

Summary
The TB Structural Genomics Consortium is a worldwide organization of collaborators whose mission is the comprehensive structural determination and analyses of Mycobacterium tuberculosis proteins to ultimately aid in tuberculosis diagnosis and treatment. Congruent to the overall vision, Consortium members have additionally established an integrated facilities core to streamline M. tuberculosis structural biology and developed bioinformatics resources for data mining. This review aims to share the latest Consortium developments with the TB community, including recent structures of proteins that play significant roles within M. tuberculosis. Atomic resolution details may unravel mechanistic insights and reveal unique and novel protein features, as well as important protein-protein and protein-ligand interactions, which ultimately leads to a better understanding of M. tuberculosis biology and may be exploited for rational, structure-based therapeutics design.
Keywords: Mycobacterium tuberculosis, Protein structure, X-ray crystallography, Structural genomics, Drug discovery
Introduction
The Tuberculosis Structural Genomics Consortium (TBSGC, http://www.webtb.org) is an international collaboration of researchers whose primary objective is to comprehensively determine the three dimensional structures of proteins from Mycobacterium tuberculosis (Mtb) with the intent of TB diagnosis and treatment. The Consortium, comprising 460 members from 93 research centers spanning 15 countries, employs state-of-the-art technologies for gene cloning, protein expression and structure determination. Since the inception of the TBSGC in 2000, approximately 250 Mtb protein structures have been elucidated by Consortium members, accounting for over one-third of Mtb structures deposited into the Protein Data Bank (PDB, http://www.rcsb.org), many of which have been previously reviewed1-4. The wealth of information from atomic resolution details of proteins, particularly in complex with their cognate substrates or cofactors as well as protein-protein interaction networks, may aid other scientists in interpreting their genetic and biochemical data and may ultimately be exploited in rational structure-based design of therapeutics for TB.
In addition to structural elucidation of Mtb proteins, the TBSGC is actively developing bioinformatics resources that can be used for data mining to complement structural information. Highlighted below are several useful and unique databases available on the TBSGC’s website:
The Gene Expression Correlation Grid or gecGrid server (http://www.webtb.org/gecGrid/) is a compilation of four Mtb H37Rv gene expression datasets5-8, which includes 553 experiments to infer approximately 7,700,000 pairwise coexpression relationships between pairs of genes9. Additionally, gecGrid is a useful tool in assessing correlation among networks/systems of genes. Gene expression correlations are represented in matrices, where each entry has an associated correlation coefficient, a measure of the level of positive (or negative) coexpression.
The Prolinks database (http://prolinks.mbi.ucla.edu) combines four algorithms (i.e., phylogenetic profile, Rosetta Stone, gene neighbor and gene cluster) to predict functional linkages between proteins from 83 organisms, including Mtb, and 10 million high confidence links10. The Phylogenetic Profile method uses the presence and absence of proteins across multiple genomes to detect functional linkages11, 12; Rosetta Stone uses a gene fusion event in a second organism to infer functional relatedness13, 14; the Gene Cluster method uses genome proximity to predict functional linkage15-17; and the Gene Neighbor method uses both gene proximity and phylogenetic distribution to infer linkage18-20. The Proteome Navigator tool allows users to browse predicted linkage networks interactively, providing accompanying annotation from additional public databases.
The ProKnow program (http://proknow.mbi.ucla.edu) constitutes a knowledgebase where features from proteins such as three-dimensional fold, sequence, motif and functional linkages are extracted and then related to annotated functions from Gene Ontology functional terms21. In the advent of structural genomics, ProKnow is a useful resource in functional assessments of proteins with structural information but unknown functions. ProKnow has been applied to the Mtb H37Rv genome and the results for all genes, their function-based similarities and links show that ProKnow is able to assign around 50% of genes in the genome with high confidence. These high confidence linkages have been incorporated into functional information on the TBSGC website.
The TBSGC has also established integrated core facilities located at Texas A&M University (TAMU), Los Alamos National Laboratory (LANL) and Lawrence Berkeley National Laboratory (LBNL) to provide technical support for Consortium members, which is presented in the first section of this review. The remaining sections highlight a compilation of recent Mtb protein structures determined by TBSGC groups, many of which are associated with metabolic pathways and are, thus, potential attractive anti-TB therapeutic targets. In particular, these include structural studies on urease (Habel and Hung), chorismate-utilizing enzymes (Johnston et al.), arginine biosynthesis enzymes (Sankaranarayanan and James), crotonase, malate synthase, fumarase and phosphoenolpyruvate carboxykinase (Krieger et al.) and heme degrader, MhuD (Chim et al.). Additionally, atomic resolution details of proteins from systems involving disulfide bond formation of secreted proteins (Chim et al.) and toxin-antitoxin gene pairs (Miallau and Eisenberg) can ultimately be exploited for rational structure-based drug design.
The TBSGC Structure Determination Pipeline (Li-Wei Hung, Chang-Yub Kim, Hongye Li, James C. Sacchettini and Thomas C. Terwilliger)
The TBSGC has established an effective pipeline as a core resource for Mtb structural biology. The purpose of the pipeline is to provide an integrated set of facilities for cloning, protein expression, purification and X-ray data collection capabilities for the entire TBSGC project. The current pipeline consists of a high-throughput cloning and expression facility at the Institute of Biosciences and Technology, TAMU, a protein production facility at LANL, and crystallization and data collection facilities at the Advanced Light Source (ALS), LBNL. These facilities are equipped with robotics and are highly automated as described previously2. These pipeline facilities work cohesively to provide Consortium members with unique opportunities to gain access to the most advanced technologies in structural biology.
The mission of the TAMU cloning facility is to establish a solid molecular biology foundation for the TBSGC. Since 2007, the cloning facility has completed construction of the entire proteome library for all open reading frames of nearly the entire Mtb H37Rv genome. Approximately 3600 genes have been cloned into the pDONR/zeo entry vector using the Gateway recombinant cloning method (Invitrogen Inc.). Each clone has an attB1-TEV-ORF-attB2 construction; the genes have a Tobacco Etch Virus (TEV) cleavage site for easy removal of any tag used for purification. Targeted genes are cloned into destination vectors and tested for protein expression and solubility levels before downstream processes.
In addition to providing expression clones of Mtb targets for the TBSGC project, the TAMU facility processed about 700 genes in two groups aiming for protein production. The first set, Top 100, was the top 100 persistence genes selected from high-priority Mtb drug targets22. The second set, Target 600, consists of 667 genes selected based on the combination of gene essentiality23 and protein sequence space coverage. All of these clones were processed through small-scale expression and solubility tests to identify suitable candidates for large-scale production. The Target 600 and Top 100 sets showed satisfactory levels of soluble protein expressions for 265 and 53 genes, respectively. A total of 56 genes from these two groups were sent to the protein production facility for large-scale protein expression. Twenty-two of these targets were successfully purified and sent to the crystallization facility for crystallization and data collection. Eight proteins were crystallized and crystal structures of four targets from these two sets have been determined.
The aim of the LANL protein production facility is to provide highly purified Mtb proteins that are suitable for protein crystallography purposes. To achieve higher yields of protein production, the LANL facility has expanded its capabilities to accommodate various expression vectors including those containing His-MBP-TEV-tags, and GST-tags, as well as vectors optimized for protein complex expression. Since 2007, the LANL facility has processed requests of over 150 Mtb targets chosen by the Consortium members. Sixty-four targets have been successfully produced and 102 samples were shipped to the targeting laboratory and/or to the LBNL facilities for crystallization and structure determination.
To improve the overall success rate through effective salvaging pathways, the protein production facility has incorporated two emerging techniques; surface entropy reduction (SER)24 and high-throughput ligand analysis25, into its production protocols. As proof of concept, six Mtb targets with poor crystallizabilities were selected for the SER process. Two of the SER-engineered targets yielded diffracting-quality crystals immediately. Crystal structures of both targets have been determined and one has been deposited (Rv3275c, PDB code 3LP6). Similarly, 32 Mtb proteins were identified to have high propensity to be nucleoside/nucleotide-binding proteins through the high throughput ligand-affinity chromatography method using Cibacron blue dye. The LANL facility purified eleven targets and obtained improved crystals of nine proteins. To date, five Mtb targets previously stalled in the pipeline have been successfully determined utilizing this approach.
The goal of the LBNL crystallization and data collection facilities is to provide high-throughput crystallization and data collection capabilities for the TBSGC project. Taking advantage of proximity to the ALS and pre-scheduled beam time, the LBNL facility offers rapid turnaround for TBSGC data collection at the 3rd-generation synchrotron X-ray source. The crystallization facility continues to improve throughput through miniaturization and automation. The standard crystallization experiments have been reduced from droplets of 500 nL to 150 nL representing a three-fold reduction of protein sample requirement. Experimental results from the LBNL facilities are synchronized daily with a centralized Laboratory Information Management System managed by UCLA. These data are available to the Consortium members in real-time through the webTB website (http://www.webtb.org). Since 2007, twenty-one Mtb targets have been crystallized at the crystallization facility resulting in six new structures. Diffraction data collected at the data collection facility have led to the structure determination of 16 de novo Mtb protein structures, and over 30 protein-ligand complex structures.
In an effort to enhance the TBSGC’s structural biology capability, the LBNL facility has incorporated high-throughput Small Angle X-ray Scattering (SAXS) into its operation. Through SAXS data, one can quickly reveal the aggregation state of protein samples in solution. This information has been an important quality indicator of protein samples for crystallization. SAXS can also provide low resolution electron density envelopes of proteins in solution which is thought to be more relevant to their biological states26. SAXS is suitable for a wide range of molecular mass and buffer composition. With the advantage of being in the proximity of a synchrotron source, the TBSGC X-ray data collection facility routinely uses SAXS to assess the solution states of protein samples. Since mid-2009, the LBNL facility has performed over 300 SAXS experiments for 68 TBSGC targets. A few SAXS results have provided valuable insights towards understanding of target proteins’ biological state (e.g. Habel et al., 2010, see following section).
The TBSGC pipeline facilities provide cost-effective means to carry out high-throughput, mid-scale cloning, protein production and structure determination for the TBSGC project. The Consortium members have been taking advantage of this unique resource to enhance their research. With the improved capacity and newly acquired technologies, the pipeline is expected to play an essential role in future research in Mtb biology and TB drug design in the TBSGC.
Solution and Crystal Structures of Mtb UreA - the Implications in Bio-molecular Assembly and Drug Discovery of Mtb Urease (Jeff E. Habel and Li-Wei Hung)
Found in a broad range of plants and bacteria, urease is a nickel-containing enzyme catalyzing the hydrolysis of urea into ammonia and carbamate (which then decomposes with water to form ammonia and carbon dioxide). Urease has been implicated as a potential virulence enhancing factor in diseases of the human gastrointestinal and urinary tract27-29. For Mtb, urease is thought to increase survival in the lung tissue possibly through pH modulation (as seen in Helicobacter pylori) and nitrogen biosynthesis30, 31.
Mtb urease is encoded within an operon whose composition suggests the formation of a heterotrimeric urease complex, consisting of α (UreC), β (UreB) and γ (UreA) subunits, similar in structure to Klebsiella aerogenes urease. Distinct from other ureases, Mtb urease contains unique properties30, including (1) a comparatively low Km of ~ 0.3mM that is thought to be a consequence of the urea deficient environment inside the macrophage and (2) remarkable temperature stability and unusual chemical tolerance, whereby enzymatic activity is retained up to 60 °C and in the presence of 4M urea, respectively, which is believed to help combat against fever within hosts.
Due to the potential virulence enhancing affects seen in microbial ureases27, 28, 32, 33, Mtb urease remains an enticing target for structure based drug design. We have targeted Mtb urease and its individual subunits for structure determination. One of the early results of this effort is the crystal and solution structures of the UreA subunit, Rv1848, under PDB code 2FVH34. While UreA contains no known enzymatic function, it remains an appealing drug target within the emerging field of protein-protein interaction (PPI) inhibitors35.
Once thought to be too intractable due to the idea that PPIs were formed from large flat surfaces, several groups have made strides to develop clinically ready molecules in the areas of diabetes targeting c-Jun N-terminal kinase 136; cancer targeting Polo-like kinase 137, 38 and chaperonin HSP9039, 40; HIV targeting CD4, tat, and cyclophilin A41-43; and immunosuppression targeting calcineurin44, 45. Mediated largely through aromatic interactions, initial ligands are being developed through fragment-based screening followed by tethering and assembly of small low-affinity inhibitors into a larger high-affinity molecule. Similarly, the structures of Mtb UreA determined by two complimentary methods indicate that Mtb UreA may represent an interesting and unique target for drug discovery through the disruption of the Mtb heterotrimeric urease complex.
The crystal structure of Mtb UreA (Rv1848) represents the first structure of an individual subunit within the (αβγ)3-type ureases reported to date and closely resembles the γ subunit of the K. aerogenes UreA structure (PDB code 2KAU). Approximately 4300 Å2 is buried in the formation of the UreA homotrimer indicating highly specific subunit interactions46, 47. The Rv1848 homotrimer (Figure 1A) bears a striking resemblance to the γ subunit quaternary structure seen in the other (αβγ)3 urease structures. When superimposed, the Rv1848 homotrimer has an RMSD of 1.13Ǻ with the K. aerogenes (αβγ)3 urease γ-subunit homotrimer. Together with nickel analysis showing six molecules per complex30, the Mtb urease complex is likely to adopt the trimer of trimers formation seen in K. aerogenes urease and other related species. Rv1848 was over expressed and crystallized in the absence of any other urease component demonstrating that Mtb UreA is able to form the homotrimer seen in the crystal structures of urease complexes independent of the UreB and catalytic UreC subunits.
Figure 1.
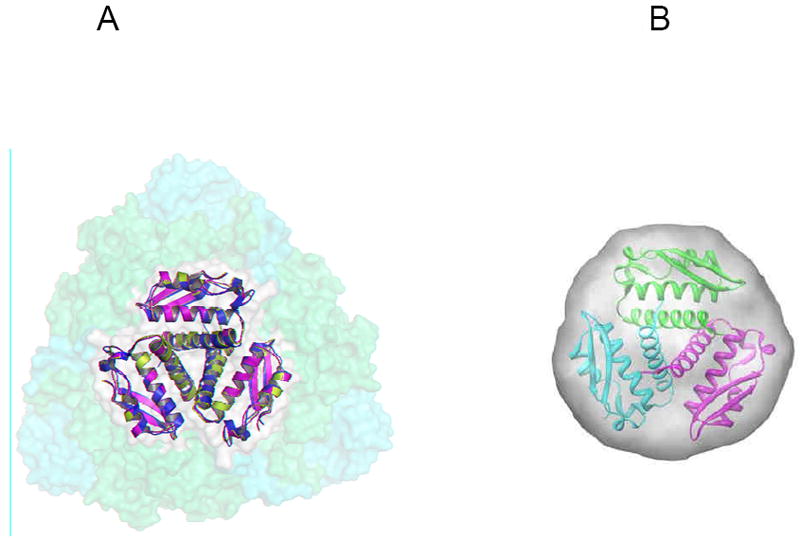
(A) Overlay of the γ subunits from Mtb (PDB code 2FVH - green), Klebsiella aerogenes (PDB code 2KAU – cyan) with 1.13Å RMSD, and Bacillus pasteurii (PDB code 1UBP- magenta) with 0.968Å RMSD superimposed onto the K. aerogenes urease trimer of trimers in semi-transparent orange (α) and yellow (β) surfaces highlighting the centralized location of the γ subunits in the urease complexes. (B) The envelope is calculated from the average of 16 GASBOR runs with P3 symmetry and 100 residues. The figures are reproduced from Reference 34.
To further confirm that the Rv1848 homotrimer seen was not a crystallization artifact, a SAXS experiment was performed on the Rv1848 protein crystallization sample solution. The SAXS experiment shows that Rv1848 has an approximate molecular weight of 33kDa in solution, three times the UreA monomer molecular weight. Ab initio molecular envelopes calculated from the SAXS data overlay nicely onto the Rv1848 homotrimer crystal structure (Figure 1B). These results suggest that the Rv1848 forms a homotrimer in solution and it may be biologically relevant. Since UreA has no known enzymatic activity, it is possible that the homotrimer of γ subunits seen at the center of urease trimer complexes is forming a scaffold for the other subunits. The in-solution auto-assembly of Rv1848 may indicate an important driving force for the urease trimer of trimers complex assembly. This potential need for oligomerization may play a role in Mtb urease’s tolerance to environmental, chemical and temperature challenges beyond simple catalytic efficiency.
