TBI from either a 137Cs or x-ray orthovoltage source. Complete immune reconstitution is achieved five weeks following infusion of 5 9 106 whole bone marrow cells into TBI recipients [34, 35]. The percentage of donor-derived cells is approximately 94.9 ± 1.1% in the spleen at this time point, as assessed by transplanting CD45.1+ bone marrow into C57Bl/6 CD45.2+ mice [34]. However, addition of 1 × 106 purified splenic T cells at the time of whole bone marrow infusion increases the rate of donor-cell reconstitution from 5–3 weeks [34, 36, 37].
Given that HSCT patients are increasingly susceptible to bacterial and viral infections of the lung throughout preand post-engraftment phases (Table 1), our research has focused primarily on pulmonary host defense against model opportunistic pathogens post-HSCT. To date, we have used the gram-negative bacteria, Pseudomonas aeruginosa, and the murine herpesvirus, gammaherpesvirus (γHV)-68, as our model opportunistic pathogens. Five weeks following syngeneic BMT in mice, we find that lung phagocytes display reduced host defense against P. aeruginosa both in vivo and in vitro compared to non-transplant controls. Furthermore, T-cell function in the lung is defective in BMT mice, contributing to reduced clearance of murine γHV-68 following intranasal challenge.
This impairment in pulmonary immunity is directly related to the highly immunosuppressive environment of the lung post-BMT. Following BMT, increased production of immunosuppressive mediators, such as prostaglandin E2 (PGE2) and TGF-β, is detected [35, 36]. In subsequent sections, we describe our models of bacterial and viral pneumonia post-BMT as well as the mechanisms that we have identified which contribute to impaired immunity in the lung following BMT.
Model of bacterial pneumonia post-BMT
P. aeruginosa infection is prevalent within the first 100 days following transplant [23, 25, 38]. P. aeruginosa is an ubiquitous pathogen, and ordinarily, exposure to P. aeruginosa through the lung airway is cleared by resident phagocytes [39]. In immunocompromised patients, however, P. aeruginosa is especially virulent and poses an increased risk in these individuals for pneumonia, bacteremia, and sepsis [40, 41]. To assess the ability of the immune reconstituted host to clear an opportunistic bacterial infection, our laboratory set up a model of acute P. aeruginosa lung infection in syngeneic BMT mice. Using syngeneic BMT, we have been able to explore how the transplant procedure alone (without GVHD or immunosuppressive drug therapy) impacts ongoing pulmonary immunity.
Impaired pulmonary host defense against P. aeruginosa post-BMT
Defective innate immunity
Following intratracheal challenge with a sublethal dose of P. aeruginosa, BMT mice display increased bacterial burden in the lung and dissemination of P. aeruginosa to the blood at 24 h relative to non-transplant controls [37]. This impairment in bacterial clearance coincides with reduced function of lung phagocytes, where BMT alveolar macrophages (AMs) show significant reduction in phagocytosis of both serum- and non-serum-opsonized bacteria [36, 37]. Furthermore, increased survival of ingested P. aeruginosa is observed in BMT AMs, suggesting defects in bacterial killing mechanisms in these phagocytes [36]. Neutrophils recruited to the lung following intratracheal injection of Pseudomonas-derived LPS also display similar defects in bacterial killing [36]. These results are consistent with experiments showing impaired bacterial killing mechanisms in AMs and neutrophils from HSCT patients [31, 32].
Cytokine and eicosanoid dysregulation
In addition to defective phagocytosis and microbial killing, BMT AMs have dysregulated production of proinflammatory cytokines and eicosanoids [42]. Post-BMT AMs display diminished TNF-α production relative to non-transplant controls. Furthermore, whole lung homogenates have reduced TNF-α levels, 24 h following P. aeruginosa lung infection. This defect in TNF-α production may explain the reduction we observe in pulmonary host-defense post-BMT, given that TNF-α can activate bacterial killing mechanisms in both macrophages and neutrophils [43–46]. Decreased production of cysteinyl leukotrienes (cys-LTs) is also observed in BMT AMs. Cys-LTs are a group of eicosanoids that can enhance proinflammatory cytokine production, phagocytosis, and microbial killing [47, 48]. Interestingly, exogenous treatment with the cys-LT, leukotriene D4, restores phagocytosis of serum- and non-serum-opsonized bacteria in BMT AMs.
Diminished production of proinflammatory mediators in the lung post-BMT may be related to elevated production of the immunosuppressive eicosanoid, prostaglandin E2 (PGE2), by alveolar epithelial cells, AMs, and recruited lung neutrophils [36]. PGE2 is known to inhibit phagocytosis, bacterial killing, chemotaxis, and proinflammatory cytokine production in phagocytes primarily via signaling through the E prostanoid 2 (EP2) receptor [49–51]. It is interesting to note that AMs from BMT mice express increased levels of the EP2 receptor making them more susceptible to the inhibitory actions of PGE2 [36]. In our model, we have also shown that pharmacologic inhibition of PGE2 production in vivo can restore host defense against P. aeruginosa pneumonia in BMT mice [36]. Furthermore, inhibition of PGE2 production can restore BMT AM and neutrophil function in vitro [36].
