

Abstract
MUC1 is a membrane-tethered mucin glycoprotein expressed on the apical surface of mucosal epithelial cells. Previous in vivo and in vitro studies established that MUC1 counter-regulates airway inflammation by suppressing TLR signaling. In this report, we elucidate the mechanism by which MUC1 inhibits TLR5 signaling. Overexpression of MUC1 in human embryonic kidney HEK293 (293) cells dramatically reduced Pseudomonas aeruginosa (Pa)-stimulated IL-8 expression, and decreased the activation of NF-κB and MAPK compared with MUC1 non-expressing cells. Overexpression of MUC1 in 293 cells, however, did not affect NF-κB or MAKP activation in response to TNF-α. Overexpression of MyD88 abrogated the ability of MUC1 to inhibit NF-κB activation, and MUC1 overexpression inhibited flagellin-induced association of TLR5/MyD88, compared with controls. The MUC1 cytoplasmic tail (MUC1 CT) associated with TLR5 in all cells tested, including 293T cells, human lung adenocarcinoma cell line A549 cells, and human and mouse primary airway epithelial cells. Activation of EGFR tyrosine kinase with TGF-α induced phosphorylation of the MUC1 CT at the Y46 EKV sequence and increased association of MUC1/TLR5. Finally, in vivo experiments demonstrated increased immunofluorescence co-localization of Muc1/TLR5 and Muc1/phosphotyrosine staining patterns in mouse airway epithelium and increased Muc1 tyrosine phosphorylation in mouse lung homogenates following Pa infection. In conclusion, EGFR tyrosine phosphorylates MUC1, leading to an increase in its association with TLR5, thereby competitively and reversibly inhibiting recruitment of MyD88 to TLR5 and downstream signaling events. This unique ability of MUC1 to control TLR5 signaling suggests its potential role in the pathogenesis of chronic inflammatory lung diseases.
INTRODUCTION
MUC1 (MUC1 in humans, Muc1 in animals) is a transmembrane glycoprotein expressed on the apical surface of mucosal epithelial cells, as well as the surface of hematopoietic cells (1). MUC1 was originally cloned in epithelial-derived human breast and pancreatic cancer cells (2,3), where it is uniformly expressed on the cell surface, as well as in the cytosol and nucleus (4). The MUC1 protein consists of two non-covalently associated polypeptide subunits, an N-terminal extracellular subunit and a C-terminal subunit. The MUC1 ectodomain is composed almost entirely of variable numbers of tandem repeats, while the C-terminal subunit consists of a 58-amino acid extracellular juxtamembranous region, a transmembrane domain, and a highly conserved cytoplasmic tail (CT) containing multiple phosphorylation sites (1). The 72-amino acid MUC1 CT contains 7 tyrosine residues whose phosphorylation status has been associated with intracellular signal transduction in cancer cells (5).
In the airways, MUC1 expressed on exposed epithelia contributes to the first line of defense against inhaled pathogens, however, the physiological role of MUC1 during airway inflammation, particularly its CT with multiple signaling motifs, is largely unknown. Previously, we reported that Muc1 knockout mice intranasally infected with P. aeruginosa (Pa) displayed greater level of airway inflammation compared with wild type littermates. The anti-inflammatory effect of MUC1 was mediated through inhibition of the proinflammatory TLR5 signaling pathway (6,7), and Pa up-regulated MUC1 expression in airway epithelial cells (8). Therefore, it was concluded that increased MUC1/Muc1 expression subsequent to production of proinflammatory mediators led to attenuation of TLR signaling via a negative feedback mechanism (7).
TLRs comprise 10- (human) or 12- (mouse) member families of innate immune receptors that initiate intracellular signaling following ligation of pathogen-associated molecular patterns (9). With the exception of TLR3, all TLRs initiate signaling by recruitment of the MyD88 adaptor protein that bridges the intracellular region of TLRs with downstream effector molecules, such as IRAK1, a serine/threonine kinase, and TRAF6, an E3 ubiquitin ligase, to activate the NF-κB and MAPK pathways, resulting in the release of proinflammatory cytokines and chemokines.
In addition to NF-κB and MAPKs, TLR5 also activates the PI3K pathway, a known mechanism for negative regulation of TLR5 signaling (10). The MUC1 CT contains a consensus binding site for PI3K (5,11), leading to the suggestion that PI3K may be responsible for MUC1-induced suppression of TLR5 signaling. However, our previous study demonstrated that whereas MUC1 activated PI3K pathway in response to flagellin, a TLR5 agonist, PI3K was not responsible for MUC1-induced suppression of TLR5 signaling (12). In this study, evidence is presented that MUC1 expression disrupts TLR5-MyD88 interaction and downstream proinflammatory signaling, and that EGFR activation promotes this anti-inflammatory effect of MUC1 through tyrosine phosphorylation of the MUC1 CT.
MATERIALS AND METHODS
Reagents
All reagents were purchased from Sigma (St. Louis, MO) unless otherwise stated. The sources of the antibodies and reagents were: phospho-p38 (Thr180/Tyr182) (3D7) rabbit monoclonal, phospho-p44/42 (Thr202/Tyr204) rabbit monoclonal, phospho-tyrosine (P-Tyr-100) mouse monoclonal, EGFR rabbit monoclonal, AG1478 (Cell Signaling Technology, Beverly, MA), MyD88 (HFL296) rabbit polyclonal, MUC1 ectodomain (GP1.4) mouse monoclonal, normal rabbit IgG, normal mouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA), TLR5 (IMG-664A) mouse monoclonal (Imgenex, San Diego, CA), horseradish peroxidase-conjugated goat anti-mouse IgG and anti-rabbit IgG secondary antibodies (KPL, Gaithersburg, MD), P. aeruginosa strain K (PAK) flagellin rabbit antiserum (Dr. Dan Wozniak, Wake Forest University, Winston-Salem, NC), MUC1-pcDNA3.1 expression plasmid and MUC1 CT (CT2) hamster antibody (Dr. Sandra J. Gendler, Mayo Clinic College of Medicine, Scottsdale, AZ), TRAF6 expression plasmid (Dr. Yong Lin, Lovelace Respiratory Research Institute, Albuquerque, NM), MyD88 and IRAK1 expression plasmids (Dr. Katherine Fitzgerald, University of Massachusetts Medical School, Worcester, MA), human recombinant TNF-α(Prospec, Rehovot, Israel). PAK flagellin was purified as described (6). Heat-inactivated PAK (1.0 × 109 colony forming units [CFU]/ml) was prepared by incubating bacteria at 60°C for 30 min and stored at −80°C. Pervanadate, a general protein tyrosine phosphatase inhibitor, was freshly prepared immediately before the use as a 100X stock solution by mixing 600 µM H2O2 and 0.2 µM Na3VO4(11).
Cells
Mouse embryonic fibroblast (MEF) cells were obtained from E13 embryos and maintained in DMEM supplemented with 10% FBS (13). Primary mouse tracheal surface epithelial (MTSE) cells were prepared from 8–12 week-old C57BL/6 mice and cultured as described (6). Normal human bronchial/tracheal epithelial (NHBE) cells were obtained from Lonza Walkersville, Inc. (Walkersville, MD) and cultured at an air/liquid interface according to the manufacturer’s protocol. A549 cells (CCL-185™), a human lung epithelial cell line, HEK293 cells (CRL-1573™) and HEK293T cells (CRL-11268™), human embryonic kidney cell lines, were purchased from American Tissue Culture Collection (Manassas, VA). A549 cells were cultured in Opti-MEM medium supplemented with 5% FBS and antibiotics. HEK293 cells were stably transfected with the pcDNA3.1 empty vector (293-pcDNA3.1 cells) or with a MUC1 cDNA cloned in pcDNA3.1using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA). HEK293T cells were used for transient transfection. Both cells were subsequently transfected with FLAG-tagged human TLR5 cloned into the HindIII and AgeI sites of the pDsRed-Monomer-Hyg-N1 vector (Clontech Laboratories, Palo Alto, CA). Stable clones were isolated in the presence of hygromycin (100 µg/ml) and referred to as 293-TLR5 and 293-TLR5/MUC1 cells, respectively, and were cultured in DMEM containing penicillin (100 U/ml), streptomycin (100 µg/ml), hygromycin (100 µg/ml), and 10% FBS.
Western blot and immunoprecipitation analyses
Western blot analysis was performed as described (12). For direct IP analysis, the cells were extracted with lysis buffer (0.5 ml/100-mm dish) and equal amounts of protein were incubated with anti-FLAG antibody (10 µg), anti-MUC1 CT (CT2) antibody (40 µl), anti-MUC1 ectodomain (GP1.4) antibody (30 µl), or isotype-matched normal IgG (Santa Cruz) at 4°C for 16 h with continuous agitation. Protein G-agarose beads (Pierce, Rockford, IL) were added to precipitate immune complexes, the beads were washed five times at 4°C with lysis buffer, and eluted by boiling in SDS-PAGE loading buffer. Immune complexes were separated by SDS-PAGE and subjected to Western blot analysis.