Given its likely importance to pathogenesis, urease remains an appealing target for tuberculosis chemotherapeutic intervention via PPI. The crystal and solution structures of Rv1848 provide structural insight into future drug design beyond conventional catalytic or substrate binding site centered targeting strategies. Compounds disrupting the UreA homotrimer PPI could disrupt proper urease holoenzyme assembly leading to potential decreases in catalytic efficiency and/or environmental heat and chemical tolerance, ultimately interfering with urease’s virulence enhancing abilities. Biochemical studies to understand the kinetics and binding dynamics of UreA homotrimer and Mtb urease holoenzyme assembly are underway.
Studies on Chorismate-utilizing Enzymes in Mtb (Jodie M. Johnston, Esther M. Bulloch, Richard J. Payne, Alexandra Manos-Turvey, Edward N. Baker and J. Shaun Lott)
Chorismate is a central metabolic intermediate that is utilized by many bacteria in the production of a variety of aromatic compounds, including the aromatic amino acids, folate, and salicylate. Because the biochemical pathways that catalyze the biosynthesis and utilization of chorismate are found in plants, bacteria and some protists but not in more complex eukaryotes, they have long been considered to be potentially useful targets for therapeutic intervention, with a strong inherent propensity for selective toxicity. An often-cited example is that the successful herbicide compound glyphosate inhibits an enzyme in the chorismate biosynthesis pathway48. In Mtb, several chorismate-utilizing enzymes have been shown to be essential for growth or virulence, including the tryptophan, folate and mycobactin biosynthesis pathways. We have previously solved the structures of the chorismate-utilizing enzyme salicylate synthase (MbtI)49 and the enzyme that catalyzes the first step in chorismate production, 3-deoxy-D-arabino-heptulosonate-7-phosphate (DAH7P) synthase (AroG)50. In this review, we highlight two recent advances in our understanding of chorismate metabolism in Mtb: the development of inhibitors of MbtI and the biochemical identification of the folate biosynthetic enzyme 4-amino-4-deoxychorismate (ADC) lyase.
a) Design of compounds to inhibit salicylate synthase
The ability of Mtb to acquire the essential micronutrient iron is primarily dependent on its production of the siderophore mycobactin T, and the first step in the biosynthesis of the siderophore is the production of salicylate. We, and others, have previously shown that MbtI (Rv2386c) is a chorismate-utilizing enzyme that produces salicylate in a two-step reaction, and have determined the enzyme’s three-dimensional structure49, 51. Like other homologs, MbtI displays a complex α/β fold, composed of two sub-domains with a deep cleft between them containing the active site residues; the similarity in the structure of these enzymes is presumed to extend to their enzyme mechanisms, although our mechanistic understanding of this enzyme family remains incomplete.
We have been working to design and synthesize inhibitors of MbtI. The observation that the use of a derivatized benzoate ring in place of the non-aromatic chorismate ring was able to produce improved inhibitors of the related salicylate synthase Irp9 from Yersinia enterolytica52, 53 inspired the use of an aromatic scaffold as a starting-point for MbtI inhibitors. The use of an aromatic scaffold greatly simplifies the chemical synthesis of new compounds, enabling more rapid synthesis and screening through candidates.
The most successful compounds were based on a 2,3-hydroxybenzoate scaffold that was intended to be an aromatic mimic of the natural reaction intermediate, isochorismate. The most potent compounds contain an enol-pyruvate sidechain with hydrophobic additions, with the best of the series (Compound 39) displaying a Ki of 11μM, as measured using a lactate-dehydrogenase-linked in vitro assay of enzyme activity54. These compounds are the most potent inhibitors of salicylate synthases yet identified, and as such represent promising lead compounds for the future synthesis of inhibitors with nanomolar or better affinity.
To understand how these inhibitors bind to MbtI, we used in silico docking using the program GOLD. This molecular modeling indicated that the compounds most likely bind in an analogous manner to the chorismate substrate, as inferred from comparison with the product-bound structure of Irp9 (PDB code 2FN1). As shown in Figure 2, the C1 carboxylate is predicted to interact with the catalytically required magnesium ion, and the methyl group attached to the enol-pyruvate side-chain is predicted to interact with a small cluster of hydrophobic residues (I207, Y385, L404). Structural analysis is currently underway to validate this predicted binding mode, and further synthesis is planned based on this compound series.
Figure 2.
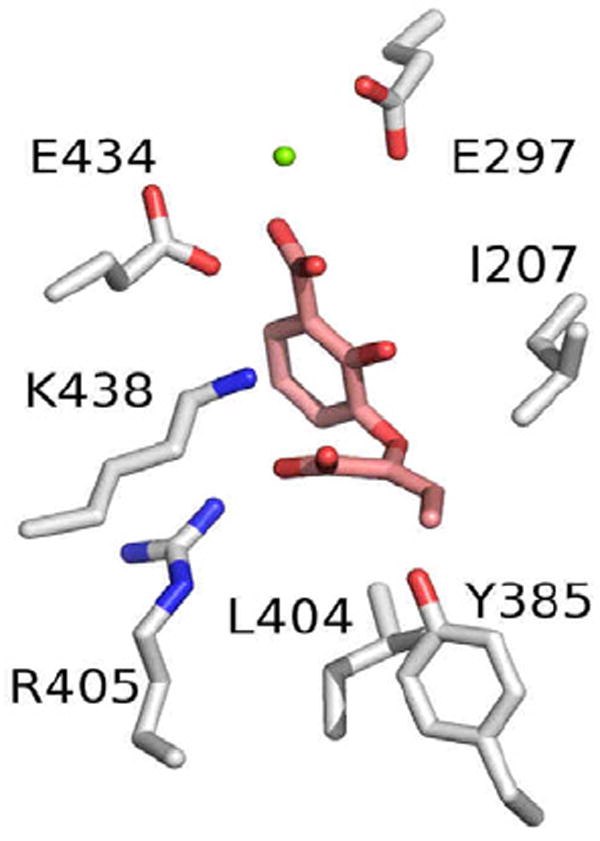
The predicted binding mode of Compound 39 in the active site of MbtI. Compound 39 is shown in pink, and the zinc atom is shown as a green sphere.
b) Identification and characterization of 4-amino-4-deoxychorismate lyase
The enzyme ADC lyase (PabC) is essential for de novo folate biosynthesis in bacteria, plants and fungi. PabC cleaves ADC, formed from chorismate by the PabA/PabB complex, to give 4-aminobenzoate (para-aminobenzoic acid, PABA) and pyruvate55-61. The PABA formed from this reaction is an important precursor of folate, which is essential for normal cell growth. The action of PabC is dependent on the cofactor pyridoxal 5′-phosphate (P5′P)55, 57. The structure of E. coli PabC shows it to be homodimeric with a two-domain monomer comprising a small α/β N-terminal domain and a larger α/β “pseudo β-barrel” C-terminal domain, with the active site located at the dimer and domain interfaces 57.
We have focused on the enzymes that catalyze the first two steps of the folate biosynthesis pathway in Mtb - PabA, PabB and PabC. Although the bioinformatic identification of PabA and PabB is unambiguous, the identity of the open reading frame (ORF) encoding PabC is not clear. We have studied the uncharacterized ORF Rv0812 as the most likely candidate for Mtb PabC. Rv0812 is annotated as a “probable amino acid transferase” (http://www.webtb.org/), with 20% amino acid sequence identity to the experimentally characterized E. coli PabC. A recent paper identified Rv0812 gene as one of the 56 preferentially expressed Mtb genes in mouse lung during infection62.
Rv0812 could be expressed in a partially soluble form in E. coli as a variety of different N- and C-terminal tagged constructs. Dynamic light scattering63 results suggested that Rv0812 is dimeric in solution, as is the case for E. coli PabC. 1H NMR and coupled spectrophotometric activity assays were subsequently carried out on the full-length, N-terminally His6-tagged construct, to test for ADC lyase activity.
In the spectrophotometric assays, ADC was generated in situ from chorismate using purified E. coli PabB enzyme, with ammonium chloride added as a source of ammonia. The conversion of ADC to PABA was monitored by UV absorption spectroscopy at 265 nm (Figure 3A). An increase in absorbance at 265 nm was seen when E. coli PabB and Rv0812 were both present but not for the various negative controls, supporting the hypothesis that Rv0812 is capable of functioning as Mtb PabC.
Figure 3.
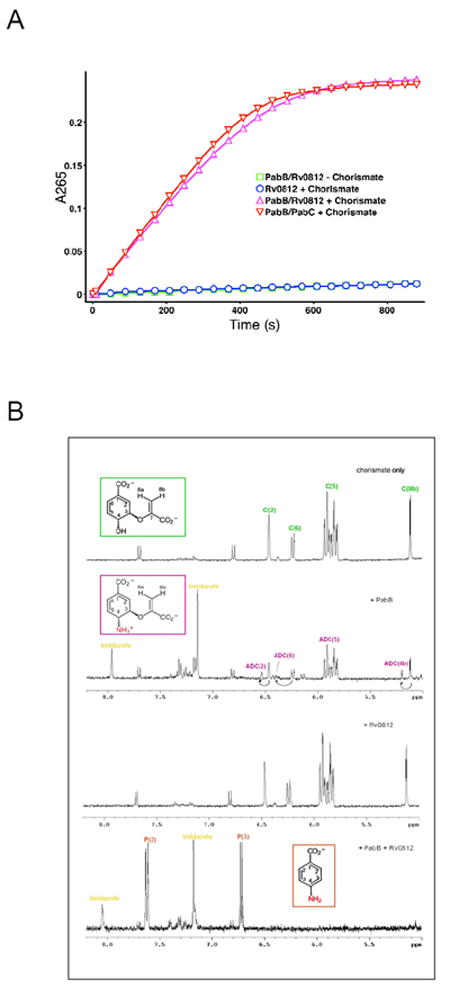
(A) Top graph - Coupled PabB and PabB/His-tagged Rv0812 assays following production of PABA (4-aminobenzoate) by measuring increase in absorbance at 265nm (reactions and controls as specified in key). (B) 1H NMR Spectra of Coupled PabB/C and PabB/Rv0812 Assays and Controls.
To investigate this further, 1H NMR spectroscopy was used to analyze the reaction. As for the spectroscopic assay, ADC was generated in situ from chorismate using purified E. coli PabB with ammonium chloride. Rv0812 was then incubated with ADC at 37°C for 4 hours prior to 1H NMR spectra being obtained (Figure 3B). Analysis of the spectra revealed the appearance of peaks consistent with the formation of 4-amino-4-deoxybenzoate in the E. coli PabB/Rv0812 reaction and in a positive control reaction containing E. coli PabB/C, which were not observed in negative control reactions.
Differential scanning fluorimetry analysis64-66 of purified Rv0812 showed that its thermostability is greatly enhanced by binding to P5′P (an increase of Tm of 10-17 °C), and that this stabilization is modestly enhanced by the addition of substrate and product analogues. However, extensive crystallization trials have so far only resulted in the production of small needle-like crystals that currently remain to be optimized for X-ray analysis.
In conclusion we have experimentally confirmed that the ORF Rv0812 encodes PabC in Mtb and have shown that the protein is greatly stabilized by binding to its known co-factor. Work continues to structurally characterize the putative Mtb PabC.
Ornithine acetyltransferase (Rv1653) and Ornithine carbamoyltransferase (Rv1656) in Arginine Biosynthesis Pathway of Mtb (Ramasamy Sankaranarayanan and Michael N.G. James)
The X-ray crystal structures of two enzymes (Ornithine Acetyltransferase, Mtb OAT (E.C.2.3.1.35) and Ornithine Carbamoyltransferase, Mtb OTC (E.C.2.1.3.3) that are involved in the arginine biosynthetic pathway of Mtb are reported here. Mtb OAT reversibly catalyzes the transfer of the acetyl group from N-acetylornithine to L-glutamate to produce N-acetylglutamate67. Although there are two other enzymes, N-acetylglutamate synthase (E.C.2.3.1.1) and N-acetylornithine deacetylase (E.C.3.5.1.16) that are present in the arginine biosynthesis pathway of Mtb, the existence of OAT as a part of an acetyl recycling pathway is still an unanswered mystery. Hence, it is our primary interest to determine the X-ray crystal structure of Mtb OAT in order to understand the catalytic mechanism of OAT in this bacterium.
Structure of Ornithine Acetyltransferase (OAT) from Mtb
The structure of ligand-free OAT was originally determined from Streptomyces clavuligerus68. Recently, we solved the 1.7 Å structure of native Mtb OAT (PDB code 3IT4), which reveals the conserved fold of the N-terminal nucleophile family of enzymes69. Additionally, we reported the first 2.4 Å binary complex of Mtb OAT bound to ornithine (ORN, PDB code 3IT6)69, a competitive inhibitor against the cognate substrate, L-glutamate. The monomer of Mtb OAT, which consists of 404 amino acid residues, splits into two halves forming a two-domain architecture; Domain I (residues 1-284) and Domain II (residues 285-404). Domain I consists of an αββα sandwich that is made up of six α-helices and ten β-strands while Domain II adopts an α/β fold that consists of four α-helices and five β-strands (Figure 4). Within the active site, which is comprised of residues from both Domains I and II, Thr200 is the nucleophile that is involved in the acetyl transfer reaction. Moreover, the last three residues at the C-terminus of the native form are poorly ordered due to the very high flexibility of this end. However, the binding of ORN stabilizes these residues. The interactions of ORN with the active site residues of Mtb OAT helps in identifying the key residues involved during the activity of this enzyme. The modeling studies of Mtb OAT with the N-acetylornithine reveals that the main-chain nitrogen atom of Gly128 and the side-chain Oγ1 of Thr127 are primarily involved in the stabilization of the oxyanion formed during the enzymatic reaction. Also, the modeling of other substrate L-glutamate in the active site of Mtb OAT based on the structure of the ORN bound complex has revealed that the side-chain nitrogen atom of Asn399 and side-chain carboxylate oxygen atom of Glu280 are the primary residues involved in the stabilization of the negatively charged side-chain carboxylate of L-glutamate.
Figure 4.
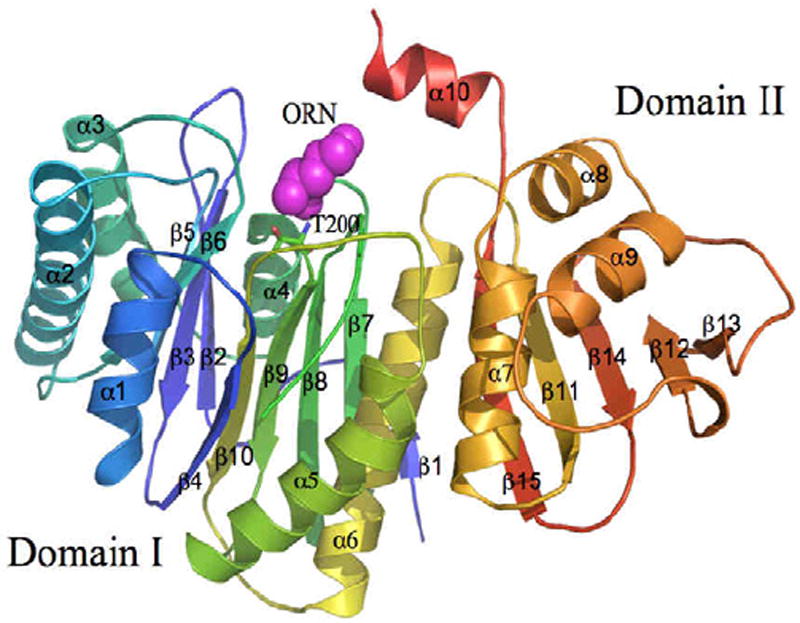
The monomer of Mtb OAT (PDB code 3IT6) is shown in cartoon representation, with a rainbow coloring scheme (the N-terminus is blue and the C-terminus is colored red). Ornithine (ORN, magenta) and active site residue, Thr200, is shown in CPK and stick representations, respectively.