Granulocyte macrophage colony-stimulating factor (GM-CSF) production is also dysregulated in the lung post-BMT. GM-CSF can induce terminal differentiation of myeloid cells, including AMs, and is a critical factor in pulmonary host defense against P. aeruginosa [52–54]. In our model, BMT AMs produce higher levels of GM-CSF under LPS-stimulated and unstimulated conditions compared to control AMs [55]. However, overall production of GM-CSF is decreased in BMT lung homogenates [55], suggesting that lung parenchymal GM-CSF production is reduced post-transplant. To determine a role for GM-CSF in modulating host defense against P. aeruginosa post-BMT, WT mice were transplanted with GM-CSF-deficient bone marrow. Bacterial clearance and phagocyte host defense function were improved in GM-CSF -/- BMT mice compared to WT BMTs [55]. In these transplants, the predominant source of GM-CSF in the lung was from structural cells, and this scenario showed improved host-defense post-BMT. Interestingly, creating the opposite chimera by transplanting WT bone marrow into GM-CSF -/- mice resulted in severe susceptibility to P. aeruginosa infection [55]. In these transplants, the lack of GM-CSF production by structural cells was clearly harmful even if the hematopoietic cells were producers. These results highlight the complexity of cytokine compartmentalization in the setting of BMT, as well as the importance of cross talk between lung parenchymal cells and AMs in the regulation of bacterial clearance following BMT.
Restored function in GM-CSF -/- BMT AMs was surprisingly associated with increased PGE2 production; however, activation of the PGE2 cyclic AMP (cAMP) signaling cascade was diminished [55]. This is likely due to the surprising reduction in expression of the EP2 receptor on BMT AMs in the absence bone marrow-derived GM-CSF [55]. How GM-CSF may regulate EP2 receptor expression and activation of the cAMP signaling cascade has not yet been determined.
Taken together, our research suggests a critical role for PGE2 and GM-CSF in the ongoing impairment of pulmonary host-defense post-BMT. Much of our current work has been focused on uncovering mechanisms for PGE2-mediated inhibition of alveolar macrophage function and identifying novel effectors in the PGE2 signaling pathway that may be targeted for potential therapeutic benefit.
Mechanisms for impaired pulmonary innate immunity in BMT mice
Post-BMT, elevated PGE2 levels in the lung inhibit a number of host defense mechanisms in phagocytes that help mediate proper uptake and clearance of bacterial pathogens. However, the cause of increased PGE2 production in the lung post-BMT remains unknown. In AMs, we have observed upregulation in expression of prostaglandin synthetic enzymes post-BMT, including cyclooxygenase (COX)-2 and PGE synthases 1 and 2 [56]. This may also explain the concomitant increases we observe in BMT AM production of prostacyclin, PGI2 [34]. Although it is not known why prostaglandin synthesis is upregulated post-BMT, our laboratory has identified two effectors in the PGE2 signaling pathway that may mediate inhibition of BMT AM function. These effectors are interleukin receptor-associated kinase (IRAK)- M and phosphatase and tensin homolog deleted on chromosome ten (PTEN).
Downstream targets of the PGE2 signaling cascade: PTEN and IRAK-M
Via the second messenger cAMP, the PGE2 signaling pathway is known to activate various downstream effectors for inhibition of phagocytosis, microbial killing, and TNF-α production in leukocytes [57]. PGE2 can signal through four G-protein-coupled receptors, EP receptors 1–4 [58]. PGE2 engagement of EP2 and EP4 is known to induce cAMP production by stimulating G-protein-adenylyl cyclase activity [59]. cAMP can then act on two effectors, exchange protein activated by cAMP (Epac)-1 and protein kinase A (PKA) to inhibit phagocytosis, microbial killing, and proinflammatory cytokine production [57]. cAMP binding to Epac-1 has been shown to increase activation of the lipid phosphatase PTEN in a Src homology phosphatase-1-dependent manner [60]. This elevation of PTEN lipid phosphatase activity inhibits phagocytosis in AMs by negatively regulating pathways that mediate phagocytic uptake of IgG-opsonized particles [60–63]. Similarly, our current research has shown that PTEN activity is increased in BMT AMs, as a result of AM overproduction of PGE2, and pharmacologic inhibition of PTEN improves innate immunity in the lung post-BMT (unpublished observation).