Luciferase reporter assays for ELAM-1and IL-8
An NF-κB dependent ELAM-1-luciferase reporter plasmid (pELAM1-luc) was generated as described elsewhere (14) by cloning a fragment (−241 to −54 bp) of human E-selectin promoter into the pGL3 reporter plasmid (Promega, Madison, WI). Briefly, cells were transiently transfected with the pIL-8-luciferase (15) or pELAM-1-luciferase reporter genes with a Renilla luciferase reporter construct using Lipofectamine™ 2000 according to the manufacture’s protocol. In addition, the cells were transfected with expression plasmids for TRAF6, IRAK1, or MyD88. The total amount of plasmid DNA was kept constant by addition of pcDNA3.1 for each transfection. At 24–36 h post-transfection, the cells were treated with appropriate stimuli for 6 h, and cell lysates were assayed for firefly and Renilla luciferase activities using the Dual-luciferase Reporter System (Promega) and a L-Max II luminometer (Molecular Devices, Sunnyvale, CA). Relative luciferase activity was determined by normalizing with Renilla luciferase activity.
Experimental Pa lung infection
C57BL/6 Muc1−/− mice and Muc1+/+ littermates have been described (16). Mice were intranasally infected with 1.0 × 107 CFU of PAK in a 40 µl suspension as described (6). Left lung lobes were homogenized in 1.0 ml of lysis buffer and subjected to IP as described above. The remaining lobes were fixed with 10% paraformaldehyde, embedded in paraffin, and 5 µm sections were made. All procedures were approved by the Institutional Animal Care and Use Committee of Temple University.
Immunofluorescence
Paraffin sections on glass slides were deparaffinized in xylene, rehydrated, blocked in normal goat serum, incubated overnight at 4°C with primary antibodies (1:100 dilution), and 1 h at room temperature with fluorescent-conjugated secondary antibodies (1:500). Nuclei were counterstained with DAPI. Coverslips were applied in antifade solution (Invitrogen), visualized by laser scanning confocal microscopy (LSM 510, Zeiss, Stuttgart, Germany), and images were analyzed using LSM 510 software.
Statistical analysis
Differences between groups were assessed by comparing mean ± SEM values using the Student’s t test and were considered significant at P< 0.05.
RESULTS
MUC1 expression attenuates TLR5-dependent IL-8 promoter activity and activation of NF-κB, p38, and ERK1/2
Airway epithelial cells sense Pa infection through recognition of flagellin by TLR5 (17). Because MUC1 expression inhibits TLR5-driven NF-κB activation and IL-8 production by airway epithelia (6,18), we sought to determine whether MUC1 suppresses TLR5 signaling and, if so, to elucidate the mechanism by building upon previous studies using the well-established 293 cell culture system (6,11,12,18–20). Stable clones of 293-pcDNA3.1, 293-TLR5, and 293-TLR5/MUC1 cells were established in MUC1 non-expressing 293 cells (Fig. 1A). Pa-induced IL-8 promoter activity was significantly enhanced in 293-TLR5 cells compared with 293-pcDNA3.1 cells (Fig. 1B). TLR5-dependent IL-8 promoter activation was completely suppressed in 293-TLR5/MUC1 cells. TLR5 regulates IL-8 gene expression through NF-κB and MAPK pathways (21). Therefore, we asked whether MUC1 attenuates TLR5-dependent activation of NF-κB and/or MAPK. Treatment of 293-TLR5 cells with flagellin increased NF-κB-dependent reporter (ELAM-1) activity compared with untreated controls (Fig. 1C). However, NF-κB activity was completely suppressed in 293-TLR5/MUC1 cells. Similarly, treatment of 293-TLR5 cells with flagellin augmented the phosphorylation of p38 and ERK1/2 compared with controls, and both effects were drastically reduced in 293-TLR5/MUC1 cells (Fig. 1D). Identical results were observed in an independent cell culture system. Here, Pa treatment of Muc1−/− MEF cells previously transfected with the pcDNA3.1 empty vector increased NF-κB activation, and enhanced p38 and ERK1/2 phosphorylation, compared with untreated controls (Figs. 1E, 1F). However, all three of these effects were substantially reduced in Muc1−/− MEF cells overexpressing human MUC1 compared with Muc1−/− MEF-pcDNA3.1 cells.
Figure 1.
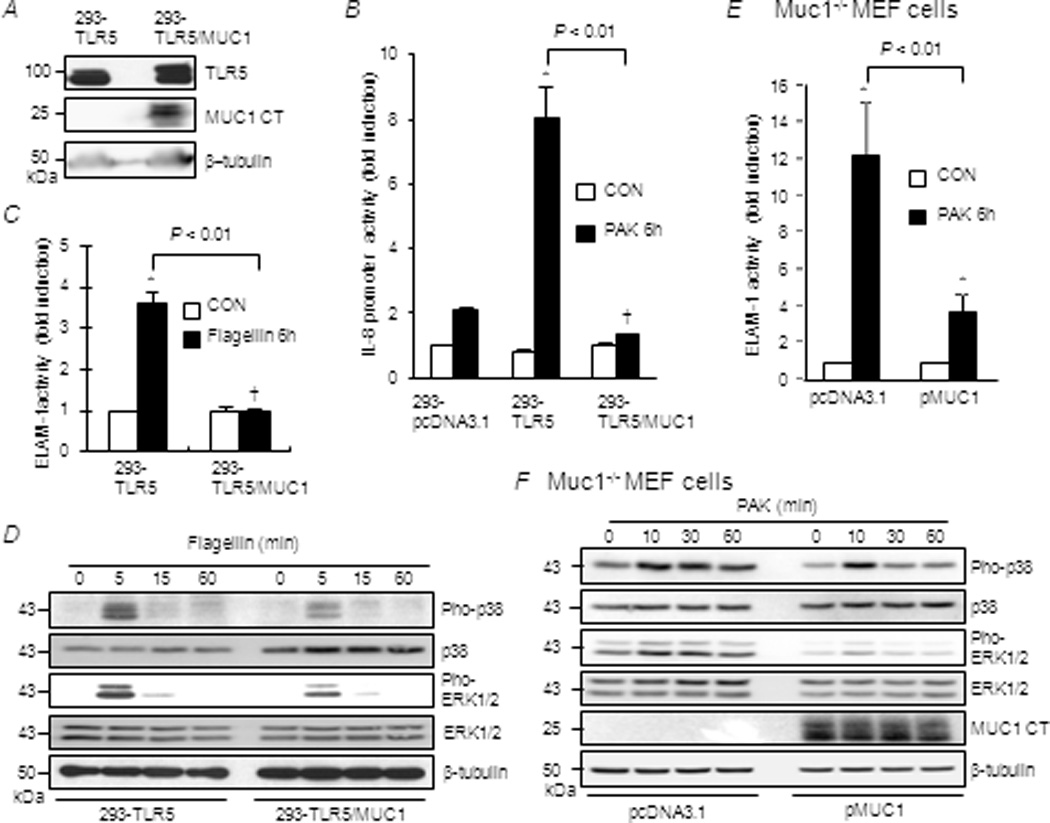
Effect of MUC1 expression on TLR5-dependent IL-8 promoter activity and activation of NF-κB, p38, and ERK1/2. (A–D) 293-pcDNA3.1, 293-TLR5, and 293-TLR5/MUC1 cells, or (E, F) Muc1−/− MEF cells transfected with pcDNA3.1 empty vector or MUC1 expression plasmid were untreated (CON) or treated for 6 h with heat-inactivated Pa strain K (PAK, 1.0 × 107 CFU/ml) or flagellin (1.0 µg/ml). Equal protein amounts of cell lysates were subjected to (A, D, F) Western blotting with the indicated antibodies, or luciferase assays for (B) IL-8 promoter activity or (C, E) NF-κB reporter activity. Each bar represents the mean ± SEM value (n= 3). *, P< 0.05 and †, P>0.05 compared with untreated controls. The results are representative of 3 independent experiments.
MUC1 expression does not affect TNF-α/TNFR signaling
To determine whether inhibition of NF-κB and MAPK by MUC1 could be replicated in another innate immune ligand/receptor system, the effects of MUC1 expression on TNF-α/TNF receptor (TNFR)-mediated activation of NF-κB and MAPK were assessed. As before, flagellin-induced NF-κB activation was suppressed in 293-TLR5/MUC1 cells compared with 293-TLR5 cells (Fig. 2A). By contrast, TNF-α-induced NF-κB activities were equal in both cell types. (Note the different numerical scales on the two ordinate axes.) Combining the flagellin and TNF-α stimuli in 293-TLR5 cells produced an NF-κB activation response that was approximately 2-fold greater than that achieved by the individual agonists. However, NF-κB activation in 293-TLR5/MUC1 cells in response to flagellin plus TNF-α was equal to that produced by TNF-α alone. Additionally, TNF-α-stimulated phosphorylations of p38 and ERK1/2 were not suppressed in 293-TLR5/MUC1 compared with 293-TLR5 cells (Fig. 2B). These combined data suggest that MUC1 attenuates activation of NF-κB and MAPK mediated by TLR5, but not by TNFR.