Structure of Ornithine Carbamoyltransferase (OTC) from Mtb
Mtb OTC reversibly catalyzes the transfer of the carbamoyl group from carbamoyl phosphate (CP) to the second substrate, ORN, to produce L-citrulline. The crystal structures of Mtb OTC in its native trimeric form (PDB code 2P2G) and in its trimeric bound form with CP and L-norvaline (NVA, PDB code 2I6U) have been determined to the resolutions of 2.7 and 2.2 Å, respectively70. NVA is a competitive inhibitor of Mtb OTC against ORN. The monomer of Mtb OTC has 307 amino-acid residues having the molecular mass of 33 kDa. Mtb OTC has an α/β topology having nine α-helices and eleven β-strands (Figure 5). The monomer is made up of two domains, namely the CP binding domain and the ORN binding domain. The active site is situated in a cleft located between the two domains. There are two loops, the 80s loop and the 240s loop that play very prominent roles in the substrate binding. The residues in the 240s loop in the native structure of Mtb OTC are highly disordered and cannot be traced, whereas, the hydrogen-bonding interactions involving ORN with the residues of 240s loop in the structure of the ORN bound complex of Mtb OTC have stabilized this loop very well. In the ORN bound complex, the 80s loop shows an appreciable displacement of ~7.5 Å when compared to its position in the native unbound form. The 240s loop shows a maximum displacement of 12.2 Å when compared to the native structure of OTC from Pyrococcus furiosus. The interactions of the ligands (CP and NVA) bound at the active site of Mtb OTC reveal that the negatively charged phosphate group of CP is stabilized by many positively charged residues such as Arg52 and Arg101. The interactions involving NVA at the active site reveals that the zwitterionic form of ORN with the NεH2 group in the side-chain is the ideal candidate to bind at the active site. The primary amine NεH2 would eventually perform the nucleophilic attack on the carbamoyl group of CP. Our modeling studies carried out with ORN fitted into the structure of the NVA bound complex also helps in identifying that one of the oxygen atoms of the phosphate group of CP takes part in the deprotonation of the doubly protonated side-chain nitrogen atom of ORN.
Figure 5.
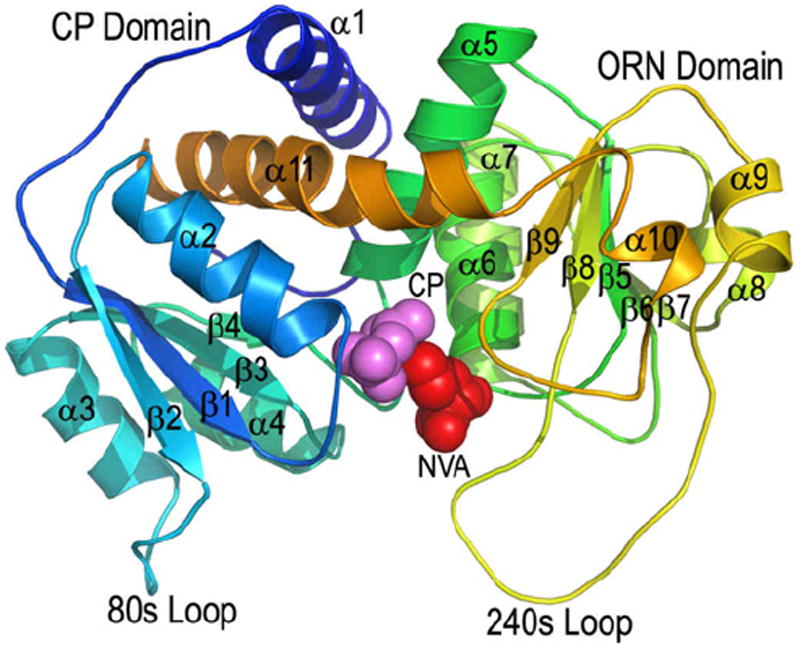
The cartoon diagram of the monomer of Mtb OTC (PDB code 2I6U). Carbamoylphosphate (CP) and Norvaline (NVA) are shown in CPK representation. This figure is reproduced from Reference 70.
Prior to the work on Mtb OTC that has been presented here, another study proposed a mechanism for human OTC in which Cys264 plays the role of proton acceptor from the NεH3 of ORN71. The Sγ of Cys264 is too far (>5.0 Å) from the Nε of ORN to be involved with a proton transfer. We propose an alternative mechanism (Figure 6) in which the neutral, deprotonated NεH2 of ORN and the positively charged α–amino nitrogen of ORN are the species of ORN that binds to the enzyme. Thus, the neutral NεH2 can nucleophilically attack the carbonyl-carbon of the carbamoyl group of CP (Figure 6). There is something of an oxyanion hole that stabilizes the negative charge on the carbonyl oxygen of the carbamoyl group of CP. The groups forming the oxyanion hole are His128, Arg101 and Arg292.
Figure 6.
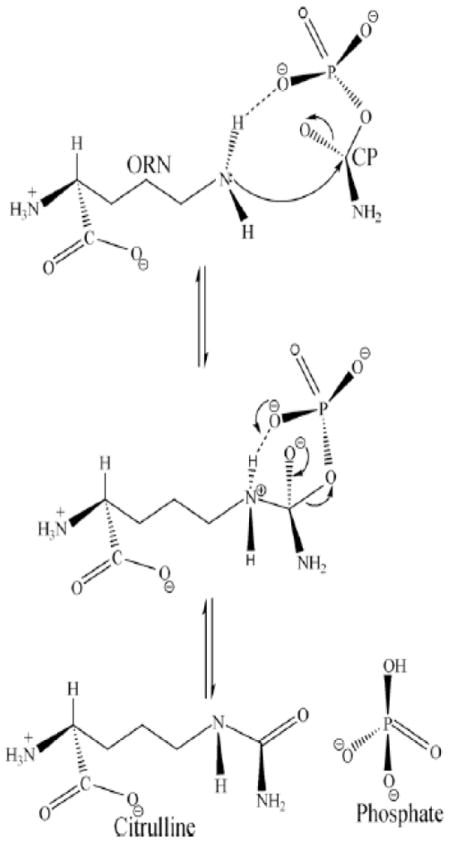
The steps proposed for the catalytic mechanism of OTC: the arrows represent the electron transfer in this mechanism. This figure is reproduced from Reference 70.
Structure of Key Enzymes in Persistence Related Metabolic Shift (Inna Krieger, John Bruning, Stephanie Swanson, Haelee Kim and James C. Sacchettini)
Mtb demonstrates remarkable metabolic versatility, allowing it to survive and persist under the hypoxic, acidic, and nutrient poor conditions inside the host macrophage. It accomplishes this by adapting its growth rate to suit its environment. During the persistent state of infection, Mtb’s growth drops to a virtually non-replicating state by significantly altering its metabolism to conserve energy and effectively use resources scavenged from the host72. Our focus has been to study the structural and biochemical function of the enzymes identified as key players in the metabolic shift that sustain the persistent state.
Experimental evidence shows that inside the macrophage, where there is little availability of complex sugars, Mtb instead utilizes fatty acids as its major carbon and energy source73, 74. The fatty acids are converted to 2- or 3-carbon units in the form of acetyl- or propionyl-CoA by ß-oxidation. ß-oxidation has long been recognized as a factor in mycobacterial virulence, required for persistence inside macrophages, and several enzymes involved in this process have been characterized74, 75. Four steps are involved in ß-oxidation. First, acyl-CoA dehydrogenase oxidizes the fatty acid creating a double bond on the acyl group. Second, the newly created double bond is hydrated at the C3 position, leaving a hydroxyl group on the acyl chain. The third step consists of further oxidation by means of hydroxyacyl-CoA-dehydrogenase, which creates a carbonyl group at the previous hydroxyl position at C3. The final step is thiolysis: acetyl-CoA is removed and a new CoA group is added to the remaining fatty acid resulting in a chain that is two carbons shorter76. Despite the available information, defining the correct location and annotation of the enzymes responsible for these steps in the Mtb genome presents a significant challenge. Structural biology is an indispensable tool for obtaining atomic detail characterization of the active sites, ligand interactions, and details of the catalytic mechanism of the enzymes in each step of ß-oxidation.
For the second step ß-oxidation, 20 enzymes are annotated as putative enoyl-CoA hydratases (crotonases) in the Mtb genome, and yet even though eukaryotic crotonase structures and kinetics have been understood for decades, the Mtb crotonase homolog is not known. In many species the prokaryotic enoyl-CoA hydratase has been proposed to be part of a multifunctional enzyme complex (fadAB operon) rather than a distinctive crotonase molecule76. We have determined the atomic level structure of the prokaryotic crotonase from Mtb, Rv1070c (manuscript in preparation). The overall fold of the prokaryotic crotonase is similar to the eukaryotic crotonase. The oligomerization state of the Mtb crotonase is that of hexamers and nonamers as compared to the trimeric structure formed in the eukaryotic structures. Additionally, packing around the acyl group of the CoA molecule in the active site of the Mtb crotonase differs from the eukaryotic crotonase, perhaps explaining its lower turnover rate. We have also co-crystallized the crotonase enzyme with a reaction intermediate in the active site providing a first look at a natural substrate (crotonyl-CoA) bound to a crotonase, which allows for a detailed interpretation of the reaction mechanism (Figure 7). Co-crystallization of the crotonase with acetoacetyl-CoA, a known crotonase inhibitor, bound to the prokaryotic crotonase, has allowed us to decipher the mechanism of inhibition. Our structural studies were complemented by kinetic assays allowing us to elucidate the turnover rate and Michaelis constants for the enzyme; we have concluded that the prokaryotic enzyme is not as efficient as the eukaryotic counterpart.
Figure 7.
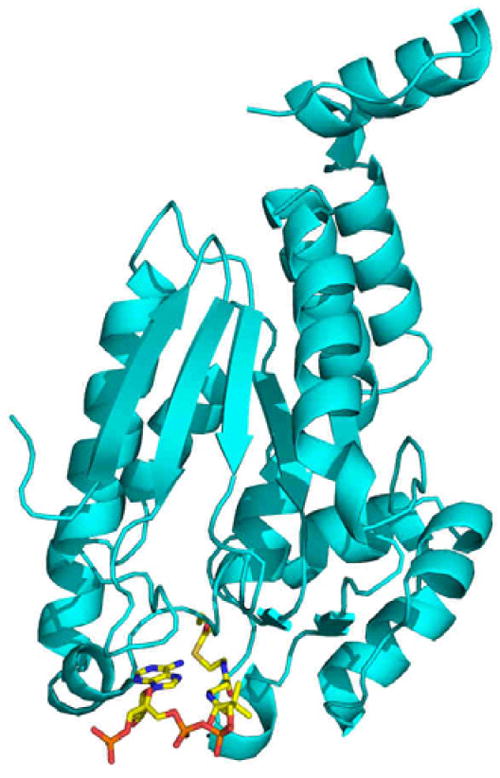
The Mtb crotonase bound to a reaction intermediate. Mtb crotonase is shown in teal in ribbon representation and a reaction intermediate derived from crotonyl-CoA is shown in stick colored by atom.
After the lipids are converted into acetyl-CoA during ß-oxidation, they can be efficiently used by the cell for energy metabolism and biosynthesis. The entry point is a short, two-enzyme anaplerotic pathway found in all prokaryotes, plants, and some lower eukaryotes, but not in vertebrates77. It is called the glyoxylate shunt because it constitutes a short cut across the tricarboxylic acid (TCA) cycle from isocitrate directly to malate, eliminating two carbon-losing steps in the form of CO2 producing reactions, and enabling incorporation of acetyl-CoA. The first enzyme of this shunt, isocitrate lyase (ICL), converts isocitrate into the TCA-intermediates succinate and glyoxylate and the latter is taken by the second shunt enzyme, malate synthase, and converted into malate using one acetyl-CoA molecule. It has been shown that the glyoxylate shunt is required for Mtb virulence; ICL knockout mutants do not survive inside macrophages to establish a persistent infection78. Significant evidence has since been added to show the crucial role of the glyoxylate shunt in the virulence of other intracellular infectious organisms such as Candida albicans79 and Pseudomonas aeruginosa80. A functional glyoxylate shunt has also been linked to the maintenance of ATP homeostasis and the viability of non-replicating persistent Mtb81.
Numerous attempts have been made to target ICL for drug design, but all efforts have failed to identify lead molecules. ICL has been declared “non-druggable” possibly due to its small and very polar active site, and the large structural rearrangements that occur upon catalysis82. The second enzyme of the shunt however, malate synthase encoded by Rv1837c, presents a well structured active site resembling a long tunnel with a catalytic Mg2+ at the bottom, with no major changes between the substrate- and product-bond forms83. Armed with the ability to use crystal structures of malate synthase for structure-based drug design, we have screened 150,000 small molecules using a malate synthase enzyme assay in collaboration with the Southern Research Institute to identify inhibitors for the enzyme. We have solved a complex structure of malate synthase with one of the most potent hits, 4-(3-methoxyphenyl)-4-oxo-2-(((3-trifluoromethyl)phenyl)amino)butanoic acid, and this structure has provided novel insight into the product and substrate exchange process (manuscript in preparation). While all previously known structures of glyoxylate- or CoA and malate-bound forms demonstrated only one opening from the active site to the solvent83, 84, the inhibitor-bound form showed the enzyme trapped in a conformation that revealed a second portal open to the active site. Movement of the loop containing residues 458-461, particularly the Phe460 side-chain flipping “open”, creates this additional portal. Figure 8 compares the inhibitor-bound enzyme surface in gray and the product-bound enzyme surface in blue, while the red arrow indicates the opening to the new portal, and the orange arrow shows the widening of the cavity behind the Mg2+ binding site. Concurrently, significant narrowing in the first, CoA accommodating, channel to the surface occurs compared to the product/substrate-bound structures. The catalytic Asp633 is pulled forward toward the coordinating waters of the Mg2+ sphere, initiating the movement of the long stretch of the protein constituting the “mouth” of the active site channel, and involved in acetyl-CoA coordination – residues from Cys619 to Asp633 move, with the largest main chain atom displacement of about 2.7 Å at the Cα of Glu632 (Figure 8). A similar opening has been observed in the structure of malate synthase form A (MSA) from E. coli – the only structure of malate synthase known with only a Mg2+ bound to it (PDB code 3CUZ)85. Despite the difference in the residues at the opening position from malate synthase form G (MSG) (MSA has Arg276 in the Phe460 position of MSG), the main chain of MSA (PDB code 3CUZ) aligns closely for the region, corresponding to residues 458-461 of MSG (Figure 8). Thus, this second opening to the solvent seen in inhibitor-bound malate synthase G must reflect the physiological state of the enzyme when unoccupied by either substrate or product. We hypothesize that two substrates of malate synthase - glyoxylate and acetyl-CoA - enter through two different openings into the active site; after the reaction, malate is released through the glyoxylate portal, and CoA through acetyl-CoA portal. Moreover, glyoxylate binding must occur before acetyl-CoA enters the second channel to allow the second portal to open sufficiently – even when CoA is not bound, the glyoxylate-bound structure (PDB code 1N8I) displays the same sufficiently wide channel as the CoA and malate-bound structure (PDB code 1N8W). Also, similar narrowing of the first CoA channel (Figure 8) is present in the MSA structure bound only with Mg2+ (PDB code 3CUZ) (2.5 Å at Met631 Cα for 3CUZ compared to 1N8W, and 1.5 Å for the inhibitor-bound MSG compared to the same model), supporting the idea that this channel widens for acetyl-CoA entry only after glyoxylate binding occurs.
Figure 8.