IRAK-M was also identified as an effector in the PGE2 signaling pathway that may mediate inhibition of host defense mechanisms in BMT AMs [56]. IRAK-M is a member of the IRAK family of serine/threonine kinases [64]. IRAK-1 and IRAK-4 interact with the MyD88 adaptor complex to mediate IL-1R/TLR signaling [64–66]. IRAK-M negatively regulates IL-1R/TLR signaling by binding to the adaptor complex and inhibiting activation of downstream signaling pathways that induce NF-κB-mediated proinflammatory cytokine production [67].
Given that BMT AMs have decreased production of the proinflammatory cytokine TNF-α, our laboratory sought to determine whether PGE2 was inducing expression of IRAK-M in BMT AMs. As predicted, BMT AMs have an approximately 3.5-fold increase in IRAK-M protein expression, which is related to increased PGE2 signaling [56]. Transplanting WT mice with IRAK-M-deficient bone marrow restores pulmonary host defense against P. aeruginosa post-BMT and improves AM bacterial killing, phagocytosis, and production of cysLTs and TNF-α [56]. No differences are observed in recruitment of neutrophils or other leukocyte effector populations to the lungs of WT and IRAK-M -/- BMT mice following P. aeruginosa infection [56]. This restoration in host defense occurs despite overproduction of PGE2 by BMT AMs [56], suggesting that the absence of IRAK-M mitigates the inhibitory effect of PGE2 signaling in AMs.
Currently, we are investigating whether PTEN, in addition to other known effectors of the PGE2 signaling pathway, is required for PGE2-mediated elevation of IRAK-M. It is plausible, however, that PTEN and IRAK-M may be downstream targets in the PGE2 signaling pathway that act independently of one another to regulate similar AM host defense mechanisms.
Model of viral pneumonia post-HSCT
Herpesvirus infections are particularly common opportunistic infections at all time points post-HSCT (see Table 1) [10, 21, 68]. Furthermore, the emergence of viral strains resistant to antiviral therapy [69, 70] warrants a better understanding of post-HSCT immune responses to viral infections. To understand how transplantation of HSCs leads to alterations in anti-viral immune responses post-transplant, despite complete immune cell reconstitution, we use the well-characterized murine herpesvirus, γHV-68, as a model pathogen. γHV-68, when administered intranasally, will establish a lytic infection principally in lung epithelial cells which lasts 7–10 days and will subsequently persist latently in epithelial cells, B cells, and macrophages [71, 72]. The immune response to this virus is complex (reviewed in [73]), but it is established that IFNγ production by CD4 + T cells is critical for control of lytic viral infection [74]. Studies of anti-viral immunity in our model have been performed in syngeneic and/or allogeneic (non-severe GVHD) transplant settings, and all experiments have been performed 5–6 weeks post-BMT, at a time when reconstitution of immune cell numbers has occurred [35].
Reduced viral clearance and enhanced pneumonitis in BMT mice
When infected intranasally with γHV-68, BMT mice are found to have increased lytic virus in the lung at day 7 post-infection [35]. This is shown by plaque assay, real time RT–PCR detection of two viral genes (DNA polymerase and gB, a viral capsid glycoprotein), and viral immunohistochemistry. At 21 days post-infection, however, there is no difference in viral load between BMT and control mice, yet there are considerable histological changes in BMT lungs at this time point, including significant alveolar airspace inflammation [75]. This latter observation supports previous data showing that herpes simplex type 1-induced pneumonitis in a murine GVHD model is independent of viral load [76]. Together, these data suggest that the anti-viral immune response in BMT mice is altered compared to non-transplanted control mice.
Mechanisms for impaired anti-viral immunity
Unlike our bacterial pneumonia model, upregulated PGE2 does not appear to be responsible for increased viral loads in BMT mice. Pharmacologic inhibition of PGE2 concurrent with viral infection does not abrogate increased viral loads, suggesting that PGE2 is not directly impairing the anti-viral immune response. Rather, impaired anti-viral immunity seems to relate to the overproduction of TGFβ in the lung, particularly from alveolar epithelial cells. The overproduction of TGFβ occurs concurrently with an expanded population of Foxp3-expressing regulatory T cells (Tregs) in lungs post-BMT. Although we initially hypothesized that Tregs might be suppressing the effector T-cell responses in this model, BMT mice treated with anti-CD25 prior to infection still had increased viral loads [35]. These data are in accordance with another study showing that adoptive transfer of Tregs in a BMT model can reduce GVHD but not alter anti-viral immunity [77]. Rather, our data suggest that TGFβ signaling may directly alter the effector T cells. Transplanting mice with bone marrow from a donor mouse that expresses a dominant negative TGFβ receptor II in T cells (T-cell DN-TGFβRII) creates a BMT mouse in which donor-derived CD4 and CD8 T cells are unresponsive to TGFβ signaling and leads to a restoration in anti-viral immunity [35].