Figure 2.
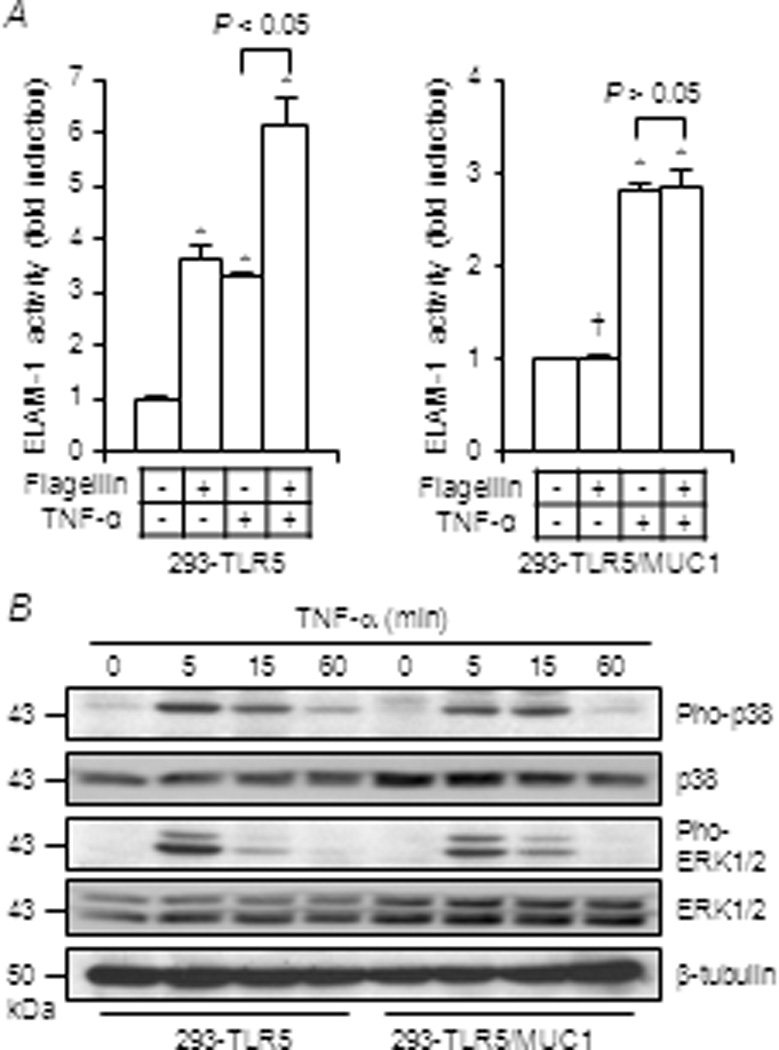
Effect of MUC1 expression on TNF-α-induced activation of NF-κB, p38, and ERK1/2. 293-TLR5 and 293-TLR5/MUC1 cells were untreated or treated for 6 h with flagellin (1.0 µg/ml), TNF-α (100 ng/ml), or flagellin plus TNF-α. Equal protein amounts of cell lysates were subjected to (A) luciferase assay to measure NF-κB reporter activity and (B) Western blotting with the indicated antibodies. Each bar represents the mean ± SEM value (n= 3). *, P< 0.05 and †, P> 0.05 compared with untreated controls. Note the different ordinate scales in (A). The results are representative of 3 independent experiments.
MUC1 interferes with flagellin-induced association ofTLR5/MyD88
Because activation of NF-κB and MAPK by TLR5 and TNFR require prior activation of TAK1, and because the signaling pathways leading to activation of TAK1 by these receptors are distinct, the data presented above suggest that crosstalk between MUC1 and TLR5 signaling occurs upstream of TAK1. Therefore, experiments were performed to identify the possible site(s) between TLR5 and TAK1 that is/are suppressed by MUC1. Engagement of TLR5 by flagellin triggers the recruitment of MyD88 to the intracellular Toll-IL1 receptor (TIR) domain of TLR5. MyD88 subsequently binds to and activates IRAK1 (22), and activated IRAK1 phosphorylates TRAF6 thereby inducing Lys63-linked auto ubiquitination and activation of TAK1 (23). Flagellin-treated 293-TLR5 and 293-TLR5/MUC1 cells exhibited equal flagellin/TLR5 binding (Fig. 3A), suggesting that MUC1 did not block binding of flagellin to its cognate receptor. Next, we determined whether MUC1-mediated suppression of TLR5 signaling was blocked following overexpression of TRAF6, IRAK1, or MyD88. While overexpression of TRAF6 or IRAK1 did not reverse the inhibitory effect of MUC1 (Fig. 3B), MyD88 overexpression completely abrogated MUC1-dependent suppression of flagellin-induced NF-κB activation (Fig. 3B). Overexpression of TRAF6, IRAK1 and MyD88 following transient transfection was confirmed by Western blotting (data not shown). Finally, treatment of 293-TLR5 cells with flagellin or heat-inactivated Pa increased association of TLR5/MyD88 compared with untreated cells (Fig. 3C). In contrast, such association was not observed in 293-TLR5/MUC1 cells. Taken together, these results suggest that MUC1 overexpression abrogates flagellin-stimulated recruitment of MyD88 to TLR5, thus inhibiting NF-κB activation, but this inhibitory effect is fully reversible upon MyD88 overexpression.
Figure 3.
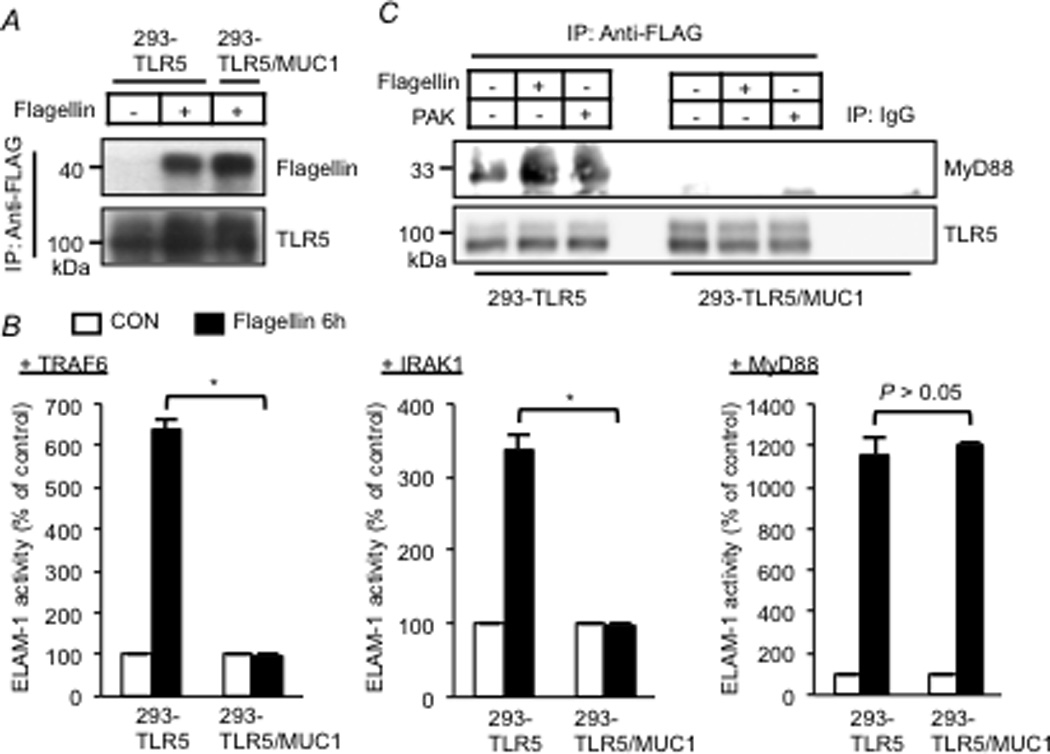
Effect of MyD88, IRAK1, or TRAF6 overexpression on MUC1-mediated suppression of TLR5 signaling. (A, C) 293-TLR5 and 293-TLR5/MUC1 cells were untreated or treated with flagellin (1.0 µg/ml) or heat-inactivated PAK (1.0 × 107 CFU/ml) for 30 min. Equal protein amounts of cell lysates were used for IP with anti-FLAG antibody (TLR5) or isotype-matched normal mouse IgG, and IPed proteins were subjected to Western blotting with the indicated antibodies. (B) 293-TLR5 and 293-TLR5/MUC1 cells were transfected with TRAF6 (left panel), IRAK1 (middle panel), or MyD88 (right panel) expression plasmids. The cells were untreated or treated for 6 h with flagellin (1.0 µg/ml). Equal protein amounts of cell lysates were subjected to luciferase assay to measure NF-κB reporter activity. Each bar represents the mean ± SEM value (n= 3). *, P< 0.05. The results are representative of 3 independent experiments.