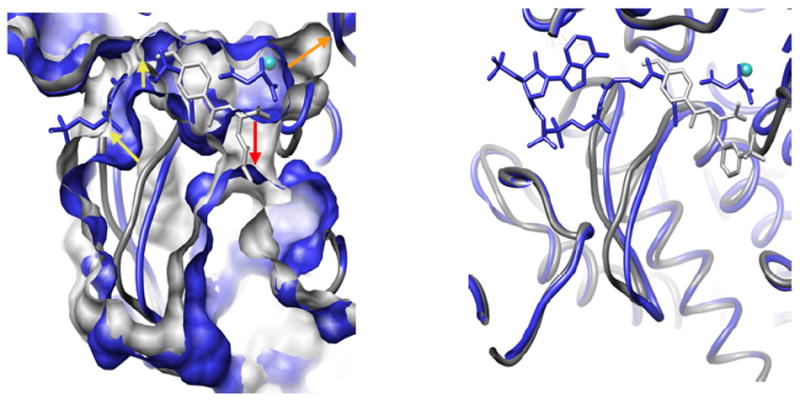
Mtb malate synthase G in complex with products (in blue) superimposed with inhibitor-bound enzyme (in gray). The enzyme is shown in ribbon, and small molecules bound to it in stick. The left panel shows surface representation of the enzyme, with arrows indicating the positions where the biggest difference between two were observed. The right panel shows the position of inhibitor binding relative to CoA and malate.
This structural information serves as an atomic detail basis to complement the study of malate synthase enzyme kinetics and the reaction mechanism, as well as providing a valuable platform for structure-based inhibitor design for TB drug discovery. We are currently working on several other lead molecules to exploit both forms of the active site - a substrate/product-bound form with only one solvent channel open, and a substrate/product-free form with an additional channel to the solvent - in structure-based drug design.
The metabolic shift to a persistent state involves not only switching to alternative sources or pathways, but also significantly changing fluxes of metabolites through the pathways functioning under any growth condition86, 87. The TCA cycle is at the center of carbon metabolism, taking in the carbon from glycolysis or ß-oxidation of fatty acids and providing the cell with ATP and NADH as well as intermediates for other biosynthetic pathways, and is also linked to the electron transport chain. This makes the TCA cycle one of the major sensing points of the cell’s well-being, and an important part of the metabolic switching observed during infection. The detailed characterization of TCA cycle enzymes and the regulation involved in their functioning is a crucial part of information required for deciphering the biology of the altered metabolic stages of Mtb.
We have solved the structure of fumarase, the enzyme that catalyzes the trans-hydration of fumarate, converting it to malate, in its apo form (manuscript in preparation). The protein is part of a superclass of enzymes that are homotetrameric and catalyze ß-elimination reactions that produce fumarate as one of the products (Figure 9). The central core of the protein consists of a twenty helical bundle that spans over 60 Å with each subunit contributing five helices. Small N- and C-terminal subdomains sit at either end of the bundle producing a dumbbell-shaped appearance in both the individual subunits and the overall tetramer. Four active sites exist in the molecule and are formed by atoms from three different subunits. Each is perched at the end of the helical bundle between the interface of the N- and C-terminal subdomains. Based on sequence and structural alignment with fumarase C from E. coli (PDB code 1FUO), all known catalytic residues are conserved including the catalytic base residue His187. One of three loops that surround the active site is disordered. In homologous structures this loop becomes ordered upon substrate binding. Recently, an allosteric site was identified in the E. coli FumC structure that sits approximately 12 Å away from the active site (A-site) at the surface of the protein. Termed the B-site, it is considered to be an extension of the active site and helps transfer substrate and product between the active site and the bulk solvent88. Part of the B-site is formed by a rare π-helix that creates a highly ordered hydrogen bond network from the bound substrate/product to an active-site water. While the π-helix is conserved in the Mtb fumarase structure along with the residues leading to the buried A-site, a short loop at the N-terminal part of the B-site is not present in the structure. Mutational analysis of two basic residues from this region in the E. coli FumC, R126A and H128N, shows the sequence change fundamentally alters the rate of the reaction by decreasing the product off rate ~160-fold. The human homolog (PDB code 3E04) also has this loop resembling the E. coli enzyme, but has a lysine residue in place of the arginine. This observation may imply a different regulation mechanism for Mtb fumarase. Future kinetic studies will hopefully shed light on the function of the B-site of Mtb fumarase and aid in the design of the selective inhibitors for the protein.
Figure 9.
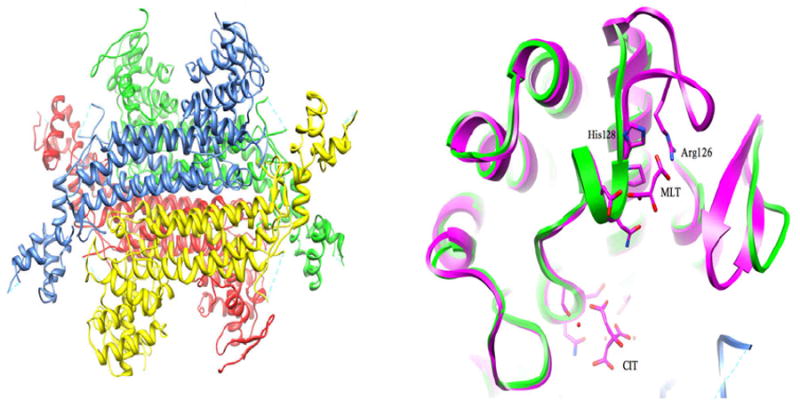
(A) Homotetramer of Mtb fumarase colored by subunit. (B) depicts the missing loop in the B-site. Mtb FumC is shown in green and E. coli FumC bound to malate (PDB code 1FUO) is in purple.
There is a one more vital part of the metabolic scheme for Mtb to sustain growth on fatty-acid derived carbon during persistence – gluconeogenesis. It has been shown that Mtb mutants lacking phosphoenolpyruvate carboxykinase (PEPCK), the enzyme catalyzing the first step of gluconeogenesis, were able to grow on dextrose as a carbon source in culture, but failed to survive in mouse lungs, and PEPCK depletion during the chronic phase of infection resulted in mycobacterial clearance89. We have determined the crystal structure of PEPCK (Rv0211), the enzyme which converts the TCA intermediate oxaloacetate into phosphoenolpyruvate in the first committed and pace-setting step of gluconeogenesis (manuscript in preparation). The overall structure of the Mtb enzyme in complex with guanidine diphosphate is similar to that of human PEPCK, however the active site cavity is larger in the Mtb enzyme than in the human enzyme. We are currently conducting a detailed examination of the differences between the Mtb and human enzymes, as well as co-crystallizing product- and substrate-bound PEPCK. This will provide an opportunity for rational drug design strategies to develop selective inhibitors of the Mtb enzyme. The structure of the enzyme active site is shown in Figure 10.
Figure 10.
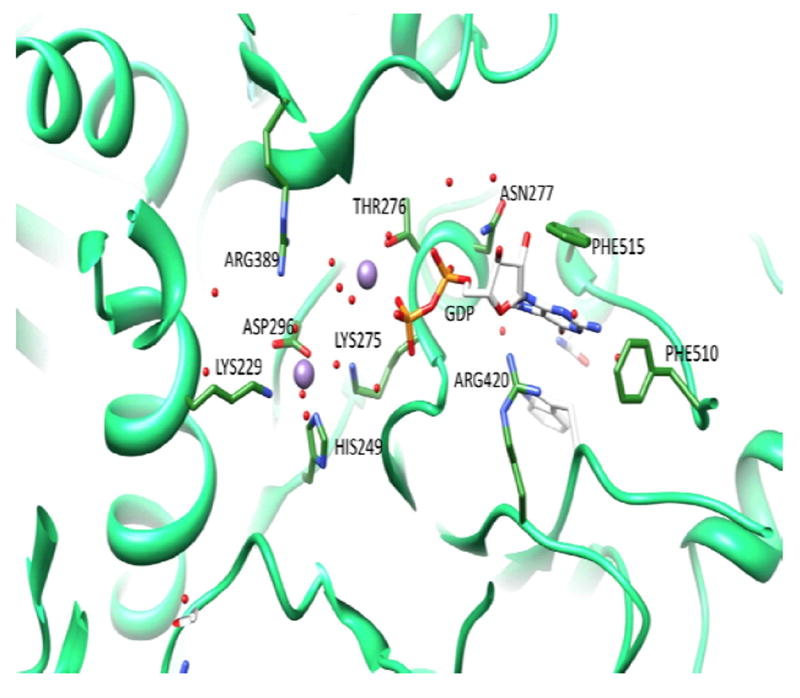
Structure of the active site of Mtb PEPCK in complex with GDP and Mn2+. GDP and active site residues interacting with Mn2+ and the cofactor are shown in stick, the two Mn2+ cations are displayed as purple spheres.
Structural and Biochemical Characterization of MhuD (Rv3592) and DsbF (Rv1677) (Nicholas Chim, Robert P. Morse and Celia W. Goulding)
Iron availability
Iron availability is essential in supporting microbial viability and growth; in Mtb, siderophores (small, high-affinity iron-chelating molecules) are secreted to sequester Fe3+ from human transferrin and lactoferrin90-92. Alternatively, pathogenic bacteria have also evolved mechanisms in which host-heme acquisition can overcome iron requirements. Staphylococcus aureus93, Serratia marcescans94, 95, Shigella dysenteriae96-98, and Neisseria spp99-101 possess distinct heme-uptake systems that are able to extract heme from host hemoglobin, transport heme into the cytosol, and eventually metabolize heme to liberate iron. For Mtb, no homologous heme-uptake system has yet been discovered although the recent identification and characterization of a heme degrader, MhuD (Rv3592) suggests the presence of one102.
MhuD shares 46% and 43% amino acid sequence identity with heme-degraders, S. aureus IsdG and IsdI, respectively103. Consequently, similar to IsdG and IsdI, biochemical characterization reveals that MhuD is able to bind and degrade heme in the presence of suitable electron donors (i.e., ascorbate or cytochrome P450 reductase) when the stoichiometry is a 1:1 protein-to-heme ratio (MhuD-heme). A conserved Asn-Trp-His triad of residues has been shown to be essential for activity whereby single alanine mutants of each residue are able to bind but not degrade heme104. Our preliminary results with MhuD Asn7Ala and His75Ala mutants concur with these observations (unpublished data). Significantly, isothermal calorimetry experiments demonstrated that MhuD, unlike IsdG and probably IsdI, is capable of binding two heme molecules per monomer (MhuD-diheme) with equal binding affinity, rendering MhuD inactive.
The 1.75 Å crystal structure of MhuD-diheme (PDB code 3HX9) shows structural homology to heme-IsdG N7A (rmsd, 2.2 Å) and CoPPIX-IsdI (rmsd, 2.1 Å), where the overall topology consists of a homodimer with a central eight anti-parallel β-barrel flanked by two α-helices on each end 104, 105, Figure 11A. The major difference occurs within the heme binding pocket: notably, each MhuD subunit contains two slightly distorted, planar stacked heme molecules which are rotated approximately 80° relatively to each other around an axis orthogonal to their planes, providing insight to an open, inactive conformation, whereas IsdG and IsdI have only one highly distorted heme molecule per monomer, suggestive of a closed, active state. At the crystallographic interface of MhuD, two subunits form a protein-protein interface and the solvent-exposed hemes from each subunit stack; as a result, this configuration may protect the dihemes from degradation while the heme in the Isd proteins are still accessible to electrons from an unknown in vivo source. Furthermore, size exclusion chromatography experiments reveal that the elution of MhuD-heme is consistent with a dimeric species whereas elution of MhuD-diheme infers various higher oligomeric states (unpublished data), providing evidence that the crystallographic protein-protein interface may occur in solution.
Figure 11.
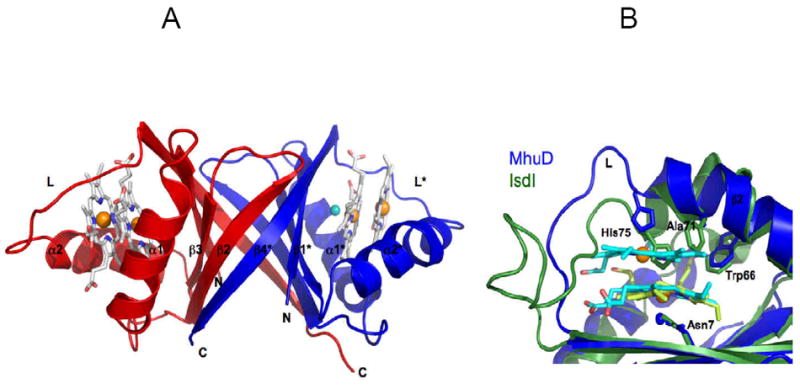
Cartoon representation of MhuD-diheme (PDB code 3HXP). (A) Each subunit in the dimer (chain A, blue; chain B, red) binds to two hemes, represented as sticks models with carbon (white), nitrogen (blue), and oxygen (red), as well as Fe (orange) and Cl- (cyan) spheres. (B) Heme binding sites of MhuD and Isd proteins. Superposition shows an extended α-helix 2 in MhuD (blue) which accommodates 2 hemes (cyan) while it is kinked by 45° in IsdI (green). The loop region, L, in MhuD makes extensive contacts to both hemes while that of IsdI is mostly extruded into solution. The side chains of the conserved Asn-His-Trp triad of residues, as well as Ala71 in MhuD and Phe72 in IsdI, are represented as stick models.
Despite the difference in stoichiometry, the Asn-Trp-His triad of residues coordinates heme in a similar fashion. For MhuD, the solvent-protected heme iron is coordinated by a chloride ion, which is, in turn, stabilized by Asn7 while the solvent-exposed heme iron is axially liganded by His75 (Figure 11B). Trp66 interacts with both heme molecules via their vinyl and methyl moieties. The corresponding asparagine and histidine interactions are also observed in IsdI (i.e., Asn6 and His76) with a single cobalt protoporphyrinIX molecule; in the IsdG Asn7Ala mutant, the heme iron is pentacoordinated, the fifth ligand provided by His77. The conserved tryptophans, among other residues, for both Isd proteins are implicated in porphyrin ring distortion. Additionally, the tryptophans in the Isd proteins are involved in edge-to-face aromatic interactions with phenylalanines (i.e., Trp67-Phe73 and Trp66-Phe72 from IsdG and IsdI, respectively), kinking α2. Interestingly, the phenylalanine is replaced by an alanine in MhuD, giving rise to an extended α2 that is proposed to be partly responsible for accommodating the second solvent-exposed heme. However, an MhuD Ala71Phe mutant was still able to bind heme in a 2:1 heme-to-protein stoichiometry (unpublished data), suggesting that loop residues subsequent to α2, designated L, making extensive interactions and stabilizing both heme molecules may further contribute to binding a second heme molecule. A more recent report of the first active heme degrader structure, heme-IsdI, shows no significant differences from inactive CoPPIX-IsdI except for the presence of a dioxygen species coordinating the conserved Asn6 instead of a chloride ion106. Taken together, these structures provide insights to 1) heme entry and product exit which involves the flexibility of α2 and L demonstrated in the open, inactive (MhuD-diheme) conformation and 2) heme catalysis by way of porphyrin ring distortion, which has been suggested to play a role in adopting a favorable conformation for nucleophilic attack105, in the closed, active (IsdG/I-metalloporphyrin) conformation.
Distinct from heme oxygenases, where heme is metabolized to biliverdin, free iron, and carbon monoxide107, IsdG and IsdI yield novel chromophore products (i.e., 15-oxo-β-bilirubin and 5-oxo-δ-bilirubin) consistent with cleavage of heme at the β- or δ-meso carbons106. The observation of mixed regioselective products reveals that heme can be loaded onto the heme degrader in two different orientations. Moreover, MhuD’s ability to bind two hemes per monomer could provide a venue for heme storage as part of a regulatory mechanism in response to iron/heme availability in Mtb. One possible scenario could be that under iron-abundant conditions, a major fraction of MhuD remains inactive as a diheme complex due to apo-MhuD’s ability to bind both heme molecules with comparable affinities. If iron supplies become limiting, MhuD is upregulated to form the MhuD-heme complex that has the ability to degrade intracellular heme to maintain pathogen viability. As heme degradation by IsdG has been shown to be essential in bacterial pathogenesis108, 109, the diheme feature of MhuD can ultimately be exploited as a potential anti-TB drug target.