Altered T-cell skewing in BMT mice
In order to assess the effects of TGFβ on T cells in our BMT mice, we analyzed cytokine production by T cells at day 7 post-infection. TGFβ is known to limit Th1 [78] and promote Th17 differentiation [79]. Indeed, at day 7 post-infection, BMT mice have decreased IFNγ-expressing CD4 cells and increased IL-17a-expressing CD4 cells in the lung, compared to non-transplant controls [35]. This alteration is correlated with a decrease in RNA expression of the Th1-promoting cytokine IL-12p35 in lung-derived CD11c + cells and IL-12p70 protein from bone marrow-derived dendritic cells (BMDCs) at baseline, prior to infection (unpublished observations). Accordingly, splenic T cells from BMT mice express decreased levels of Eomesodermin and Tbet, transcription factors associated with IFNγ production. Expression of these transcription factors in BMT splenic T cells decreases with increased irradiation dose, highlighting again the importance of TBI-conditioned structural cells in regulating the phenotype of donor-derived hematopoietic cells (unpublished observations). BMT mice transplanted with T-cell DN-TGFbRII bone marrow have restored T-cell IFNγ production [35]. Taken together, these data support a model wherein BMT mice are skewed away from a Th1 environment in both the lung and periphery at baseline, perhaps through actions of TGFβ and altered antigen presenting cell function, leading to a diminished anti-viral immune response.
Conclusion
Data from our laboratory suggest that both bacterial and viral pulmonary infections that occur following HSCT may be related to specific alterations in immune parameters that occur as a result of transplantation. Specific cellular alterations that we have found in our BMT models are summarized in Table 2. In our models, the lung environment is immunosuppressive post-BMT, even following immune cell reconstitution. Differential mechanisms appear to be responsible for impaired immunity to bacterial and viral infections, but both are due to the procedure of transplantation. Our work provides insight into understanding how the transplantation procedure, outside of immunosuppressive drug therapy and severe GVHD, leads to immunodeficiency post-transplant. Our next steps are to determine whether alterations that characterize these murine models are also present in humans post-HSCT. It is our hope that targeted therapies such as cyclooxygenase inhibitors or treatments that alter TGFβ signaling might offer novel treatments to prevent the devastating consequences of infection in HSCT patients.
Table 2.
Cellular alterations post-BMT
| Cell Type | Baseline alterations post-HSCT | Further alterations observed post-infection |
|---|---|---|
| Alveolar macrophages | ↑PGE2, IRAK-M, GM-CSF, PTEN activity ↓TNFα, CysLTs |
In response to P. aeruginosa: ↓ Phagocytosis& killing |
| Lung neutrophils | ↑PGE2 | ↓Bacterial killing |
| CD4T cells | ↓Responsein MLR ↑Lung Tregs |
In response to γHV-68: ↓Th1 ↑Th17 |
| Alveolarepithelial cell: | ↑TGFβ ↑PGE2, GM-CSF |
In response to P. aeruginosa: ↓ GM-CSF, CCL2 |
| Bone marrow-derived dendritic cells | ↑ IL-10 ↓ IL-12p70 ↑Responsein MLR |
This table summarizes alterations in specific cell types that we have observed in our BMT model both pre- and post-infection
Acknowledgments
This work was supported by NIH grant AI065543 awarded to BBM. SMC was supported by the Herman and Dorothy Miller Fund for Immunology research.
Footnotes
Stephanie M. Coomes and Leah L. N. Hubbard contributed equally to this work.
Contributor Information
Stephanie M. Coomes, Graduate Program in Immunology, University of Michigan, Ann Arbor, MI 48109-2200, USA
Leah L. N. Hubbard, Graduate Program in Immunology, University of Michigan, Ann Arbor, MI 48109-2200, USA
Bethany B. Moore, Department of Internal Medicine, Division of Pulmonary and Critical Care Medicine, University of Michigan, 4053 BSRB, 109 Zina Pitcher Pl., Ann Arbor, MI 48109-2200, USA Bmoore@umich.edu Department of Microbiology and Immunology, University of Michigan, Ann Arbor, MI 48109-2200, USA.