MUC1 associates with TLR5
Two possible mechanisms were considered for MUC1’s ability to reversibly block the interaction between MyD88 and TLR5 – binding of MUC1 to MyD88, and/or binding of MUC1 to TLR5. Reciprocal co-immunoprecipitation (co-IP) studies revealed that MUC1 constitutively forms a protein complex with TLR5 but not with MyD88 (data not shown) in lysates of 293T cells transiently transfected with MUC1 and TLR5 (Fig. 4A). Deletion of the MUC1 CT domain (MUC1 ΔCT) almost completely abated its association with TLR5 (Fig. 4B), suggesting that MUC1 CT domain is necessary to interact with TLR5. Constitutive interaction between endogenously expressed MUC1 and TLR5 was also demonstrated in lysates of A549 cells, as well as in primary MTSE cells and NHBE cells (Fig. 5A). In addition, association of MUC1/TLR5 in NHBE cells was increased 5-fold over basal levels following treatment with Pa (Fig. 5B). In summary, these results indicate that MUC1 binds to TLR5 through its CT domain.
Figure 4.
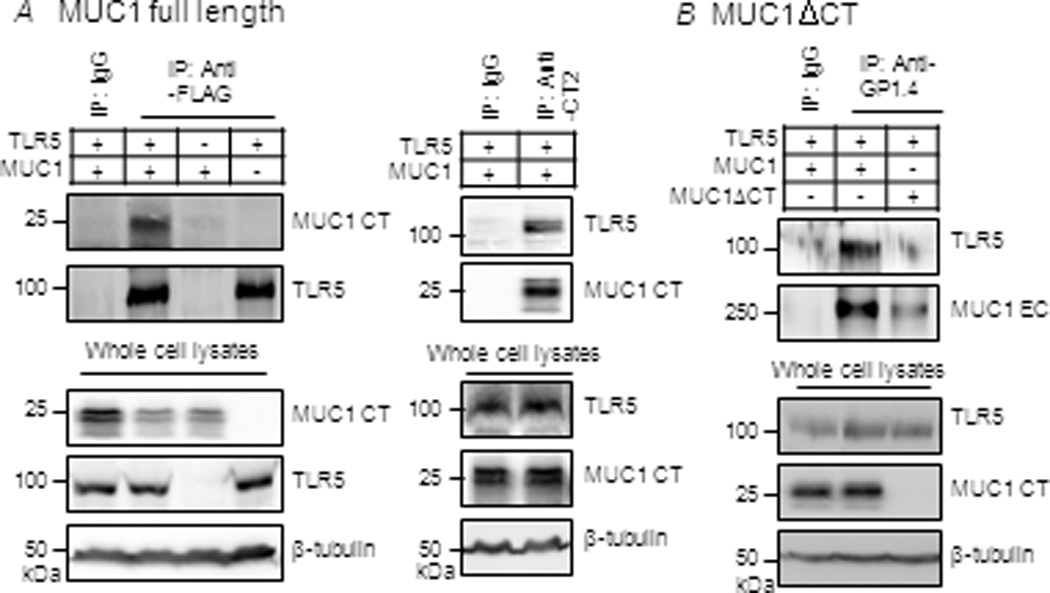
Association of MUC1 and TLR5 in 293T cells. 293T cells were untransfected or transiently transfected with expression plasmids for TLR5-FLAG and/or(A) full length MUC1 or (B) MUC1ΔCT. At 24 h post-transfection, equal protein amounts of cell lysates were used for IP with isotype-matched normal mouse IgG, anti-FLAG (TLR5-FLAG), anti-CT2 (full length MUC1), or anti-MUC1 ectodomain (GP1.4)(MUC1ΔCT). IPed proteins were subjected to Western blotting with the indicated antibodies. Protein expression levels of TLR5 and MUC1 were verified in the same lysates used for IP. The results are representative of 2–3 independent experiments.
Figure 5.
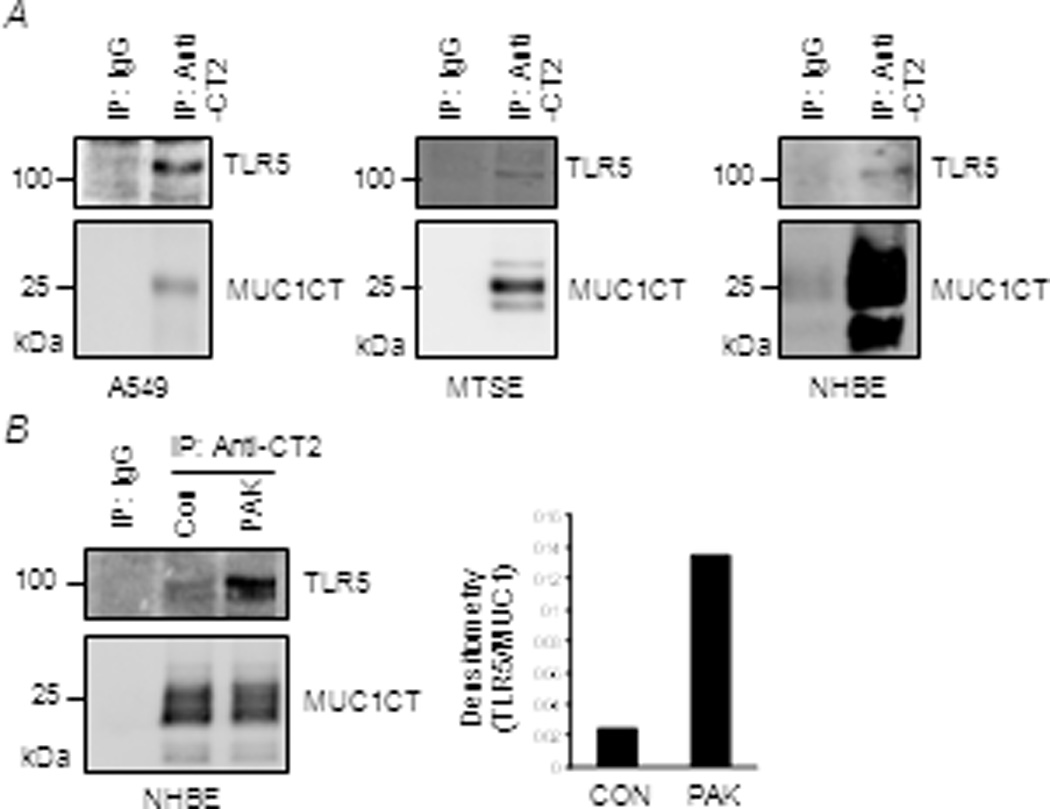
Association of MUC1 and TLR5 in human airway epithelial cells. (A)Equal protein amounts of lysates from A549, MTSE, and NHBE cells were used for IP with isotype-matched normal IgG or anti-CT2 to precipitate MUC1 and IPed proteins were subjected to Western blotting with the indicated antibodies. (B) NHBE cells were untreated (CON) or treated for 48 h with heat-inactivated PAK (1.0 × 107 CFU/ml). Equal protein amounts of cell lysates were IPed as in (A) (left panel). The density of each TLR5 band that was normalized to the density of the corresponding MUC1 CT band is presented. The results are representative of 2–3 independent experiments.