Mtb disulfide oxidoreductase system: Mtb DsbF (Rv1677) and Mtb DsbE (Rv2878c)
Of the approximately 4,000 genes within the Mtb genome, it has been proposed that roughly 4% of translated proteins are secreted, of which more than 60% are predicted to contain at least one disulfide bond110. Just as E. coli has well-characterized disulfide bond forming (Dsb) proteins residing in the periplasm to ensure proper oxidation of disulfide bonds in secreted proteins111, a similar disulfide oxidoreductase system must exist in Mtb. To-date, only three Dsb proteins have been identified in Mtb: Mtb DsbE (Rv2878c), its homolog Mtb DsbF (Rv1677), and its potential redox protein partner, Mtb DsbD (Rv2874). Mtb DsbE and Mtb DsbF are proposed to be secreted to the recently characterized mycobacterial periplasm112, 113 while Mtb DsbD is a predicted inner membrane protein with eight transmembrane helices and two soluble domains on the N and C termini 110.
Mtb DsbE and Mtb DsbF share 55% sequence identity and recent X-ray structures reveal that Mtb DsbE (PDB code 1LU4)110, and Mtb DsbF (PDB code 3IOS)114, are superimposable with a root-mean-square deviation of 0.89 Å (Figure 12). Similar to other Dsb proteins, Mtb DsbE and Mtb DsbF have thioredoxin folds, consisting of a central β-sheet (β4, β3, β6, and β7) sandwiched between 3 α-helices (α3, α4, and α5). Both Mtb protein structures, however, have additional secondary structural elements (β5, α5, and α6), including a smaller N-terminal domain made up of two 310-helices (α1 and α2) capped by a β-hairpin (β1 and β2). Despite very similar overall topologies, two distinct features distinguish Mtb DsbE from Mtb DsbF: 1) The active site motif, CxxC, of Mtb DsbE (i.e., Cys36 and Cys39) is in its reduced state whereas the corresponding active site cysteines in Mtb DsbF (i.e., Cys81 and Cys84) exist in both oxidized and reduced conformations. This difference is, in part, attributed to an amino acid pair conserved in most Dsb proteins: a strong hydrogen bond between Trp30 and Glu42 (2.8 Å) in Mtb DsbE stabilizes its reduced state whereas the corresponding interaction (Trp75-Glu87) in Mtb DsbF is disrupted where, instead, a weak hydrogen bond network is coordinated by the amino acid pair with Tyr142 and a water molecule. 2) In Mtb DsbE, the loop between β7 and α6 is extended by three additional amino acid residues, which leads to a partial occlusion of the active site, and hence, limits solvent accessibility compared to that of Mtb DsbF active site. Interestingly, in the crystal packing, this extended loop in the Mtb DsbE structure forms extensive contacts and may play a role in forming a potential homodimeric interface. On the contrary, the crystal packing within the Mtb DsbF structure reveals no potential dimeric interface and hence the active site is solvent exposed in the crystal lattice.
Figure 12.
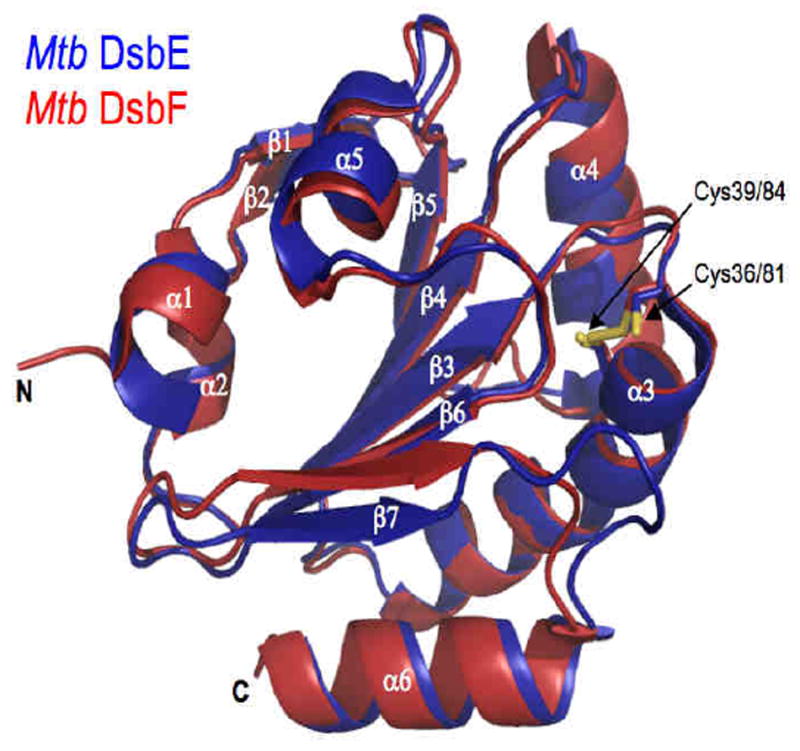
Cartoon representations of superimposed Mtb DsbE (blue, PDB code 1LU4) and Mtb DsbF (red, PDB code 3HX9) with active site cysteines (yellow) shown as stick models. In Mtb DsbE, Cys36 and Cys39 are reduced whereas Cys81 and Cy84 in Mtb DsbF are modeled in both reduced and oxidized states.
Biochemical characterization demonstrates that the reduced states of Mtb DsbE and Mtb DsbF are significantly more stable than the oxidized states (i.e., 12.4 kJ/mol and 24 kJ/mol more stable for Mtb DsbE and Mtb DsbF, respectively). Additionally, the redox potentials [relative to glutathione] of Mtb DsbE (-128 mV) and Mtb DsbF (-89 mV) are consistent with known oxidase, E. coli DsbA (between -89 and -119 mV)115, 116 and in contrast to known reductase, E. coli thioredoxin (-269 mV) 117. Most notably, Mtb DsbE and Mtb DsbF have been shown to be capable of oxidatively folding reduced, unfolded hirudin; coupled with their extracellular/cell-wall localization, this strongly suggests their role in aiding disulfide bond formation in other secreted Mtb proteins.
Although Mtb DsbE and Mtb DsbF function analogously, gene expression data from four microarray data sets6-8, 118 indicate that their gene expressions are negatively correlated, suggesting that both proteins are involved in distinct Dsb pathways. Further gene expression analyses reveal that Mtb DsbD positively correlates with Mtb DsbE. Interestingly, Mtb DsbE expression is also correlated with SigL (Rv0735), a sigma factor that has shown to be required for Mtb pathogenesis and the upregulation of secreted/membrane proteins, including Mtb DsbE119, thereby implicating Mtb DsbE’s potential role in virulence. Mtb DsbF, on the other hand, has been shown to transiently interact with a predicted peroxiredoxin protein, Rv1676, whose gene is in the same operon as Rv1677. Mtb DsbF and Rv1676 have correlated expression with the antioxidant Ahp system and, thus, could play a role in oxidative stress. Additionally, the gene expression patterns of the Mtb DsbF system co-expresses with the cytochrome c oxidase and cytochrome c reductase systems whereas it has anticorrelated expression patterns with the cytochrome bd1 oxidase system120-122. Based on this observation and that Mtb DsbF is a homolog of E. coli DsbE, we propose that Mtb DsbF may be involved in cytochrome c maturation. A cluster of proteins, which includes ResA, CcdA, and CcsA, has been suggested to be involved in cytochrome c maturation; these genes are also found in the Mtb genome123, 124. Consequently, we observed the expression patterns of the Mtb DsbF system, a selection of cytochrome P450 proteins125, and ResA, CcdA, and CcsA. Their expression profiles indicate that ResA, CcdA and CcsA appear to have positive expression with cytochrome P450 proteins and exhibit anticorrelated expression with respect to Mtb DsbF and cytochrome c systems. Hence, we hypothesize that Mtb DsbF and its protein partner, Rv1676, may participate in cytochrome c maturation, whereas the cluster of proteins, ResA, CcsA, and CcdA, may participate in the maturation pathway of cytochrome P450 proteins in Mtb.
Some of the Dsb proteins from E. coli, despite similar biochemical and structural characteristics, function in different pathways, as our results suggest for Mtb DsbE and Mtb DsbF. The differing oxidation state of the CxxC motif within the crystal structures of Mtb DsbE and Mtb DsbF are intriguing, and highlights the potential different functional roles they may play in the physiology of Mtb. The Dsb proteins in E. coli have been extensively studied, and though E. coli DsbC and E. coli DsbG are disulfide bond isomerases, and E. coli DsbE is a reductase, all three of these periplasmic proteins are maintained in their active reduced form by the same transmembrane protein E. coli DsbD126. One of the main differences between these proteins is that E. coli DsbC and E. coli DsbG are both homodimers127, whereas E. coli DsbE is monomeric and tethered to the cytoplasmic membrane128. The fact that Mtb DsbE has a potential homodimeric interface and Mtb DsbF is monomeric may contribute to their proposed functional differences in periplasm of Mtb. Furthermore, Mtb DsbE is predicted to have a signal peptide only 33 amino acid residues long and has been observed in Mtb culture filtrate129. On the contrary, the predicted N-terminal signal peptide region of Mtb DsbF is 52 amino acid residues long and this protein has not been observed in culture filtrate to-date, which suggests that Mtb DsbF may be tethered to the membrane as observed for E. coli DsbE130. Hence, the potential tethering to the membrane and the monomeric nature of Mtb DsbF may account, in part, for the difference in function under different cellular conditions in comparison to Mtb DsbE. This highlights the increasing need for understanding components of biological systems in terms of their context as well as simple homology relationships. Additionally, Mtb DsbE and Mtb DsbF are attractive therapeutic targets as inhibiting them may disrupt the proper oxidation of Mtb secreted proteins, which are often virulent factors.
Toxin-Antitoxin Complexes from Mtb (Linda Miallau and David Eisenberg)
Toxin-antitoxin (TA) gene pairs were initially discovered on plasmids and were found to be essential for the maintenance of foreign genetic elements in host cells through the process termed “plasmid addiction”131. Bacteria acquire plasmids that encode both toxin and antitoxin genes in an operon. Under normal physiological conditions both proteins are expressed and form a tight complex incapable of inhibiting bacterial growth. However, when the plasmid encoding those systems is lost, for instance during cell division, the unstable antitoxin is rapidly degraded by proteases and the free toxin kills the cured bacterial host cell132. Hence the cell is “addicted” to the plasmid.
Subsequently, TA gene pairs were discovered on chromosomes of diverse but mostly slow growing prokaryotes, which suggests functions other than plasmid addiction133. The most probable function supported by experimental evidence is the cessation of growth under conditions of stress134. Thus, under favorable growth conditions, toxin and antitoxin assemble in a stable and inoffensive complex. However, when the bacteria are stressed, for instance by increased temperature, oxidative shock, or the presence of antibiotics, proteases are overexpressed and degrade the labile antitoxin. The liberated toxin then exerts a bacteriostatic effect primarily through mRNA degradation although there are other mechanisms of toxin-mediated cessation of bacterial growth. Depending on the particular TA pair, the mode of action can vary: E. coli MazF cleaves free cytosolic mRNA at ACA sequences135 while RelE toxins are ribosome-dependent RNases which bind to the ribosomal A-site and degrade incoming mRNA and tmRNA136, 137.
TA systems are unusually abundant in the major human pathogen Mtb, which encodes approximately 90 TA pairs (unpublished data available at: http://www.ncl.ac.uk/camb/staff/profile/kenn.gerdes) including one member from the HigBA system, two ParDE138 systems, three RelBE139 systems, nine MazEF140 systems and 45 VapBC141, 142 systems. Since the activation of some of the toxins results in reduced cell growth and eventually dormancy, it is possible that TA pairs may play an important role in the persistence of Mtb and in its capacity to survive in a dormant state in the phagosome after infection of macrophages132, 143.
Despite the abundance of TA pairs in the Mtb genome there is little information on their molecular structures and functions. We have employed X-ray crystallography and other biophysical and biochemical techniques to characterize Mtb TA pairs to understand how the toxins exert their bacteriostatic effects and the mechanisms by which antitoxins neutralize their cognate toxins. We have determined the structures of two VapBC systems (VapBC-3 and VapBC-5) and two RelBE systems (RelFG and RelJK) and our results suggest that the toxins are ribonucleases.
VapBC TA Systems
We determined the first structure of a VapBC complex (encoded by Rv0626-Rv0627; PDB code 3DBO), Mtb VapBC-5, to 1.9 Å resolution, using single wavelength anomalous dispersion (SAD)144. The VapC-5 toxin structure and the C-terminal part of its cognate antitoxin, VapB-5, are well ordered in our structure while the N-terminal DNA-binding domain of VapB-5 is completely disordered (Figure 13A, Left panel). It has been shown that VapC toxins have a deleterious effect on cell growth and that VapC toxicity is inhibited by the specific binding of cognate VapB antitoxins143.
Figure 13.
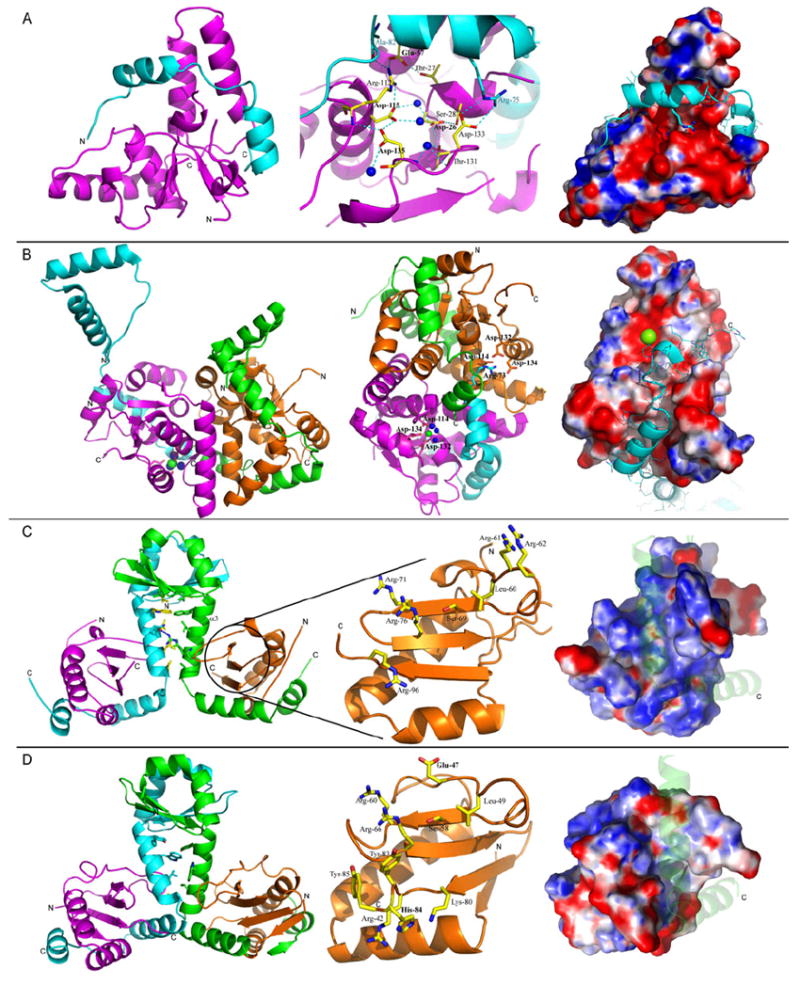
Structure of Toxin-Antitoxin (TA) complexes from Mtb solved by the UCLA group. In all figures, Mtb toxin proteins are shown in magenta and orange and antitoxin proteins are in cyan and green. Water molecules are shown as blue spheres and the magnesium ion is shown as a green sphere. Nitrogen and oxygen atoms belonging to amino acid sidechains are shown in blue and red, respectively. N- and C-termini are indicated by N or C, respectively. Surface potential is colored blue (basic) and red (acidic). (A) The structure of VapBC-5 (Rv0626-Rv0627). (Left) Ribbon diagram of Mtb VapBC-5 heterodimer. (Middle) Putative active site of Mtb VapC-5. Residues involved in the active site formation are shown as yellow sticks. (Right) Surface potential representation of the VapC-5 toxin in the same orientation as in the Left panel, in complex with VapB-5 antitoxin (shown in ribbon representation). (B) The structure of VapBC-3 (Rv0300-Rv0301). (Left) Ribbon diagram of Mtb VapBC-3 heterotetramer. (Middle) Ribbon diagram of the heterotetramer showing the magnesium ion bound to the toxin (green). The arginine residue from the neighboring antitoxin (cyan) that prevents the binding of a magnesium ion in the second toxin (orange) is shown in cyan. (Right) Surface potential representation of the VapC-3 toxin in complex with VapB-3 antitoxin (shown as a cyan ribbon). (C) The structure of RelFG (Rv2865-Rv2866). (Left) Ribbon diagram of Mtb RelFG heterotetramer. Hydrophobic amino acid side chains from RelF are shown as yellow and green sticks. The single hydrogen bond formed between RelF of one heterodimer and RelG of the symmetry related molecule is represented as a blue dotted line. (Middle) Ribbon diagram of RelG and stick representation of residues that could participate in the binding and cleavage of RNA. (Right) Surface potential representation of the toxin RelG in complex with RelF (shown as a transparent ribbon). (D) The structure of RelJK (Rv3357-Rv3358). (Left) Ribbon diagram of Mtb RelJK heterotetramer. Hydrophobic amino acid side chains from RelJ are shown as green and cyan sticks. (Middle) Ribbon diagram of RelK and stick representation of residues that could participate in the binding and cleavage of RNA. (Right) Surface potential representation of the toxin RelK in complex with RelJ (shown as a transparent ribbon).