References
- 1.Thomas ED, Lochte HL, Jr, Cannon JH, Sahler OD, Ferrebee JW. Supralethal whole body irradiation and isologous marrow transplantation in man. J Clin Invest. 1959;38:1709–16. doi: 10.1172/JCI103949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas ED, Lochte HL, Jr, Lu WC, Ferrebee JW. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med. 1957;257:491–6. doi: 10.1056/NEJM195709122571102. [DOI] [PubMed] [Google Scholar]
- 3.Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med. 2006;354:1813–26. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- 4.Horowitz MM, Loberiza FR, Bredeson CN, Rizzo JD, Nugent ML. Transplant registries: guiding clinical decisions and improving outcomes. Oncology (Williston Park) 2001;15:649–59. Discussion 63–4, 66. [PubMed] [Google Scholar]
- 5.Aschan J. Risk assessment in haematopoietic stem cell transplantation: conditioning. Best Pract Res Clin Haematol. 2007;20:295–310. doi: 10.1016/j.beha.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Milanetti F, Abinun M, Voltarelli JC, Burt RK. Autologous hematopoietic stem cell transplantation for childhood autoimmune disease. Pediatr Clin North Am. 2010;57:239–71. doi: 10.1016/j.pcl.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Zhong XY, Zhang B, Asadollahi R, Low SH, Holzgreve W. Umbilical cord blood stem cells: what to expect. Ann N Y Acad Sci. 2010;1205:17–22. doi: 10.1111/j.1749-6632.2010.05659.x. [DOI] [PubMed] [Google Scholar]
- 8.Bensinger WI, Weaver CH, Appelbaum FR, et al. Transplantation of allogeneic peripheral blood stem cells mobilized by recombinant human granulocyte colony-stimulating factor. Blood. 1995;85:1655–8. [PubMed] [Google Scholar]
- 9.Fry TJ, Willasch A, Bader P. The graft-versus-tumor effect in pediatric malignancy. Pediatr Clin North Am. 2010;57:67–81. doi: 10.1016/j.pcl.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 10.Soubani AO, Miller KB, Hassoun PM. Pulmonary complications of bone marrow transplantation. Chest. 1996;109:1066–77. doi: 10.1378/chest.109.4.1066. [DOI] [PubMed] [Google Scholar]
- 11.Sharma S, Nadrous HF, Peters SG, et al. Pulmonary complications in adult blood and marrow transplant recipients: autopsy findings. Chest. 2005;128:1385–92. doi: 10.1378/chest.128.3.1385. [DOI] [PubMed] [Google Scholar]
- 12.Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373:1550–61. doi: 10.1016/S0140-6736(09)60237-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curtis RE, Rowlings PA, Deeg HJ, et al. Solid cancers after bone marrow transplantation. N Engl J Med. 1997;336:897–904. doi: 10.1056/NEJM199703273361301. [DOI] [PubMed] [Google Scholar]
- 14.Afessa B, Litzow MR, Tefferi A. Bronchiolitis obliterans and other late onset non-infectious pulmonary complications in hematopoietic stem cell transplantation. Bone Marrow Transplant. 2001;28:425–34. doi: 10.1038/sj.bmt.1703142. [DOI] [PubMed] [Google Scholar]
- 15.Martin-Pena A, Aguilar-Guisado M, Espigado I, Parody R, Miguel Cisneros J. Prospective study of infectious complications in allogeneic hematopoietic stem cell transplant recipients. Clin Transplant. 2010 doi: 10.1111/j.1399-0012.2010.01286.x. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 16.Gil L, Styczynski J, Komarnicki M. Infectious complication in 314 patients after high-dose therapy and autologous hematopoietic stem cell transplantation: risk factors analysis and outcome. Infection. 2007;35:421–7. doi: 10.1007/s15010-007-6350-2. [DOI] [PubMed] [Google Scholar]
- 17.Auner HW, Sill H, Mulabecirovic A, Linkesch W, Krause R. Infectious complications after autologous hematopoietic stem cell transplantation: comparison of patients with acute myeloid leukemia, malignant lymphoma, and multiple myeloma. Ann Hematol. 2002;81:374–7. doi: 10.1007/s00277-002-0484-1. [DOI] [PubMed] [Google Scholar]
- 18.Wingard JR, Hsu J, Hiemenz JW. Hematopoietic stem cell transplantation: an overview of infection risks and epidemiology. Infect Dis Clin North Am. 2010;24:257–72. doi: 10.1016/j.idc.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 19.Kotloff RM, Ahya VN, Crawford SW. Pulmonary complications of solid organ and hematopoietic stem cell transplantation. Am J Respir Crit Care Med. 2004;170:22–48. doi: 10.1164/rccm.200309-1322SO. [DOI] [PubMed] [Google Scholar]
- 20.Boeckh M, Nichols WG, Papanicolaou G, Rubin R, Wingard JR, Zaia J. Cytomegalovirus in hematopoietic stem cell transplant recipients: current status, known challenges, and future strategies. Biol Blood Marrow Transplant. 2003;9:543–58. doi: 10.1016/s1083-8791(03)00287-8. [DOI] [PubMed] [Google Scholar]