Activation of EGFR induces MUC1 CT tyrosine phosphorylation and increases association of MUC1/TLR5
Tyrosine residues in the MUC1 CT are targets of cytosolic and receptor protein kinases and these phosphotyrosines serve as docking sites for oncoproteins (24). For example, EGFR phosphorylates the MUC1 CT at a Y46 EKV sequence that, upon phosphorylation, serves as a binding site for the kinase (25). Therefore, we investigated constitutive and EGFR-induced CT tyrosine phosphorylation and the role of phosphorylation in MUC1/TLR5 association. Treatment of A549 cells with pervanadate, a broad-spectrum tyrosine phosphatase inhibitor, increased constitutive MUC1 CT tyrosine phosphorylation and augmented MUC1/TLR5 association compared with untreated cells (Fig. 6A). To investigate the possible role of the Y46 residue in MUC1 CT phosphorylation and association of MUC1/TLR5, 293T cells were co-transfected with TLR5 and a CD8/MUC1 chimeric protein (26), either one with the CD8 extracellular (EC) and transmembrane (TM) domains and the MUC1 CT (CD8/MUC1-WT) or CD8/MUC1-Y46 F in which Tyrosine-46 residue was mutated to phenylalanine (11,26) (Fig. 6B). Pervanadate treatment increased both constitutive tyrosine phosphorylation of CD8/MUC1-WT (Fig. 6C, lower panel) and association of CD8/MUC1-WT and TLR5 (Fig. 6C, upper panel) compared with untreated cells. However, CD8/MUC1-Y46 F exhibited significant decreases in both constitutive tyrosine phosphorylation and association with TLR5 compared with CD8/MUC1-WT (Fig. 6C). Next, we determined whether Y46 is important for the suppressive effect of MUC1 on PAK-induced NF-κB activation as shown in Fig. 1. Overexpression of CD8/MUC1-WT and CD8/MUC1-Y46 F decreased PAK-induced ELAM-1 activity by 65% and 25% respectively, compared with pcDNA3.1 control, suggesting that Y46 of MUC1 CT plays an important role in suppressing PAK-induced TLR5 signaling (Fig. 6D). Even in the absence of pervanadate, treatment of A549 cells with an EGFR agonist, TGF-α, increased tyrosine phosphorylation of the MUC1 CT and enhanced association of MUC1/TLR5 as well as MUC1/EGFR (Fig. 6E). Identical results were observed using EGF as the stimulus (data not shown). Similarly, TGF-α stimulated tyrosine phosphorylation of CD8/MUC1-WT (Fig. 6F, lower panel) and increased association of CD8/MUC1-WT with TLR5 (Fig. 6F, upper panel) compared with untreated cells. However, TGF-α treatment failed to increase association of CD8/MUC1-Y46 F with TLR5 (Fig. 6G). In addition, treatment of CD8/MUC1-WT-transfected cells with a selective EGFR inhibitor, AG1478, abrogated TGF-α-induced association of CD8/MUC1-WT/TLR5 (Fig. 6H). In conclusion, these data suggest that EGFR-dependent Y46 phosphorylation of the MUC1 CT increases association of MUC1 CT with TLR5, hence presumably enhancesMUC1-mediated TLR5 suppression.
Figure 6.
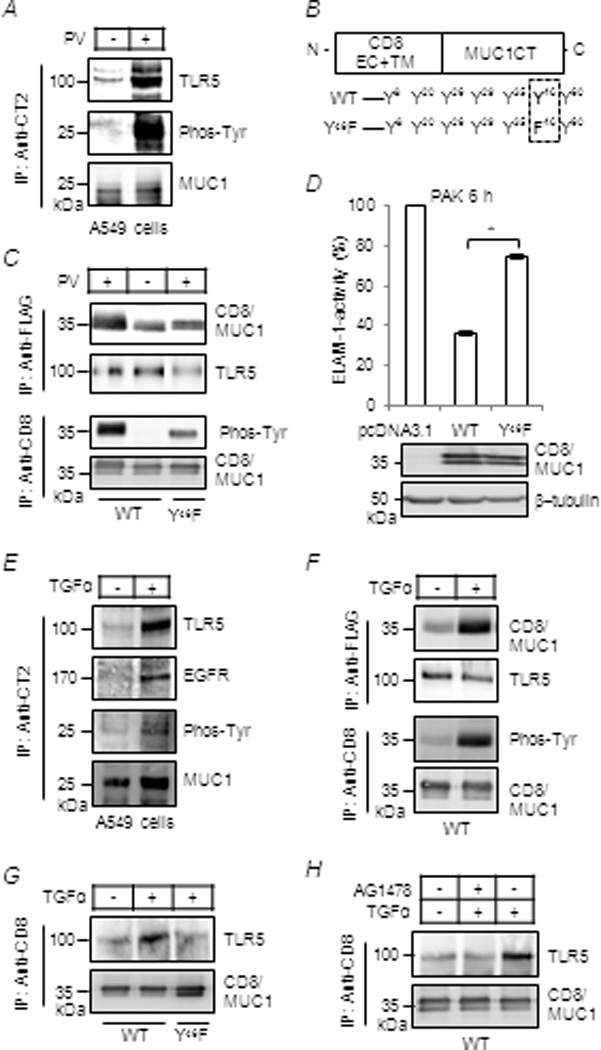
Phosphorylation of the MUC1 CT at Y46 by EGFR increases MUC1/TLR5 association.(A, C–H) A549 cells or 293T cells, transiently transfected with expression plasmids for TLR5 plus CD8/MUC1-WT or CD8/MUC1-Y46 F, were untreated or treated for 30 min with pervanadate (0.2 µM), AG1478 (100 nM) or TGF-α (100 ng/ml). Equal amounts of cell lysates were used for IP with anti-CT2, anti-CD8 (CD8/MUC1), or anti-FLAG (TLR5-FLAG), and IPed proteins were subjected to Western blotting with the indicated antibodies. (D) For ELAM-1-luciferase reporter assay, cells were untreated or treated with PAK and assayed as in Figure 1. Luciferase activity of pcDNA3.1-transfectants treated with PAK was set as 100%. Each bar represents the mean ± SEM value (n= 4). *, P< 0.01. Transfection efficiency of CD8/MUC1-WT and CD8/MUC1-Y46 F was verified by Western blotting. (B) A schematic illustration of the CD8/MUC1 chimeric protein. The results are representative of 3 independent experiments.
Immunofluorescence colocalization of Muc1 and TLR5 and tyrosine phosphorylation of the Muc1 CT in vivo
Pa infection of mice has been shown to activate EGFR in airway epithelium (27). Therefore, we asked whether Pa airway infection of mice stimulates tyrosine phosphorylation of the Muc1 CT in vivo and increases Muc1/TLR5 interaction. Mice were infected intranasally with Pa and lung homogenates were subjected to Muc1 CT IP and phosphotyrosine immunoblot analysis at 0, 4, and 24 h post-infection. Airway infection was confirmed by increased inflammatory cell infiltrate into the lungs at 4 h and 24 h (data not shown). Muc1 CT tyrosine phosphorylation was increased at 24 h post-infection compared with 0 and 4 h (Fig. 7A). Identical results were observed in the reciprocal approach when lung homogenates were subjected to phosphotyrosine IP followed by Muc1 CT immunoblotting (Fig. 7B). As a negative control, a 25 kDa tyrosine-phosphorylated CT protein band was not detected in the lungs of Pa-infected Muc1−/− mice (Fig. 7C). Finally, laser scanning confocal immunofluorescence microscopy was used to assess the Muc1, TLR5, and phosphotyrosine staining patterns in lungs of uninfected and Pa-infected mice. In uninfected animals, no evidence of Muc1/TLR5 or Muc1/phosphotyrosine co-localization was observed (Fig. 8). However, epithelial cell apical co-localization of the Muc1/TLR5 and Muc1/phosphotyrosine immunostaining patterns were seen in Pa-infected mice at 24 h post-infection.
Figure 7.
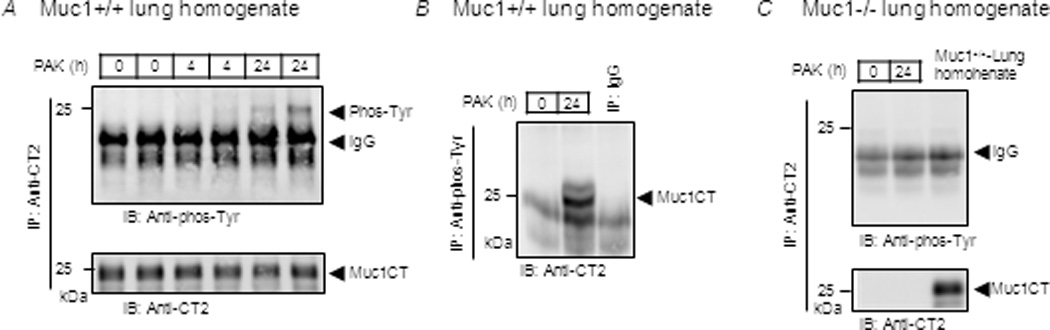
Pa infection increases tyrosine phosphorylation of the Muc1 CT in vivo. Muc1+/+ and Muc1−/− mice were intranasally infected with PAK (1.0 × 107 CFU/mouse). At 0, 4, and 24 h post-infection, lung homogenates were prepared and equal amounts of protein were used for IP with (A, C) anti-CT2 antibody or (B) anti-phosphotyrosine antibody. IPed proteins were subjected to Western blotting with the indicated antibodies. Each lane represents a lung homogenate from an individual mouse. The results are representative of 2–3 independent experiments.
Figure 8.

Co-localization of Muc1 and TLR5 in mouse airway epithelium in vivo. Paraffin embedded mouse lung sections from uninfected (CON) and PAK intranasally-infected Muc1+/+ mice were processed for immunostaining with anti-CT2 and (A) anti-TLR5 or (B) anti-phosphotyrosine antibodies, followed by incubation with fluorescein-conjugated secondary antibodies and DAPI to counter stain nuclei. Scale bars represent 20 µm. Arrows indicate areas of co-localization.