The crystal structure of the Mtb VapBC-5 toxin-antitoxin complex suggests a possible mechanism for VapB-5 antitoxin inhibition of the toxic activity of VapC-5, involving distortion of the catalytic site of VapC-5. This mechanism is suggested by the structural homology between the active sites of VapC and the FEN-1 endo- and exonuclease, in which residues in a conserved acidic pocket bind Mg2+ ions that are necessary for catalysis145. Upon VapB-5 binding by VapC-5, the main chain carbonyl oxygen of VapB-5 Ala82 reorients the side chain of VapC-5 Arg112, which locks VapC-5 Glu57 in an unfavorable conformation that prevents it from binding the Mg2+ ion. Moreover, VapB-5 Arg75 can obstruct the side chain of VapC-5 Asp133, pushing it out of the active site/catalytic cavity and thus preventing the binding of the second Mg2+ ion required for catalysis (Figure 13A, Middle panel). The inhibition of nuclease activity by the antitoxin would thus be due to the direct or indirect seclusion of the active site residues in catalytically unfavorable conformations. VapC-5 would then become active upon the release of VapB-5, which is triggered by a still unknown signal. The RNA substrate could then be bound in a groove formed between the core and clip domains of VapC-5 (Figure 13A, Right).
The main result regarding the enzymatic activity of VapBC-5 is that this complex clearly shows low nuclease activity on dsRNA substrates in vitro as well as magnesium dependence consistent with a two-metal-ion catalytic mechanism. Based on these results, we propose that Mtb VapC-5 is most likely both an endo- and exoribonuclease that acts on free RNA in a manner similar to that of the FEN-1 endo- and exonuclease.
The second structure of an Mtb VapBC complex (VapBC-3 encoded by Rv0300-Rv0301; PDB code 3H87; to be published) was solved to a resolution of 1.49 Å by SAD (Figure 13B, Left and Middle). The asymmetric unit contains a 2:2 heterotetramer although a SAXS experiment has shown that it is likely to form an octamer in solution. The fold of the toxin is consistent with ribonuclease activity although structurally different from VapC-5. The N-terminal part of the antitoxin forms a Ribbon-Helix-Helix DNA binding motif through dimerization of antitoxins from adjacent asymmetric units. Thus, it is possible that the physiological assembly is a tetramer different from the one seen in the asymmetric unit but similar to those previously observed for the RelBE-like systems with the N-terminal domain of the antitoxin involved in dimerization of the heterodimers and the C-terminal domain involved in the neutralization of the toxin.
In the VapBC-3 heterotetramer, the two toxin-antitoxin dimers are asymmetric. In one dimer, the antitoxin is tightly wrapped around the toxin and its C-terminal toxin neutralization domain is disordered which allows the toxin from the neighboring dimer to bind a magnesium ion (Figure 13B, Left and Middle panels). The magnesium ion is coordinated by residues Asp114, Asp132 and Asp134 as well as three water molecules (Figure 13B, Middle panel). In the other dimer the C-terminus of the antitoxin is well ordered and reorients an arginine residue (Arg73) in the magnesium binding site of the neighboring toxin preventing magnesium binding (Figure 13B, Middle). In both dimers, the C-terminal domain of the antitoxin binds to a deep groove in the toxin. Thus, as in VapBC-5, the antitoxin may inhibit the toxin through two modes: by occupying the substrate binding site of the toxin; and possibly by sequestering the catalytic residues involved in magnesium binding. Although our structure shows a bound magnesium ion, no catalytic mechanism can be determined from the structure alone and further investigation is necessary.
The structures of VapC-5 and VapC-3 reveal that there is considerable diversity in the structure of Mtb VapC toxins. However, we believe that both function as RNases, which is supported by the structural homology of the active site(s) to the FEN-1 nuclease, the bound magnesium in VapC-3, and the magnesium dependence of the VapC-5 nuclease activity. As the DNA-binding domain of the VapB-5 antitoxin is disordered in our structure, it is difficult to compare the overall fold of the VapB-3 and VapB-5 antitoxins although they adopt a similar strategy for toxin neutralization.
RelBE TA Systems
RelBE systems have been extensively studied in E. coli137, 139, 146, 147 but the role of RelBE systems in regulating Mtb physiology is poorly understood and appears to be very complex148. RelBE-like systems are of particular interest as RelE is expressed during infection of the macrophage by Mtb and may play a role in the phenomenon of persistence145. Moreover, overexpression of RelG increases Mtb antibiotic resistance while its deletion decreases the survival rate of the bacteria149. In order to understand the structural mechanism used by Mtb RelBE systems to target RNA, we have determined the crystal structures of RelFG and RelJK for which no structural information was previously available.
The structure of Mtb RelFG (PDB code 3G5O, to be published) was solved to 2.0 Å resolution by SeMet SAD. The structure reveals a heterotetramer composed of a dimer of an antitoxin:toxin heterodimer. The heterodimers form the tetramer around C2 symmetry primarily through the globular N-terminal domain of the antitoxin and an extended hydrophobic zipper that involves helix α-3 of the antitoxin. The structure of RelG is a three stranded β-sheet, that harbors residues potentially involved in the binding and cleavage of RNA, surrounded by two α-helices and a 310 helix (Figure 13C, Middle panel). Thirty percent of the total surface area of RelG is involved in the dimerization interface with RelF.
Based on structural comparison, the RelF antitoxin belongs to the YefM family whose cognate toxin is YoeB. The RelG toxin has the same fold as YoeB. However, functionally important YoeB residues that have previously been described150, 151 are not conserved in RelG. Recently, the structure of E. coli RelE bound to the 70S ribosome was solved and revealed that a C-terminal tyrosine residue is crucial for E. coli RelE activity152. As this residue is absent in Mtb RelG, it is likely that RelG uses a different strategy to cleave RNA.
Superposition of Mtb RelG with structural homologs, Pyrococcus horikoshii RelE and Methanococcus jannaschii RelE, demonstrates that only a few residues are conserved in all three homologs and interact with the antitoxins despite the antitoxins belonging to structurally distinct families. We postulate that in addition to the RelE residues previously identified by mutagenesis to be functionally important (RelG Leu-60, Ser-69 and Arg-76), Arg-71, Arg-92 and Lys-58 from RelG may also play a role in the catalytic mechanism of the RelG toxin (Figure 13C, Middle panel). This also suggests that the RelF antitoxin may inhibit RelG toxicity by direct interaction with key residues required for RNase activity. This hypothesis is supported by the surface potential calculation, which shows a single highly positively charged groove occupied by the antitoxin (Figure 13C, Right panel).
The structure of RelJK, which is still in refinement, has a similar secondary structure as RelFG although the tertiary structure and quaternary arrangement of RelJ and RelK are slightly different (Figure 13D, Left panel). The RelJ antitoxin has a flexible C-terminal domain due to a hinge motion of the flexible elongated helices, which bind to RelK in a similar manner as to RelFG. Superposition of structural homologs indicates that RelK may cleave RNA in the same manner as E. coli YoeB150, 151. Thus the catalytic mechanism of RelK could involve Glu-47 which acts as a general base and His84 that acts as the general acid and would require a large rearrangement of the C-terminal domain of RelK upon antitoxin release in order to bring Glu47 and His84 in favorable conformations for catalysis. Similarly to YoeB, Arg60 and/or Arg65 could be involved in binding the RNA reactive phosphate group151.
The differences in the putative catalytic residues between RelG and RelK suggested by the crystal structures are consistent with the evolutionary relationships that exist between Mtb RelE toxins and the E. coli YoeB and RelE toxins (Figure 14). The amino acid sequence of Mtb RelK is evolutionary closer to E. coli YoeB than it is to Mtb RelG. Similarly, Mtb RelG is closer to Mtb RelE than it is to E. coli RelE suggesting that Mtb RelG uses a different strategy than those used by Mtb RelK and E. coli RelE to cleave RNA.
Figure 14.

Horizontal cladogram representing the evolutionary distance of Mtb RelE, RelG and RelK and E. coli YoeB and RelE.
The structures of the four TA systems from Mtb determined by the UCLA group share common features. The toxins, in all four cases, are built around a β-sheet core that is surrounded by α-helices while the antitoxins are organized with a globular N-terminal DNA-binding domain and an elongated C-terminal tail that binds to and inhibits the toxin.
The crystal structures of Mtb toxin-antitoxin pairs also reveal the mechanism by which the antitoxins inhibit their toxins. In the case of the VapBC systems, it appears that residues from the antitoxin sequester residues from the toxin involved in binding of the crucial magnesium ions. In the RelJK TA pairs, the antitoxin constrains the toxin in an unfavorable conformation for catalysis. However, all TA pairs share a common feature: the toxic activities of the toxins are neutralized by the binding of the elongated antitoxins into a narrow and positively charged groove. This suggests that the antitoxins most likely compete with the binding of the single stranded RNA substrates of the toxins.
It is not surprising that structurally similar toxins that belong to the same family would use different mechanisms for RNA cleavage. Indeed, differences in the active site may reflect differences in substrate specificity. The different active sites suggest that different TA systems may be triggered by different regulatory controls and therefore by different stresses. Thus, the variety of TA pairs would allow the bacteria to adapt to a wide variety of hostile environments, which would be especially important within the phagolysosome after phagocytosis by macrophages. Thus the proliferation and differentiation of TA systems may be a key component of the ability of Mtb to respond and adapt to the stresses it encounters throughout its life cycle.
Conclusions
In the last decade, the TBSGC project has yielded useful bioinformatics resources as well as structural information. The richness of information available in molecular structures also provides the starting point for many new research avenues and future drug design projects unanticipated in the present day. For example, it is possible that a future finding will implicate one of the biosynthetic enzymes highlighted in this review as pivotal during Mtb pathogenesis, prompting a large-scale drug development project. We hope that this review succeeds in its intention of providing the progress of the TBSGC and we look forward to working with other members of the Mtb community.
Acknowledgments
The TBSGC is supported by NIH/NIAID grant number PO1 AIO68135 (J.C.S.) The TAMU research has been supported by Robert A. Welch Foundation grant A-0015 (J.C.S) and the UCI research has been supported by NIH. The UCLA authors wish to acknowledge Duilio Cascio, Dan Anderson, Andrew Min, Mark Arbing and the PETC laboratory. The UCLA work has been supported by NIH.