- 21.Ljungman P. Prevention and treatment of viral infections in stem cell transplant recipients. Br J Haematol. 2002;118:44–57. doi: 10.1046/j.1365-2141.2002.03515.x. [DOI] [PubMed] [Google Scholar]
- 22.de Pagter PJ, Schuurman R, Meijer E, van Baarle D, Sanders EA, Boelens JJ. Human herpesvirus type 6 reactivation after haematopoietic stem cell transplantation. J Clin Virol. 2008;43:361–6. doi: 10.1016/j.jcv.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 23.Gasink LB, Blumberg EA. Bacterial and mycobacterial pneumonia in transplant recipients. Clin Chest Med. 2005;26:647–59. vii. doi: 10.1016/j.ccm.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Asano-Mori Y. Fungal infections after hematopoietic stem cell transplantation. Int J Hematol. 2010;91:576–87. doi: 10.1007/s12185-010-0574-0. [DOI] [PubMed] [Google Scholar]
- 25.Afessa B, Peters SG. Major complications following hematopoietic stem cell transplantation. Semin Respir Crit Care Med. 2006;27:297–309. doi: 10.1055/s-2006-945530. [DOI] [PubMed] [Google Scholar]
- 26.Burns LJ. Late effects after autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2009;15:21–4. doi: 10.1016/j.bbmt.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 27.Jantunen E, Itala M, Siitonen T, et al. Late non-relapse mortality among adult autologous stem cell transplant recipients: a nationwide analysis of 1, 482 patients transplanted in 1990–2003. Eur J Haematol. 2006;77:114–9. doi: 10.1111/j.1600-0609.2006.00685.x. [DOI] [PubMed] [Google Scholar]
- 28.Auletta JJ, Lazarus HM. Immune restoration following hematopoietic stem cell transplantation: an evolving target. Bone Marrow Transplant. 2005;35:835–57. doi: 10.1038/sj.bmt.1704966. [DOI] [PubMed] [Google Scholar]
- 29.Guillaume T, Rubinstein DB, Symann M. Immune reconstitution and immunotherapy after autologous hematopoietic stem cell transplantation. Blood. 1998;92:1471–90. [PubMed] [Google Scholar]
- 30.Lum LG. The kinetics of immune reconstitution after human marrow transplantation. Blood. 1987;69:369–80. [PubMed] [Google Scholar]
- 31.Zimmerli W, Zarth A, Gratwohl A, Speck B. Neutrophil function and pyogenic infections in bone marrow transplant recipients. Blood. 1991;77:393–9. [PubMed] [Google Scholar]
- 32.Winston DJ, Territo MC, Ho WG, Miller MJ, Gale RP, Golde DW. Alveolar macrophage dysfunction in human bone marrow transplant recipients. Am J Med. 1982;73:859–66. doi: 10.1016/0002-9343(82)90777-x. [DOI] [PubMed] [Google Scholar]
- 33.Mir MA, Battiwalla M. Immune deficits in allogeneic hematopoietic stem cell transplant (HSCT) recipients. Mycopathologia. 2009;168:271–82. doi: 10.1007/s11046-009-9181-0. [DOI] [PubMed] [Google Scholar]
- 34.Hubbard LL, Ballinger MN, Wilke CA, Moore BB. Comparison of conditioning regimens for alveolar macrophage reconstitution and innate immune function post bone marrow transplant. Exp Lung Res. 2008;34:263–75. doi: 10.1080/01902140802022518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coomes SM, Wilke CA, Moore TA, Moore BB. Induction of TGF-beta 1, not regulatory T cells, impairs antiviral immunity in the lung following bone marrow transplant. J Immunol. 2010;184:5130–40. doi: 10.4049/jimmunol.0901871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ballinger MN, Aronoff DM, McMillan TR, et al. Critical role of prostaglandin E2 overproduction in impaired pulmonary host response following bone marrow transplantation. J Immunol. 2006;177:5499–508. doi: 10.4049/jimmunol.177.8.5499. [DOI] [PubMed] [Google Scholar]
- 37.Ojielo CI, Cooke K, Mancuso P, et al. Defective phagocytosis and clearance of Pseudomonas aeruginosa in the lung following bone marrow transplantation. J Immunol. 2003;171:4416–24. doi: 10.4049/jimmunol.171.8.4416. [DOI] [PubMed] [Google Scholar]
- 38.Leung AN, Gosselin MV, Napper CH, et al. Pulmonary infections after bone marrow transplantation: clinical and radiographic findings. Radiology. 1999;210:699–710. doi: 10.1148/radiology.210.3.r99mr39699. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi H, Kobayashi O, Kawai S. Pathogenesis and clinical manifestations of chronic colonization by pseudomonas aeruginosa and its biofilms in the airway tract. J Infect Chemother. 2009;15:125–42. doi: 10.1007/s10156-008-0691-3. [DOI] [PubMed] [Google Scholar]