DISCUSSION
MUC1/Muc1 expression counter-regulates airway inflammation during Pa infection (6,8), and this anti-inflammatory activity is attributed to its ability to suppress TLR5 signaling (6,12,18). However, the mechanism of crosstalk between TLR5 and MUC1/Muc1 is unknown. In this report, we now demonstrate the suppressive effect of MUC1 expression on TLR5-dependent IL-8 promoter activity (Fig. 1B), as well as NF-κB and MAPK activation (Figs. 1C–1F), that correlated with reduced TLR5/MyD88 association (Fig. 3D) and increased MUC1/TLR5 association (Figs. 4A, 4C, 5A, 5B). NF-κB inhibition was completely reversed upon MyD88 overexpression (Fig. 3C). Increased MUC1 CT phosphorylation, likely at the Y46 EKV site, and greater MUC1/TLR5 association were associated with TGF-α-dependent EGFR activation (Figs. 6B–6E). In vivo experiments established increased Muc1 CT tyrosine phosphorylation in mouse lung homogenates following Pa infection (Fig. 7) and greater Muc1/TLR5 and Muc1/phosphotyrosine immunofluorescence co-localization in infected mouse airway epithelium (Fig. 8). Together, these results suggest a mechanism whereby EGFR tyrosine-phosphorylates the MUC1 CT, thus increasing its association with TLR5 and competitively and reversibly inhibiting recruitment of MyD88 to TLR5 and subsequent proinflammatory signal transduction.
Flagellin binding to the TLR5 ectodomain induces receptor homodimerization resulting in a protein conformational change in its CT domain and allowing recruitment of the MyD88 adapter protein (22). The data presented in this study indicate that MUC1 mediates its anti-inflammatory effects at the level of the TLR5 intracellular domain, and are entirely consistent with the previous publication demonstrating that transfection of RAW264.7 cells with MUC1ΔCT, but not MUC1ΔEC, blocked its ability to inhibit TLR-driven TNF-α production (18). This effect was achieved not only with the MyD88-dependent TLR2, TLR4, TLR7, and TLR9, but also with TLR3 which signals through TRIF rather than MyD88. Given that all TLRs and many adaptor proteins share the conserved TIR domain mediating homo- and heterotypic interactions, it seems very likely that MUC1 suppresses TLR signaling by associating with receptor TIR domains, thus acting as a steric hindrance/decoy receptor and excluding recruitment of MyD88 and TRIF to their respective receptors. Interestingly, the MUC1 CT contains an amino acid sequence (R17 DTYHP) that is homologous to the consensus RDXΦ1Φ2G motif (where Φ represents a hydrophobic residue, and X any residue) of the TIR domain and that has been shown to be responsible for heteromeric interaction between TLR2/4 and MyD88 (28). However, the possible role of the MUC1 CT “TIR domain-like” sequence in TLR signal transduction remains speculative.
EGFR regulates innate immune responses in the airways, including mucin secretion by goblet cells, and chemokine production and proliferation by epithelial cells (29). As with all EGFR ligands, TGF-α is synthesized in a latent form as a membrane-tethered precursor protein on the surface of airway epithelial cells. Proteolytic cleavage of pro-TGF-α by TNF-α converting enzyme (TACE) precedes EGFR activation and proinflammatory signaling (30). Like TGF-α, TACE is initially synthesized in an inactive form that is activated by a variety of diverse stimuli, including airway bacterial pathogens (31), cigarette smoke (32), and reactive oxygen species (ROS) (33), the latter of which are up-regulated by TLRs and dual oxidase (33). On the basis of the current results, a second function can now be ascribed to activated EGFR apart from proinflammatory signaling, namely tyrosine phosphorylation of the MUC1 CT and MUC1/TLR5 protein interaction. Of note, airway epithelial cell expression levels of both MUC1 and EGFR are increased by a common proinflammatory cytokine, TNF-α (8,29,34,35). It is tempting to speculate that simultaneous up-regulation of MUC1 and EGFR in the vicinity of an ongoing inflammatory response facilitates sequential steps of EGFR-mediated tyrosine phosphorylation of MUC1, MUC1/TLR5 interaction, and counter-regulation of airway inflammation.
In addition to its anti-inflammatory properties mediated through its CT, a growing body of evidence suggests that the MUC1 extracellular regions contribute to the pathogenesis of microorganisms that colonize and infect mucosal surfaces (36). MUC1/Muc1is an extracellular adhesion site for Pa on airway epithelia (19,37,38), and for Escherichia coli and Salmonella enteric on intestinal epithelia (39,40). Helicobacter pylori binds to the MUC1 ectodomain on gastric epithelial cells and MUC1 ectodomain shedding acts as a releasable decoy to block infection by this pathogen (36,41). McAuley et al. (42) demonstrated that Muc1−/− mice are more susceptible to infection by gastrointestinal Campylobacter jejuni compared with Muc1+/+ littermates. Increased bacterial colonization by C. jejuni was accompanied by severe epithelial damage and exaggerated penetration through the intestinal barrier, eventually resulting in systemic infection. As originally proposed by Gendler (43), the heterodimeric nature of the MUC1 protein may provide a mechanism for rapid ectodomain shedding that concurrently signals to the cell interior, through its CT, the presence of an invading pathogen. While Muc1 acts as a decoy receptor for invading bacteria in the intestinal tract, it concurrently plays an anti-inflammatory role in the respiratory tract by a discrete mechanism not involving its ectodomain. These multidimensional effects may relate to the functional difference between the two organs - the former constituting an impenetrable barrier against commensal bacteria, whereas the latter dealing with the timely resolution of inflammation in order to maintain its vital function of gas exchange.
It is also apparent that the MUC1 CT exhibits functional activities apart from its ability to directly block TLR signaling. Ahmad et al. (44,45) demonstrated that the MUC1 C-terminal subunit promotes TNF-α-induced activation of NF-κB in human breast cancer cells. In contrast, we reported that the MUC1 CT binds to the IKKγ subunit to inhibit H. pylori-dependent NF-κB activation in a human gastric cancer cell line (46). It is currently unknown whether IKK/NF-κB are recruited to the inner leaflet of the plasma membrane by the MUC1 CT, or whether the membrane-bound CT is released into the cytoplasm to interact with IKK/NF-κB. Nonetheless, apically-polarized MUC1 presumably favors its ability to interact with TLR5 and, hence, selectively suppress TLR5-induced NF-κB activity per se. Interestingly, exposure of polarized airway epithelial cells to cigarette smoke re-distributed apical MUC1 into the cytosol, suggesting that exogenous insults can affect the subcellular localization of MUC1 and, by implication, its functional properties in the lung (47). Elucidation of the factors controlling MUC1 cellular and functional heterogeneity deserves further investigation.
In conclusion, we propose the following sequence of events during airway Pa infection in the context of the anti-inflammatory role of MUC1/Muc1 (Fig. 9). Host defense against the pathogen is mediated primarily by mucociliary clearance and phagocytosis. During the early stage of infection, TLR5 on respiratory epithelial cells (and, perhaps, resident macrophages) sense Pa through its interaction with flagellin (step 1) and triggers MyD88-dependent signaling (step 2) to induce inflammatory mediators which result in recruitment of leukocytes into the site of infection to clear the bacteria. The inflammatory products generated during this process, such as neutrophil elastase and TNF-α (8,34,48) (step 3), up-regulate MUC1/Muc1 expression during the late stage of infection following clearance of the pathogen from the airways. Activation of EGFR by TLRs and/or alternative mechanisms, stimulates phosphorylation of the MUC1 CT at Y46 (step 4), leading to MUC1/TLR5 interaction (step 5), thus interfering with the recruitment of MyD88 to TLR5 and down-regulating the inflammatory response. Ongoing studies in our laboratory are directed at testing this hypothetical model.
Figure 9.