Abbreviations
- Mtb
Mycobacterium tuberculosis
- TB
Tuberculosis
- TBSGC
TB Structural Genomics Consortium
- Rv number
Sanger center notation for each gene in Mtb
- PDB
Protein Data Bank
- SAXS
Small Angle X-ray Scattering
- PPI
Protein-Protein Interaction
- ORF
Open Reading Frame
- NMR
Nuclear Magnetic Resonance
- ADC
4-amino-4-deoxychorismate
- PABA
para-aminobenzoic acid
- OAT
Ornithine Acetyltransferase
- OTC
Ornithine Carbamoyltransferase
- ORN
L-Ornithine
- NVA
L-norvaline
- CP
Carbamoyl Phosphate
- TCA
Tricarboxylic Acid
- ICL
Isocitrate Lyase
- CoA
Coenzyme A
- MSA
Malate Synthase form A
- MSG
Malate Synthase form G
- ATP
Adenosine-5’-triphosphate
- NAD
Nicotinamide adenine dinucleotide
- PEPCK
Phosphoenolpyruvate Carboxykinase
- MhuD
Mycobacterial Heme Utilization, Degrader
- Isd
Iron Surface Determinant
- Dsb
Disulfide bond-forming
- TA
Toxin-Antitoxin
- SAD
Single-wavelength Anomalous Dispersion
- SeMet
Selenomethionine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chim N, McMath LM, Beeby M, Goulding CW. Advances in Mycobacterium tuberculosis structural genomics: investigating potential chinks in the armor of a deadly pathogen. Infect Disord Drug Targets. 2009;9:475–92. doi: 10.2174/187152609789105731. [DOI] [PubMed] [Google Scholar]
- 2.Murillo AC, Li HY, Alber T, Baker EN, Berger JM, Cherney LT, Cherney MM, Cho YS, Eisenberg D, Garen CR, Goulding CW, Hung LW, Ioerger TR, Jacobs WR, James MN, Kim C, Krieger I, Lott JS, Sankaranarayanan R, Segelke BW, Terwilliger TC, Wang F, Wang S, Sacchettini JC. High throughput crystallography of TB drug targets. Infect Disord Drug Targets. 2007;7:127–39. doi: 10.2174/187152607781001853. [DOI] [PubMed] [Google Scholar]
- 3.Terwilliger TC, Park MS, Waldo GS, Berendzen J, Hung LW, Kim CY, Smith CV, Sacchettini JC, Bellinzoni M, Bossi R, De Rossi E, Mattevi A, Milano A, Riccardi G, Rizzi M, Roberts MM, Coker AR, Fossati G, Mascagni P, Coates AR, Wood SP, Goulding CW, Apostol MI, Anderson DH, Gill HS, Eisenberg DS, Taneja B, Mande S, Pohl E, Lamzin V, Tucker P, Wilmanns M, Colovos C, Meyer-Klaucke W, Munro AW, McLean KJ, Marshall KR, Leys D, Yang JK, Yoon HJ, Lee BI, Lee MG, Kwak JE, Han BW, Lee JY, Baek SH, Suh SW, Komen MM, Arcus VL, Baker EN, Lott JS, Jacobs W, Jr, Alber T, Rupp B. The TB structural genomics consortium: a resource for Mycobacterium tuberculosis biology. Tuberculosis (Edinb) 2003;83:223–49. doi: 10.1016/s1472-9792(03)00051-9. [DOI] [PubMed] [Google Scholar]
- 4.Goulding CW, Apostol M, Anderson DH, Gill HS, Smith CV, Kuo MR, Yang JK, Waldo GS, Suh SW, Chauhan R, Kale A, Bachhawat N, Mande SC, Johnston JM, Lott JS, Baker EN, Arcus VL, Leys D, McLean KJ, Munro AW, Berendzen J, Sharma V, Park MS, Eisenberg D, Sacchettini J, Alber T, Rupp B, Jacobs W, Jr, Terwilliger TC. The TB structural genomics consortium: providing a structural foundation for drug discovery. Curr Drug Targets Infect Disord. 2002;2:121–41. doi: 10.2174/1568005023342551. [DOI] [PubMed] [Google Scholar]
- 5.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Edgar R. NCBI GEO: mining tens of millions of expression profiles--database and tools update. Nucleic Acids Res. 2007;35:D760–5. doi: 10.1093/nar/gkl887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boshoff HI, Reed MB, Barry CE, 3rd, Mizrahi V. DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell. 2003;113:183–93. doi: 10.1016/s0092-8674(03)00270-8. [DOI] [PubMed] [Google Scholar]
- 7.Gao Q, Kripke K, Arinc Z, Voskuil M, Small P. Comparative expression studies of a complex phenotype: cord formation in Mycobacterium tuberculosis. Tuberculosis (Edinb) 2004;84:188–96. doi: 10.1016/j.tube.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 8.Gao Q, Kripke KE, Saldanha AJ, Yan W, Holmes S, Small PM. Gene expression diversity among Mycobacterium tuberculosis clinical isolates. Microbiology. 2005;151:5–14. doi: 10.1099/mic.0.27539-0. [DOI] [PubMed] [Google Scholar]
- 9.Riley R, Pellegrini M, Eisenberg D. Identifying cognate binding pairs among a large set of paralogs: the case of PE/PPE proteins of Mycobacterium tuberculosis. PLoS Comput Biol. 2008;4:e1000174. doi: 10.1371/journal.pcbi.1000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowers PM, Pellegrini M, Thompson MJ, Fierro J, Yeates TO, Eisenberg D. Prolinks: a database of protein functional linkages derived from coevolution. Genome Biol. 2004;5:R35. doi: 10.1186/gb-2004-5-5-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huynen MA, Bork P. Measuring genome evolution. Proc Natl Acad Sci U S A. 1998;95:5849–56. doi: 10.1073/pnas.95.11.5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci U S A. 1999;96:4285–8. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA. Protein interaction maps for complete genomes based on gene fusion events. Nature. 1999;402:86–90. doi: 10.1038/47056. [DOI] [PubMed] [Google Scholar]
- 14.Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein-protein interactions from genome sequences. Science. 1999;285:751–3. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 15.Dandekar T, Snel B, Huynen M, Bork P. Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem Sci. 1998;23:324–8. doi: 10.1016/s0968-0004(98)01274-2. [DOI] [PubMed] [Google Scholar]
- 16.Overbeek R, Fonstein M, D’Souza M, Pusch GD, Maltsev N. The use of gene clusters to infer functional coupling. Proc Natl Acad Sci U S A. 1999;96:2896–901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Overbeek R, Fonstein M, D’Souza M, Pusch GD, Maltsev N. Use of contiguity on the chromosome to predict functional coupling. Silico Biol. 1999;1:93–108. [PubMed] [Google Scholar]
- 18.Ermolaeva MD, White O, Salzberg SL. Prediction of operons in microbial genomes. Nucleic Acids Res. 2001;29:1216–21. doi: 10.1093/nar/29.5.1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreno-Hagelsieb G, Collado-Vides J. A powerful non-homology method for the prediction of operons in prokaryotes. Bioinformatics. 2002;18(Suppl 1):S329–36. doi: 10.1093/bioinformatics/18.suppl_1.s329. [DOI] [PubMed] [Google Scholar]
- 20.Strong M, Mallick P, Pellegrini M, Thompson MJ, Eisenberg D. Inference of protein function and protein linkages in Mycobacterium tuberculosis based on prokaryotic genome organization: a combined computational approach. Genome Biol. 2003;4:R59. doi: 10.1186/gb-2003-4-9-r59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pal D, Eisenberg D. Inference of protein function from protein structure. Structure. 2005;13:121–30. doi: 10.1016/j.str.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 22.Hasan S, Daugelat S, Rao PS, Schreiber M. Prioritizing genomic drug targets in pathogens: application to Mycobacterium tuberculosis. PLoS Comput Biol. 2006;2:e61. doi: 10.1371/journal.pcbi.0020061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sassetti CM, Boyd DH, Rubin EJ. Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci U S A. 2001;98:12712–7. doi: 10.1073/pnas.231275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Derewenda ZS. Rational protein crystallization by mutational surface engineering. Structure. 2004;12:529–35. doi: 10.1016/j.str.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Kim CY, Webster C, Roberts JK, Moon JH, Alipio Lyon EZ, Kim H, Yu M, Hung LW, Terwilliger TC. Analysis of nucleoside-binding proteins by ligand-specific elution from dye resin: application to Mycobacterium tuberculosis aldehyde dehydrogenases. J Struct Funct Genomics. 2009;10:291–301. doi: 10.1007/s10969-009-9073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–12. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eaton KA, Brooks CL, Morgan DR, Krakowka S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect Immun. 1991;59:2470–5. doi: 10.1128/iai.59.7.2470-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones BD, Lockatell CV, Johnson DE, Warren JW, Mobley HL. Construction of a urease-negative mutant of Proteus mirabilis: analysis of virulence in a mouse model of ascending urinary tract infection. Infect Immun. 1990;58:1120–3. doi: 10.1128/iai.58.4.1120-1123.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones BD, Mobley HL. Proteus mirabilis urease: nucleotide sequence determination and comparison with jack bean urease. J Bacteriol. 1989;171:6414–22. doi: 10.1128/jb.171.12.6414-6422.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clemens DL, Lee BY, Horwitz MA. Purification, characterization, and genetic analysis of Mycobacterium tuberculosis urease, a potentially critical determinant of host-pathogen interaction. J Bacteriol. 1995;177:5644–52. doi: 10.1128/jb.177.19.5644-5652.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gordon AH, Hart PD, Young MR. Ammonia inhibits phagosome-lysosome fusion in macrophages. Nature. 1980;286:79–80. doi: 10.1038/286079a0. [DOI] [PubMed] [Google Scholar]
- 32.Cox GM, Mukherjee J, Cole GT, Casadevall A, Perfect JR. Urease as a virulence factor in experimental cryptococcosis. Infect Immun. 2000;68:443–8. doi: 10.1128/iai.68.2.443-448.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsuda M, Karita M, Morshed MG, Okita K, Nakazawa T. A urease-negative mutant of Helicobacter pylori constructed by allelic exchange mutagenesis lacks the ability to colonize the nude mouse stomach. Infect Immun. 1994;62:3586–9. doi: 10.1128/iai.62.8.3586-3589.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Habel JE, Bursey EH, Rho BS, Kim CY, Segelke BW, Rupp B, Park MS, Terwilliger TC, Hung LW. Structure of Rv1848 (UreA), the Mycobacterium tuberculosis urease gamma subunit. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:781–6. doi: 10.1107/S1744309110019536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joce C, Stahl JA, Shridhar M, Hutchinson MR, Watkins LR, Fedichev PO, Yin H. Application of a novel in silico high-throughput screen to identify selective inhibitors for protein-protein interactions. Bioorg Med Chem Lett. 2010 doi: 10.1016/j.bmcl.2010.07.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaneto H, Nakatani Y, Miyatsuka T, Kawamori D, Matsuoka TA, Matsuhisa M, Kajimoto Y, Ichijo H, Yamasaki Y, Hori M. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nat Med. 2004;10:1128–32. doi: 10.1038/nm1111. [DOI] [PubMed] [Google Scholar]
- 37.Reindl W, Yuan J, Kramer A, Strebhardt K, Berg T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol. 2008;15:459–66. doi: 10.1016/j.chembiol.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 38.Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006;6:321–30. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 39.Neckers L. Heat shock protein 90: the cancer chaperone. J Biosci. 2007;32:517–30. doi: 10.1007/s12038-007-0051-y. [DOI] [PubMed] [Google Scholar]
- 40.Yi F, Regan L. A novel class of small molecule inhibitors of Hsp90. ACS Chem Biol. 2008;3:645–54. doi: 10.1021/cb800162x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Billich A, Hammerschmid F, Peichl P, Wenger R, Zenke G, Quesniaux V, Rosenwirth B. Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus (HIV) type 1: interference with HIV protein-cyclophilin A interactions. J Virol. 1995;69:2451–61. doi: 10.1128/jvi.69.4.2451-2461.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fatkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, Saag MS, Goebel FD, Rockstroh JK, Dezube BJ, Jenkins TM, Medhurst C, Sullivan JF, Ridgway C, Abel S, James IT, Youle M, van der Ryst E. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med. 2005;11:1170–2. doi: 10.1038/nm1319. [DOI] [PubMed] [Google Scholar]
- 43.Marchio S, Alfano M, Primo L, Gramaglia D, Butini L, Gennero L, De Vivo E, Arap W, Giacca M, Pasqualini R, Bussolino F. Cell surface-associated Tat modulates HIV-1 infection and spreading through a specific interaction with gp120 viral envelope protein. Blood. 2005;105:2802–11. doi: 10.1182/blood-2004-06-2212. [DOI] [PubMed] [Google Scholar]
- 44.Roehrl MH, Kang S, Aramburu J, Wagner G, Rao A, Hogan PG. Selective inhibition of calcineurin-NFAT signaling by blocking protein-protein interaction with small organic molecules. Proc Natl Acad Sci U S A. 2004;101:7554–9. doi: 10.1073/pnas.0401835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roehrl MH, Wang JY, Wagner G. Discovery of small-molecule inhibitors of the NFAT--calcineurin interaction by competitive high-throughput fluorescence polarization screening. Biochemistry. 2004;43:16067–75. doi: 10.1021/bi048232o. [DOI] [PubMed] [Google Scholar]
- 46.Bahadur RP, Chakrabarti P, Rodier F, Janin J. A dissection of specific and non-specific protein-protein interfaces. J Mol Biol. 2004;336:943–55. doi: 10.1016/j.jmb.2003.12.073. [DOI] [PubMed] [Google Scholar]
- 47.Ponstingl H, Henrick K, Thornton JM. Discriminating between homodimeric and monomeric proteins in the crystalline state. Proteins. 2000;41:47–57. doi: 10.1002/1097-0134(20001001)41:1<47::aid-prot80>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 48.Schonbrunn E, Eschenburg S, Shuttleworth WA, Schloss JV, Amrhein N, Evans JN, Kabsch W. Interaction of the herbicide glyphosate with its target enzyme 5-enolpyruvylshikimate 3-phosphate synthase in atomic detail. Proc Natl Acad Sci U S A. 2001;98:1376–80. doi: 10.1073/pnas.98.4.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harrison AJ, Yu M, Gardenborg T, Middleditch M, Ramsay RJ, Baker EN, Lott JS. The structure of MbtI from Mycobacterium tuberculosis, the first enzyme in the biosynthesis of the siderophore mycobactin, reveals it to be a salicylate synthase. J Bacteriol. 2006;188:6081–91. doi: 10.1128/JB.00338-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Webby CJ, Baker HM, Lott JS, Baker EN, Parker EJ. The structure of 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase from Mycobacterium tuberculosis reveals a common catalytic scaffold and ancestry for type I and type II enzymes. J Mol Biol. 2005;354:927–39. doi: 10.1016/j.jmb.2005.09.093. [DOI] [PubMed] [Google Scholar]
- 51.Zwahlen J, Kolappan S, Zhou R, Kisker C, Tonge PJ. Structure and mechanism of MbtI, the salicylate synthase from Mycobacterium tuberculosis. Biochemistry. 2007;46:954–64. doi: 10.1021/bi060852x. [DOI] [PubMed] [Google Scholar]
- 52.Payne RJ, Bulloch EM, Abell AD, Abell C. Design and synthesis of aromatic inhibitors of anthranilate synthase. Org Biomol Chem. 2005;3:3629–35. doi: 10.1039/b510633h. [DOI] [PubMed] [Google Scholar]
- 53.Payne RJ, Toscano MD, Bulloch EM, Abell AD, Abell C. Design and synthesis of aromatic inhibitors of anthranilate synthase. Org Biomol Chem. 2005;3:2271–81. doi: 10.1039/b503802b. [DOI] [PubMed] [Google Scholar]
- 54.Manos-Turvey A, Bulloch EM, Rutledge PJ, Baker EN, Lott JS, Payne RJ. Inhibition studies of Mycobacterium tuberculosis salicylate synthase (MbtI) ChemMedChem. 2010;5:1067–79. doi: 10.1002/cmdc.201000137. [DOI] [PubMed] [Google Scholar]
- 55.Green JM, Merkel WK, Nichols BP. Characterization and sequence of Escherichia coli pabC, the gene encoding aminodeoxychorismate lyase, a pyridoxal phosphate-containing enzyme. J Bacteriol. 1992;174:5317–23. doi: 10.1128/jb.174.16.5317-5323.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Green JM, Nichols BP. p-Aminobenzoate biosynthesis in Escherichia coli. Purification of aminodeoxychorismate lyase and cloning of pabC. J Biol Chem. 1991;266:12971–5. [PubMed] [Google Scholar]
- 57.Nakai T, Mizutani H, Miyahara I, Hirotsu K, Takeda S, Jhee KH, Yoshimura T, Esaki N. Three-dimensional structure of 4-amino-4-deoxychorismate lyase from Escherichia coli. J Biochem. 2000;128:29–38. doi: 10.1093/oxfordjournals.jbchem.a022727. [DOI] [PubMed] [Google Scholar]
- 58.Nichols BP, Green JM. Cloning and sequencing of Escherichia coli ubiC and purification of chorismate lyase. J Bacteriol. 1992;174:5309–16. doi: 10.1128/jb.174.16.5309-5316.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nichols BP, Seibold AM, Doktor SZ. para-aminobenzoate synthesis from chorismate occurs in two steps. J Biol Chem. 1989;264:8597–601. [PubMed] [Google Scholar]
- 60.Parsons JF, Jensen PY, Pachikara AS, Howard AJ, Eisenstein E, Ladner JE. Structure of Escherichia coli aminodeoxychorismate synthase: architectural conservation and diversity in chorismate-utilizing enzymes. Biochemistry. 2002;41:2198–208. doi: 10.1021/bi015791b. [DOI] [PubMed] [Google Scholar]