- 40.Marty FM, Rubin RH. The prevention of infection post-transplant: the role of prophylaxis, preemptive and empiric therapy. Transpl Int. 2006;19:2–11. doi: 10.1111/j.1432-2277.2005.00218.x. [DOI] [PubMed] [Google Scholar]
- 41.Micek ST, Lloyd AE, Ritchie DJ, Reichley RM, Fraser VJ, Kollef MH. Pseudomonas aeruginosa bloodstream infection: importance of appropriate initial antimicrobial treatment. Antimicrob Agents Chemother. 2005;49:1306–11. doi: 10.1128/AAC.49.4.1306-1311.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ballinger MN, McMillan TR, Moore BB. Eicosanoid regulation of pulmonary innate immunity post-hematopoietic stem cell transplantation. Arch Immunol Ther Exp (Warsz) 2007;55:1–12. doi: 10.1007/s00005-007-0001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leenen PJ, Canono BP, Drevets DA, Voerman JS, Campbell PA. TNF-alpha and IFN-gamma stimulate a macrophage precursor cell line to kill Listeria monocytogenes in a nitric oxide-independent manner. J Immunol. 1994;153:5141–7. [PubMed] [Google Scholar]
- 44.Munoz-Fernandez MA, Fernandez MA, Fresno M. Activation of human macrophages for the killing of intracellular trypanosoma cruzi by TNF-alpha and IFN-gamma through a nitric oxide-dependent mechanism. Immunol Lett. 1992;33:35–40. doi: 10.1016/0165-2478(92)90090-b. [DOI] [PubMed] [Google Scholar]
- 45.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–82. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 46.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 47.Kanaoka Y, Boyce JA. Cysteinyl leukotrienes and their receptors: cellular distribution and function in immune and inflammatory responses. J Immunol. 2004;173:1503–10. doi: 10.4049/jimmunol.173.3.1503. [DOI] [PubMed] [Google Scholar]
- 48.Peters-Golden M, Canetti C, Mancuso P, Coffey MJ. Leukotrienes: underappreciated mediators of innate immune responses. J Immunol. 2005;174:589–94. doi: 10.4049/jimmunol.174.2.589. [DOI] [PubMed] [Google Scholar]
- 49.Armstrong RA. Investigation of the inhibitory effects of PGE2 and selective EP agonists on chemotaxis of human neutrophils. Br J Pharmacol. 1995;116:2903–8. doi: 10.1111/j.1476-5381.1995.tb15943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J Immunol. 2004;173:559–65. doi: 10.4049/jimmunol.173.1.559. [DOI] [PubMed] [Google Scholar]
- 51.Kunkel SL, Spengler M, May MA, Spengler R, Larrick J, Remick D. Prostaglandin E2 regulates macrophage-derived tumor necrosis factor gene expression. J Biol Chem. 1988;263:5380–4. [PubMed] [Google Scholar]
- 52.Ballinger MN, Paine R, 3rd, Serezani CH, et al. Role of granulocyte macrophage colony-stimulating factor during gram-negative lung infection with pseudomonas aeruginosa. Am J Respir Cell Mol Biol. 2006;34:766–74. doi: 10.1165/rcmb.2005-0246OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berclaz PY, Shibata Y, Whitsett JA, Trapnell BC. GM-CSF, via PU.1, regulates alveolar macrophage Fcgamma R-mediated phagocytosis and the IL-18/IFN-gamma -mediated molecular connection between innate and adaptive immunity in the lung. Blood. 2002;100:4193–200. doi: 10.1182/blood-2002-04-1102. [DOI] [PubMed] [Google Scholar]
- 54.Fleetwood AJ, Cook AD, Hamilton JA. Functions of granulocyte-macrophage colony-stimulating factor. Crit Rev Immunol. 2005;25:405–28. doi: 10.1615/critrevimmunol.v25.i5.50. [DOI] [PubMed] [Google Scholar]
- 55.Ballinger MN, Hubbard LL, McMillan TR, et al. Paradoxical role of alveolar macrophage-derived granulocyte-macrophage colony-stimulating factor in pulmonary host defense post-bone marrow transplantation. Am J Physiol Lung Cell Mol Physiol. 2008;295:L114–22. doi: 10.1152/ajplung.00309.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hubbard LL, Ballinger MN, Thomas PE, et al. A role for IL-1 Receptor-associated kinase-M in Prostaglandin E(2)-induced immunosuppression post-bone marrow transplantation. J Immunol. 2010;184:6299–308. doi: 10.4049/jimmunol.0902828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J Immunol. 2005;174:595–9. doi: 10.4049/jimmunol.174.2.595. [DOI] [PubMed] [Google Scholar]
- 58.Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–7. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 59.Regan JW. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003;74:143–53. doi: 10.1016/j.lfs.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 60.Canetti C, Serezani CH, Atrasz RG, White ES, Aronoff DM, Peters-Golden M. Activation of phosphatase and tensin homolog on chromosome 10 mediates the inhibition of FcgammaR phagocytosis by prostaglandin E2 in alveolar macrophages. J Immunol. 2007;179:8350–6. doi: 10.4049/jimmunol.179.12.8350. [DOI] [PubMed] [Google Scholar]