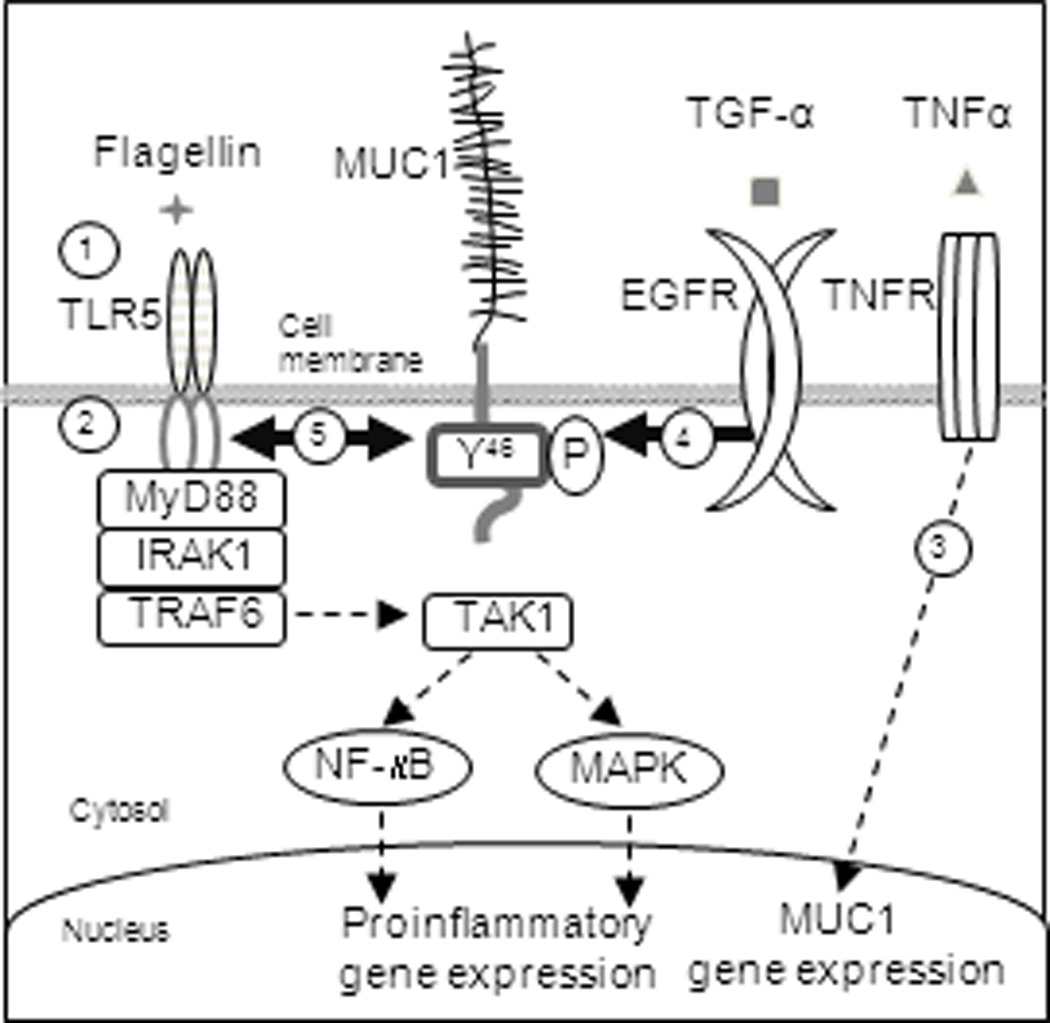
Schematic illustration of the proposed mechanism through which MUC1 negatively regulates TLR5 signaling. Step 1, TLR5 on epithelial cells sense bacteria-derived flagellin. Step 2, activated TLR5 triggers MyD88-dependent signaling to induce the release of inflammatory mediators which results in recruitment of leukocytes into the site of infection to clear the bacteria. Step 3, inflammatory products, such as neutrophil elastase and TNF-α, up-regulate MUC1 expression. Step 4, EGFR activated by TLRs or other means phosphorylates the MUC1 CT at Y46. Step 5, MUC1 binds to TLR5 which interferes with the recruitment of MyD88 to TLR5.
ACKNOWLEDGMENTS
We are grateful to Dr. Katie Fitzgerald (University of Massachusetts), Dr. Yong Lin (Lovelace Respiratory Research Institute), Dr. Sandra Gendler (Mayo Clinic College of Medicine), and Dr. Dan Wozniak (Wake Forest University) for kindly providing reagents.
This study was supported by U.S. Public Health Service grants HL-47125 and HL-81825 (KCK) and AI-83463 (EPL).
Abbreviations used in this article
- 293
human embryonic kidney HEK 293
- MUC1 CT
MUC1 cytoplasmic tail
- EC
extracellular
- TM
transmembrane
- MyD88
myeloid differentiation primary response gene 88
- EGFR
epidermal growth factor receptor
- IRAK1
Interleukin 1 receptor-associated kinase 1
- TIR
Toll-IL1 receptor
- TRAF6
TNF receptor-associated factor 6
- Pa
Pseudomonas aeruginosa
- PAK
Pa strain K
- MEF
mouse embryonic fibroblast
- MTSE
primary mouse tracheal surface epithelial
- NHBE
normal human bronchial/tracheal epithelial
- TAK1
TGF-beta-activated kinase 1
- IP
immunoprecipitation
Reference List
- 1.Hattrup CL, Gendler SJ. Structure and function of the cell surface (tethered) mucins. Annu.Rev.Physiol. 2008;70:431–457. doi: 10.1146/annurev.physiol.70.113006.100659. [DOI] [PubMed] [Google Scholar]
- 2.Gendler SJ, Lancaster CA, Taylor-Papadimitriou J, Duhig T, Peat N, Burchell J, Pemberton L, Lalani EN, Wilson D. Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. J.Biol.Chem. 1990;265:15286–15293. [PubMed] [Google Scholar]
- 3.Lan MS, Batra SK, Qi WN, Metzgar RS, Hollingsworth MA. Cloning and sequencing of a human pancreatic tumor mucin cDNA. J.Biol.Chem. 1990;265:15294–15299. [PubMed] [Google Scholar]
- 4.Wen Y, Caffrey TC, Wheelock MJ, Johnson KR, Hollingsworth MA. Nuclear association of the cytoplasmic tail of MUC1 and beta-catenin. J.Biol.Chem. 2003;278:38029–38039. doi: 10.1074/jbc.M304333200. [DOI] [PubMed] [Google Scholar]
- 5.Zrihan-Licht S, Baruch A, Elroy-Stein O, Keydar I, Wreschner DH. Tyrosine phosphorylation of the MUC1 breast cancer membrane proteins. Cytokine receptor-like molecules. FEBS Lett. 1994;356:130–136. doi: 10.1016/0014-5793(94)01251-2. [DOI] [PubMed] [Google Scholar]
- 6.Lu W, Hisatsune A, Koga T, Kato K, Kuwahara I, Lillehoj EP, Chen W, Cross AS, Gendler SJ, Gewirtz AT, Kim KC. Cutting edge: enhanced pulmonary clearance of Pseudomonas aeruginosa by Muc1 knockout mice. J.Immunol. 2006;176:3890–3894. doi: 10.4049/jimmunol.176.7.3890. [DOI] [PubMed] [Google Scholar]
- 7.Kim KC, Lillehoj EP. MUC1 mucin: a peacemaker in the lung. Am.J.Respir.Cell Mol.Biol. 2008;39:644–647. doi: 10.1165/rcmb.2008-0169TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi S, Park YS, Koga T, Treloar A, Kim KC. TNF-{alpha} is a Key Regulator of MUC1, an Anti-inflammatory Molecule During Airway Pseudomonas aeruginosa Infection. Am.J.Respir.Cell Mol.Biol. 2010 doi: 10.1165/rcmb.2009-0323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 10.Yu Y, Nagai S, Wu H, Neish AS, Koyasu S, Gewirtz AT. TLR5-mediated phosphoinositide 3-kinase activation negatively regulates flagellin-induced proinflammatory gene expression. J.Immunol. 2006;176:6194–6201. doi: 10.4049/jimmunol.176.10.6194. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Lillehoj EP, Kim KC. Identification of four sites of stimulated tyrosine phosphorylation in the MUC1 cytoplasmic tail. Biochem.Biophys.Res.Commun. 2003;310:341–346. doi: 10.1016/j.bbrc.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 12.Kato K, Lu W, Kai H, Kim KC. Phosphoinositide 3-kinase is activated by MUC1 but not responsible for MUC1-induced suppression of Toll-like receptor 5 signaling. Am.J.Physiol Lung Cell Mol.Physiol. 2007;293:L686–L692. doi: 10.1152/ajplung.00423.2006. [DOI] [PubMed] [Google Scholar]
- 13.Vasir B, Wu Z, Crawford K, Rosenblatt J, Zarwan C, Bissonnette A, Kufe D, Avigan D. Fusions of dendritic cells with breast carcinoma stimulate the expansion of regulatory T cells while concomitant exposure to IL-12, CpG oligodeoxynucleotides, and anti-CD3/CD28 promotes the expansion of activated tumor reactive cells. J.Immunol. 2008;181:808–821. doi: 10.4049/jimmunol.181.1.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J.Biol.Chem. 1999;274:10689–10692. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 15.Kuwahara I, Lillehoj EP, Lu W, Singh IS, Isohama Y, Miyata T, Kim KC. Neutrophil elastase induces IL-8 gene transcription and protein release through p38/NF-{kappa}B activation via EGFR transactivation in a lung epithelial cell line. Am.J.Physiol Lung Cell Mol.Physiol. 2006;291:L407–L416. doi: 10.1152/ajplung.00471.2005. [DOI] [PubMed] [Google Scholar]
- 16.Spicer AP, Rowse GJ, Lidner TK, Gendler SJ. Delayed mammary tumor progression in Muc-1 null mice. J.Biol.Chem. 1995;270:30093–30101. doi: 10.1074/jbc.270.50.30093. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z, Louboutin JP, Weiner DJ, Goldberg JB, Wilson JM. Human airway epithelial cells sense Pseudomonas aeruginosa infection via recognition of flagellin by Toll-like receptor 5. Infect.Immun. 2005;73:7151–7160. doi: 10.1128/IAI.73.11.7151-7160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueno K, Koga T, Kato K, Golenbock DT, Gendler SJ, Kai H, Kim KC. MUC1 mucin is a negative regulator of toll-like receptor signaling. Am.J.Respir.Cell Mol.Biol. 2008;38:263–268. doi: 10.1165/rcmb.2007-0336RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kato K, Lillehoj EP, Kai H, Kim KC. MUC1 expression by human airway epithelial cells mediates pseudomonas aeruginosa adhesion. Front Biosci.(Elite.Ed) 2010;2:68–77. doi: 10.2741/e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, Lillehoj EP, Kim KC. MUC1 tyrosine phosphorylation activates the extracellular signal-regulated kinase. Biochem.Biophys.Res.Commun. 2004;321:448–454. doi: 10.1016/j.bbrc.2004.06.167. [DOI] [PubMed] [Google Scholar]