- 61.Cao X, Wei G, Fang H, et al. The inositol 3-phosphatase PTEN negatively regulates Fc gamma receptor signaling, but supports Toll-like receptor 4 signaling in murine peritoneal macrophages. J Immunol. 2004;172:4851–7. doi: 10.4049/jimmunol.172.8.4851. [DOI] [PubMed] [Google Scholar]
- 62.Cox D, Dale BM, Kashiwada M, Helgason CD, Greenberg S. A regulatory role for Src homology 2 domain-containing inositol 5′-phosphatase (SHIP) in phagocytosis mediated by Fc gamma receptors and complement receptor 3 (alpha(M)beta(2); CD11b/CD18). J Exp Med. 2001;193:61–71. doi: 10.1084/jem.193.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stiles BL. Phosphatase and tensin homologue deleted on chromosome 10: extending its PTENtacles. Int J Biochem Cell Biol. 2009;41:757–61. doi: 10.1016/j.biocel.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Janssens S, Beyaert R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol Cell. 2003;11:293–302. doi: 10.1016/s1097-2765(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 65.Gan L, Li L. Regulations and roles of the interleukin-1 receptor associated kinases (IRAKs) in innate and adaptive immunity. Immunol Res. 2006;35:295–302. doi: 10.1385/IR:35:3:295. [DOI] [PubMed] [Google Scholar]
- 66.Li X, Qin J. Modulation of Toll-interleukin 1 receptor mediated signaling. J Mol Med. 2005;83:258–66. doi: 10.1007/s00109-004-0622-4. [DOI] [PubMed] [Google Scholar]
- 67.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 68.Konoplev S, Champlin RE, Giralt S, et al. Cytomegalovirus pneumonia in adult autologous blood and marrow transplant recipients. Bone Marrow Transplant. 2001;27:877–81. doi: 10.1038/sj.bmt.1702877. [DOI] [PubMed] [Google Scholar]
- 69.Kontoyiannis DP, Lewis RE, Marr K. The burden of bacterial and viral infections in hematopoietic stem cell transplant. Biol Blood Marrow Transplant. 2009;15:128–33. doi: 10.1016/j.bbmt.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 70.Gohring K, Feuchtinger T, Mikeler E, et al. Dynamics of the emergence of a human cytomegalovirus UL97 mutant strain conferring ganciclovir resistance in a pediatric stem-cell transplant recipient. J Mol Diagn. 2009;11:364–8. doi: 10.2353/jmoldx.2009.080153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Flano E, Husain SM, Sample JT, Woodland DL, Blackman MA. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J Immunol. 2000;165:1074–81. doi: 10.4049/jimmunol.165.2.1074. [DOI] [PubMed] [Google Scholar]
- 72.Stewart JP, Usherwood EJ, Ross A, Dyson H, Nash T. Lung epithelial cells are a major site of murine gammaherpesvirus persistence. J Exp Med. 1998;187:1941–51. doi: 10.1084/jem.187.12.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Doherty PC, Christensen JP, Belz GT, Stevenson PG, Sangster MY. Dissecting the host response to a gamma-herpesvirus. Philos Trans R Soc Lond B Biol Sci. 2001;356:581–93. doi: 10.1098/rstb.2000.0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sparks-Thissen RL, Braaten DC, Hildner K, Murphy TL, Murphy KM. Virgin HWt. CD4 T cell control of acute and latent murine gammaherpesvirus infection requires IFNgamma. Virology. 2005;338:201–8. doi: 10.1016/j.virol.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 75.Coomes SM, Moore BB. Pleiotropic effects of transforming growth factor-beta in hematopoietic stem-cell transplantation. Transplantation. 2010 doi: 10.1097/TP.0b013e3181efd018. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adler H, Beland JL, Kozlow W, Del-Pan NC, Kobzik L, Rimm IJ. A role for transforming growth factor-beta1 in the increased pneumonitis in murine allogeneic bone marrow transplant recipients with graft-versus-host disease after pulmonary herpes simplex virus type 1 infection. Blood. 1998;92:2581–9. [PubMed] [Google Scholar]
- 77.Nguyen VH, Shashidhar S, Chang DS, et al. The impact of regulatory T cells on T-cell immunity following hematopoietic cell transplantation. Blood. 2008;111:945–53. doi: 10.1182/blood-2007-07-103895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ludviksson BR, Seegers D, Resnick AS, Strober W. The effect of TGF-beta1 on immune responses of naive versus memory CD4 + Th1/Th2 T cells. Eur J Immunol. 2000;30:2101–11. doi: 10.1002/1521-4141(200007)30:7<2101::AID-IMMU2101>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 79.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]