- 21.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J.Immunol. 2001;167:1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 23.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat.Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat.Rev.Cancer. 2009;9:874–885. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Ren J, Yu W, Li Q, Kuwahara H, Yin L, Carraway KL, III, Kufe D. The epidermal growth factor receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and beta-catenin. J.Biol.Chem. 2001;276:35239–35242. doi: 10.1074/jbc.C100359200. [DOI] [PubMed] [Google Scholar]
- 26.Meerzaman D, Xing PX, Kim KC. Construction and characterization of a chimeric receptor containing the cytoplasmic domain of MUC1 mucin. Am.J.Physiol Lung Cell Mol.Physiol. 2000;278:L625–L629. doi: 10.1152/ajplung.2000.278.3.L625. [DOI] [PubMed] [Google Scholar]
- 27.Martin C, Thevenot G, Danel S, Chapron J, Tazi A, Macey J, Dusser DJ, Fajac I, Burgel PR. Pseudomonas aeruginosa induces VEGF synthesis in airway epithelium in vitro and in vivo. Eur.Respir.J. 2011 doi: 10.1183/09031936.00134910. [DOI] [PubMed] [Google Scholar]
- 28.Xu Y, Tao X, Shen B, Horng T, Medzhitov R, Manley JL, Tong L. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. 2000;408:111–115. doi: 10.1038/35040600. [DOI] [PubMed] [Google Scholar]
- 29.Burgel PR, Nadel JA. Epidermal growth factor receptor-mediated innate immune responses and their roles in airway diseases. Eur.Respir.J. 2008;32:1068–1081. doi: 10.1183/09031936.00172007. [DOI] [PubMed] [Google Scholar]
- 30.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 31.Shao MX, Ueki IF, Nadel JA. Tumor necrosis factor alpha-converting enzyme mediates MUC5AC mucin expression in cultured human airway epithelial cells. Proc.Natl.Acad.Sci.U.S.A. 2003;100:11618–11623. doi: 10.1073/pnas.1534804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shao MX, Nakanaga T, Nadel JA. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-alpha-converting enzyme in human airway epithelial (NCI-H292) cells. Am.J.Physiol Lung Cell Mol.Physiol. 2004;287:L420–L427. doi: 10.1152/ajplung.00019.2004. [DOI] [PubMed] [Google Scholar]
- 33.Koff JL, Shao MX, Ueki IF, Nadel JA. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am.J.Physiol Lung Cell Mol.Physiol. 2008;294:L1068–L1075. doi: 10.1152/ajplung.00025.2008. [DOI] [PubMed] [Google Scholar]
- 34.Koga T, Kuwahara I, Lillehoj EP, Lu W, Miyata T, Isohama Y, Kim KC. TNF-alpha induces MUC1 gene transcription in lung epithelial cells: its signaling pathway and biological implication. Am.J.Physiol Lung Cell Mol.Physiol. 2007;293:L693–L701. doi: 10.1152/ajplung.00491.2006. [DOI] [PubMed] [Google Scholar]
- 35.Kyo Y, Kato K, Park YS, Gajghate S, Umehara T, Lillehoj EP, Suzaki H, Kim KC. Anti-inflammatory role of MUC1 mucin during nontypeable Haemophilus influenzae infection. Am.J.Respir.Cell Mol.Biol. 2011 doi: 10.1165/rcmb.2011-0142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linden SK, Sutton P, Karlsson NG, Korolik V, McGuckin MA. Mucins in the mucosal barrier to infection. Mucosal.Immunol. 2008;1:183–197. doi: 10.1038/mi.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lillehoj EP, Hyun SW, Kim BT, Zhang XG, Lee DI, Rowland S, Kim KC. Muc1 mucins on the cell surface are adhesion sites for Pseudomonas aeruginosa. Am.J.Physiol Lung Cell Mol.Physiol. 2001;280:L181–L187. doi: 10.1152/ajplung.2001.280.1.L181. [DOI] [PubMed] [Google Scholar]
- 38.Lillehoj EP, Kim BT, Kim KC. Identification of Pseudomonas aeruginosa flagellin as an adhesin for Muc1 mucin. Am.J.Physiol Lung Cell Mol.Physiol. 2002;282:L751–L756. doi: 10.1152/ajplung.00383.2001. [DOI] [PubMed] [Google Scholar]
- 39.Parker P, Sando L, Pearson R, Kongsuwan K, Tellam RL, Smith S. Bovine Muc1 inhibits binding of enteric bacteria to Caco-2 cells. Glycoconj.J. 2010;27:89–97. doi: 10.1007/s10719-009-9269-2. [DOI] [PubMed] [Google Scholar]
- 40.Sando L, Pearson R, Gray C, Parker P, Hawken R, Thomson PC, Meadows JR, Kongsuwan K, Smith S, Tellam RL. Bovine Muc1 is a highly polymorphic gene encoding an extensively glycosylated mucin that binds bacteria. J.Dairy Sci. 2009;92:5276–5291. doi: 10.3168/jds.2009-2216. [DOI] [PubMed] [Google Scholar]
- 41.Linden SK, Sheng YH, Every AL, Miles KM, Skoog EC, Florin TH, Sutton P, McGuckin MA. MUC1 limits Helicobacter pylori infection both by steric hindrance and by acting as a releasable decoy. PLoS.Pathog. 2009;5 doi: 10.1371/journal.ppat.1000617. e1000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McAuley JL, Linden SK, Png CW, King RM, Pennington HL, Gendler SJ, Florin TH, Hill GR, Korolik V, McGuckin MA. MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. J.Clin.Invest. 2007;117:2313–2324. doi: 10.1172/JCI26705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gendler SJ. MUC1, the renaissance molecule. J.Mammary.Gland.Biol.Neoplasia. 2001;6:339–353. doi: 10.1023/a:1011379725811. [DOI] [PubMed] [Google Scholar]
- 44.Ahmad R, Raina D, Trivedi V, Ren J, Rajabi H, Kharbanda S, Kufe D. MUC1 oncoprotein activates the IkappaB kinase beta complex and constitutive NF-kappaB signalling. Nat.Cell Biol. 2007;9:1419–1427. doi: 10.1038/ncb1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahmad R, Raina D, Joshi MD, Kawano T, Ren J, Kharbanda S, Kufe D. MUC1-C oncoprotein functions as a direct activator of the nuclear factor-kappaB p65 transcription factor. Cancer Res. 2009;69:7013–7021. doi: 10.1158/0008-5472.CAN-09-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guang W, Ding H, Czinn SJ, Kim KC, Blanchard TC, Lillehoj EP. MUC1 cell surface mucin attenuates epithelial inflammation in response to a common mucosal pathogen. J.Biol.Chem. 2010;285:20547–20557. doi: 10.1074/jbc.M110.121319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen YT, Gallup M, Nikulina K, Lazarev S, Zlock L, Finkbeiner W, McNamara N. Cigarette smoke induces epidermal growth factor receptor-dependent redistribution of apical MUC1 and junctional beta-catenin in polarized human airway epithelial cells. Am.J.Pathol. 2010;177:1255–1264. doi: 10.2353/ajpath.2010.091129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuwahara I, Lillehoj EP, Hisatsune A, Lu W, Isohama Y, Miyata T, Kim KC. Neutrophil elastase stimulates MUC1 gene expression through increased Sp1 binding to the MUC1 promoter. Am.J.Physiol.Lung Cell.Mol.Physiol. 2005;289:L355–L362. doi: 10.1152/ajplung.00040.2005. [DOI] [PubMed] [Google Scholar]