- 61.Ye QZ, Liu J, Walsh CT. p-Aminobenzoate synthesis in Escherichia coli: purification and characterization of PabB as aminodeoxychorismate synthase and enzyme X as aminodeoxychorismate lyase. Proc Natl Acad Sci U S A. 1990;87:9391–5. doi: 10.1073/pnas.87.23.9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dubnau E, Chan J, Mohan VP, Smith I. responses of mycobacterium tuberculosis to growth in the mouse lung. Infect Immun. 2005;73:3754–7. doi: 10.1128/IAI.73.6.3754-3757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilson WW. Light scattering as a diagnostic for protein crystal growth--a practical approach. J Struct Biol. 2003;142:56–65. doi: 10.1016/s1047-8477(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 64.Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, Ellestad G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 2004;332:153–9. doi: 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 65.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–21. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 66.Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ, Jr, Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, Hui R, Marsden BD, Nordlund P, Sundstrom M, Weigelt J, Edwards AM. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci U S A. 2006;103:15835–40. doi: 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cunin R, Glansdorff N, Pierard A, Stalon V. Biosynthesis and metabolism of arginine in bacteria. Microbiol Rev. 1986;50:314–52. doi: 10.1128/mr.50.3.314-352.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Elkins JM, Kershaw NJ, Schofield CJ. X-ray crystal structure of ornithine acetyltransferase from the clavulanic acid biosynthesis gene cluster. Biochem J. 2005;385:565–73. doi: 10.1042/BJ20040814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sankaranarayanan R, Cherney MM, Garen C, Garen G, Niu C, Yuan M, James MN. The molecular structure of ornithine acetyltransferase from Mycobacterium tuberculosis bound to ornithine, a competitive inhibitor. J Mol Biol. 2010;397:979–90. doi: 10.1016/j.jmb.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 70.Sankaranarayanan R, Cherney MM, Cherney LT, Garen CR, Moradian F, James MN. The crystal structures of ornithine carbamoyltransferase from Mycobacterium tuberculosis and its ternary complex with carbamoyl phosphate and L-norvaline reveal the enzyme’s catalytic mechanism. J Mol Biol. 2008;375:1052–63. doi: 10.1016/j.jmb.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 71.Shi D, Morizono H, Ha Y, Aoyagi M, Tuchman M, Allewell NM. 1.85-A resolution crystal structure of human ornithine transcarbamoylase complexed with N-phosphonacetyl-L-ornithine. Catalytic mechanism and correlation with inherited deficiency. J Biol Chem. 1998;273:34247–54. doi: 10.1074/jbc.273.51.34247. [DOI] [PubMed] [Google Scholar]
- 72.Wayne LG, Sohaskey CD. Nonreplicating persistence of mycobacterium tuberculosis. Annu Rev Microbiol. 2001;55:139–63. doi: 10.1146/annurev.micro.55.1.139. [DOI] [PubMed] [Google Scholar]
- 73.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol. 2002;43:717–31. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 74.Munoz-Elias EJ, McKinney JD. Carbon metabolism of intracellular bacteria. Cell Microbiol. 2006;8:10–22. doi: 10.1111/j.1462-5822.2005.00648.x. [DOI] [PubMed] [Google Scholar]
- 75.Wheeler PR. Analyzing lipid metabolism: activation and beta-oxidation of fatty acids. Methods Mol Biol. 2009;465:47–59. doi: 10.1007/978-1-59745-207-6_4. [DOI] [PubMed] [Google Scholar]
- 76.Kunau WH, Dommes V, Schulz H. beta-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: a century of continued progress. Prog Lipid Res. 1995;34:267–342. doi: 10.1016/0163-7827(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 77.Sauer U, Eikmanns BJ. The PEP-pyruvate-oxaloacetate node as the switch point for carbon flux distribution in bacteria. FEMS Microbiol Rev. 2005;29:765–94. doi: 10.1016/j.femsre.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 78.McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR, Jr, Russell DG. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–8. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 79.Lorenz MC, Fink GR. The glyoxylate cycle is required for fungal virulence. Nature. 2001;412:83–6. doi: 10.1038/35083594. [DOI] [PubMed] [Google Scholar]
- 80.Lindsey TL, Hagins JM, Sokol PA, Silo-Suh LA. Virulence determinants from a cystic fibrosis isolate of Pseudomonas aeruginosa include isocitrate lyase. Microbiology. 2008;154:1616–27. doi: 10.1099/mic.0.2007/014506-0. [DOI] [PubMed] [Google Scholar]
- 81.Gengenbacher M, Rao SP, Pethe K, Dick T. Nutrient-starved, non-replicating Mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiology. 2010;156:81–7. doi: 10.1099/mic.0.033084-0. [DOI] [PubMed] [Google Scholar]
- 82.Sharma V, Sharma S, Hoener zu Bentrup K, McKinney JD, Russell DG, Jacobs WR, Jr, Sacchettini JC. Structure of isocitrate lyase, a persistence factor of Mycobacterium tuberculosis. Nat Struct Biol. 2000;7:663–8. doi: 10.1038/77964. [DOI] [PubMed] [Google Scholar]
- 83.Smith CV, Huang CC, Miczak A, Russell DG, Sacchettini JC, Honer zu Bentrup K. Biochemical and structural studies of malate synthase from Mycobacterium tuberculosis. J Biol Chem. 2003;278:1735–43. doi: 10.1074/jbc.M209248200. [DOI] [PubMed] [Google Scholar]
- 84.Anstrom DM, Remington SJ. The product complex of M. tuberculosis malate synthase revisited. Protein Sci. 2006;15:2002–7. doi: 10.1110/ps.062300206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lohman JR, Olson AC, Remington SJ. Atomic resolution structures of Escherichia coli and Bacillus anthracis malate synthase A: comparison with isoform G and implications for structure-based drug discovery. Protein Sci. 2008;17:1935–45. doi: 10.1110/ps.036269.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boshoff HI, Barry CE., 3rd Tuberculosis - metabolism and respiration in the absence of growth. Nat Rev Microbiol. 2005;3:70–80. doi: 10.1038/nrmicro1065. [DOI] [PubMed] [Google Scholar]
- 87.Fischer E, Sauer U. A novel metabolic cycle catalyzes glucose oxidation and anaplerosis in hungry Escherichia coli. J Biol Chem. 2003;278:46446–51. doi: 10.1074/jbc.M307968200. [DOI] [PubMed] [Google Scholar]
- 88.Rose IA, Weaver TM. The role of the allosteric B site in the fumarase reaction. Proc Natl Acad Sci U S A. 2004;101:3393–7. doi: 10.1073/pnas.0307524101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Marrero J, Rhee KY, Schnappinger D, Pethe K, Ehrt S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc Natl Acad Sci U S A. 2010;107:9819–24. doi: 10.1073/pnas.1000715107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gobin J, Moore CH, Reeve JR, Jr, Wong DK, Gibson BW, Horwitz MA. Iron acquisition by Mycobacterium tuberculosis: isolation and characterization of a family of iron-binding exochelins. Proc Natl Acad Sci U S A. 1995;92:5189–93. doi: 10.1073/pnas.92.11.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gobin J, Wong DK, Gibson BW, Horwitz MA. Characterization of exochelins of the Mycobacterium bovis type strain and BCG substrains. Infect Immun. 1999;67:2035–9. doi: 10.1128/iai.67.4.2035-2039.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wong DK, Gobin J, Horwitz MA, Gibson BW. Characterization of exochelins of Mycobacterium avium: evidence for saturated and unsaturated and for acid and ester forms. J Bacteriol. 1996;178:6394–8. doi: 10.1128/jb.178.21.6394-6398.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reniere ML, Torres VJ, Skaar EP. Intracellular metalloporphyrin metabolism in Staphylococcus aureus. Biometals. 2007 doi: 10.1007/s10534-006-9032-0. [DOI] [PubMed] [Google Scholar]
- 94.Cescau S, Cwerman H, Letoffe S, Delepelaire P, Wandersman C, Biville F. Heme acquisition by hemophores. Biometals. 2007 doi: 10.1007/s10534-006-9050-y. [DOI] [PubMed] [Google Scholar]
- 95.Letoffe S, Ghigo JM, Wandersman C. Iron acquisition from heme and hemoglobin by a Serratia marcescens extracellular protein. Proc Natl Acad Sci U S A. 1994;91:9876–80. doi: 10.1073/pnas.91.21.9876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mills M, Payne SM. Genetics and regulation of heme iron transport in Shigella dysenteriae and detection of an analogous system in Escherichia coli O157:H7. J Bacteriol. 1995;177:3004–9. doi: 10.1128/jb.177.11.3004-3009.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mills M, Payne SM. Identification of shuA, the gene encoding the heme receptor of Shigella dysenteriae, and analysis of invasion and intracellular multiplication of a shuA mutant. Infect Immun. 1997;65:5358–63. doi: 10.1128/iai.65.12.5358-5363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wilks A, Burkhard KA. Heme and virulence: how bacterial pathogens regulate, transport and utilize heme. Nat Prod Rep. 2007;24:511–22. doi: 10.1039/b604193k. [DOI] [PubMed] [Google Scholar]
- 99.Stojiljkovic I, Hwa V, de Saint Martin L, O’Gaora P, Nassif X, Heffron F, So M. The Neisseria meningitidis haemoglobin receptor: its role in iron utilization and virulence. Mol Microbiol. 1995;15:531–41. doi: 10.1111/j.1365-2958.1995.tb02266.x. [DOI] [PubMed] [Google Scholar]
- 100.Stojiljkovic I, Larson J, Hwa V, Anic S, So M. HmbR outer membrane receptors of pathogenic Neisseria spp.: iron-regulated, hemoglobin-binding proteins with a high level of primary structure conservation. J Bacteriol. 1996;178:4670–8. doi: 10.1128/jb.178.15.4670-4678.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Turner PC, Thomas CE, Elkins C, Clary S, Sparling PF. Neisseria gonorrhoeae heme biosynthetic mutants utilize heme and hemoglobin as a heme source but fail to grow within epithelial cells. Infect Immun. 1998;66:5215–23. doi: 10.1128/iai.66.11.5215-5223.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chim N, Iniguez A, Nguyen TQ, Goulding CW. Unusual diheme conformation of the heme-degrading protein from Mycobacterium tuberculosis. J Mol Biol. 2010;395:595–608. doi: 10.1016/j.jmb.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Skaar EP, Gaspar AH, Schneewind O. IsdG and IsdI, heme-degrading enzymes in the cytoplasm of Staphylococcus aureus. J Biol Chem. 2004;279:436–43. doi: 10.1074/jbc.M307952200. [DOI] [PubMed] [Google Scholar]
- 104.Wu R, Skaar EP, Zhang R, Joachimiak G, Gornicki P, Schneewind O, Joachimiak A. Staphylococcus aureus IsdG and IsdI, heme-degrading enzymes with structural similarity to monooxygenases. J Biol Chem. 2005;280:2840–6. doi: 10.1074/jbc.M409526200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee WC, Reniere ML, Skaar EP, Murphy ME. Ruffling of metalloporphyrins bound to IsdG and IsdI, two heme-degrading enzymes in Staphylococcus aureus. J Biol Chem. 2008;283:30957–63. doi: 10.1074/jbc.M709486200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Reniere ML, Ukpabi GN, Harry SR, Stec DF, Krull R, Wright DW, Bachmann BO, Murphy ME, Skaar EP. The IsdG-family of haem oxygenases degrades haem to a novel chromophore. Mol Microbiol. 2010;75:1529–38. doi: 10.1111/j.1365-2958.2010.07076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase. Characterization of the enzyme. J Biol Chem. 1969;244:6388–94. [PubMed] [Google Scholar]
- 108.Reniere ML, Skaar EP. Staphylococcus aureus haem oxygenases are differentially regulated by iron and haem. Mol Microbiol. 2008;69:1304–15. doi: 10.1111/j.1365-2958.2008.06363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Skaar EP, Gaspar AH, Schneewind O. Bacillus anthracis IsdG, a heme-degrading monooxygenase. J Bacteriol. 2006;188:1071–80. doi: 10.1128/JB.188.3.1071-1080.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goulding CW, Apostol MI, Gleiter S, Parseghian A, Bardwell J, Gennaro M, Eisenberg D. Gram-positive DsbE proteins function differently from Gram-negative DsbE homologs. A structure to function analysis of DsbE from Mycobacterium tuberculosis. J Biol Chem. 2004;279:3516–24. doi: 10.1074/jbc.M311833200. [DOI] [PubMed] [Google Scholar]
- 111.Missiakas D, Raina S. Protein folding in the bacterial periplasm. J Bacteriol. 1997;179:2465–71. doi: 10.1128/jb.179.8.2465-2471.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hoffmann C, Leis A, Niederweis M, Plitzko JM, Engelhardt H. Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc Natl Acad Sci U S A. 2008;105:3963–7. doi: 10.1073/pnas.0709530105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zuber B, Chami M, Houssin C, Dubochet J, Griffiths G, Daffe M. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J Bacteriol. 2008;190:5672–80. doi: 10.1128/JB.01919-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chim N, Riley R, The J, Im S, Segelke B, Lekin T, Yu M, Hung LW, Terwilliger T, Whitelegge JP, Goulding CW. An extracellular disulfide bond forming protein (DsbF) from Mycobacterium tuberculosis: structural, biochemical, and gene expression analysis. J Mol Biol. 2010;396:1211–26. doi: 10.1016/j.jmb.2009.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wunderlich M, Glockshuber R. Redox properties of protein disulfide isomerase (DsbA) from Escherichia coli. Protein Sci. 1993;2:717–26. doi: 10.1002/pro.5560020503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zapun A, Bardwell JC, Creighton TE. The reactive and destabilizing disulfide bond of DsbA, a protein required for protein disulfide bond formation in vivo. Biochemistry. 1993;32:5083–92. doi: 10.1021/bi00070a016. [DOI] [PubMed] [Google Scholar]
- 117.Aslund F, Berndt KD, Holmgren A. Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein-protein redox equilibria. J Biol Chem. 1997;272:30780–6. doi: 10.1074/jbc.272.49.30780. [DOI] [PubMed] [Google Scholar]
- 118.Boshoff HI, Myers TG, Copp BR, McNeil MR, Wilson MA, Barry CE., 3rd The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J Biol Chem. 2004;279:40174–84. doi: 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
- 119.Hahn MY, Raman S, Anaya M, Husson RN. The Mycobacterium tuberculosis extracytoplasmic-function sigma factor SigL regulates polyketide synthases and secreted or membrane proteins and is required for virulence. J Bacteriol. 2005;187:7062–71. doi: 10.1128/JB.187.20.7062-7071.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 121.Matsoso LG, Kana BD, Crellin PK, Lea-Smith DJ, Pelosi A, Powell D, Dawes SS, Rubin H, Coppel RL, Mizrahi V. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J Bacteriol. 2005;187:6300–8. doi: 10.1128/JB.187.18.6300-6308.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shi L, Sohaskey CD, Kana BD, Dawes S, North RJ, Mizrahi V, Gennaro ML. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc Natl Acad Sci U S A. 2005;102:15629–34. doi: 10.1073/pnas.0507850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lewin A, Crow A, Oubrie A, Le Brun NE. Molecular basis for specificity of the extracytoplasmic thioredoxin ResA. J Biol Chem. 2006;281:35467–77. doi: 10.1074/jbc.M607047200. [DOI] [PubMed] [Google Scholar]
- 124.Xie Z, Merchant S. A novel pathway for cytochromes c biogenesis in chloroplasts. Biochim Biophys Acta. 1998;1365:309–18. doi: 10.1016/s0005-2728(98)00085-1. [DOI] [PubMed] [Google Scholar]
- 125.Munro AW, McLean KJ, Marshall KR, Warman AJ, Lewis G, Roitel O, Sutcliffe MJ, Kemp CA, Modi S, Scrutton NS, Leys D. Cytochromes P450: novel drug targets in the war against multidrug-resistant Mycobacterium tuberculosis. Biochem Soc Trans. 2003;31:625–30. doi: 10.1042/bst0310625. [DOI] [PubMed] [Google Scholar]
- 126.Kadokura H, Katzen F, Beckwith J. Protein disulfide bond formation in prokaryotes. Annu Rev Biochem. 2003;72:111–35. doi: 10.1146/annurev.biochem.72.121801.161459. [DOI] [PubMed] [Google Scholar]
- 127.Yeh SM, Koon N, Squire C, Metcalf P. Structures of the dimerization domains of the Escherichia coli disulfide-bond isomerase enzymes DsbC and DsbG. Acta Crystallogr D Biol Crystallogr. 2007;63:465–71. doi: 10.1107/S0907444907003320. [DOI] [PubMed] [Google Scholar]
- 128.Li Q, Hu H, Xu G. Biochemical characterization of the thioredoxin domain of Escherichia coli DsbE protein reveals a weak reductant. Biochem Biophys Res Commun. 2001;283:849–53. doi: 10.1006/bbrc.2001.4876. [DOI] [PubMed] [Google Scholar]
- 129.Johnson S, Brusasca P, Lyashchenko K, Spencer JS, Wiker HG, Bifani P, Shashkina E, Kreiswirth B, Harboe M, Schluger N, Gomez M, Gennaro ML. Characterization of the secreted MPT53 antigen of Mycobacterium tuberculosis. Infect Immun. 2001;69:5936–9. doi: 10.1128/IAI.69.9.5936-5939.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fabianek RA, Hennecke H, Thony-Meyer L. The active-site cysteines of the periplasmic thioredoxin-like protein CcmG of Escherichia coli are important but not essential for cytochrome c maturation in vivo. J Bacteriol. 1998;180:1947–50. doi: 10.1128/jb.180.7.1947-1950.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ogura T, Hiraga S. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci U S A. 1983;80:4784–8. doi: 10.1073/pnas.80.15.4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gerdes K, Rasmussen PB, Molin S. Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc Natl Acad Sci U S A. 1986;83:3116–20. doi: 10.1073/pnas.83.10.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Magnuson RD. Hypothetical functions of toxin-antitoxin systems. J Bacteriol. 2007;189:6089–92. doi: 10.1128/JB.00958-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Buts L, Lah J, Dao-Thi MH, Wyns L, Loris R. Toxin-antitoxin modules as bacterial metabolic stress managers. Trends Biochem Sci. 2005;30:672–9. doi: 10.1016/j.tibs.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 135.Zhang Y, Zhang J, Hoeflich KP, Ikura M, Qing G, Inouye M. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol Cell. 2003;12:913–23. doi: 10.1016/s1097-2765(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 136.Christensen SK, Gerdes K. RelE toxins from bacteria and Archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Mol Microbiol. 2003;48:1389–400. doi: 10.1046/j.1365-2958.2003.03512.x. [DOI] [PubMed] [Google Scholar]
- 137.Christensen SK, Mikkelsen M, Pedersen K, Gerdes K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc Natl Acad Sci U S A. 2001;98:14328–33. doi: 10.1073/pnas.251327898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Oberer M, Zangger K, Gruber K, Keller W. The solution structure of ParD, the antidote of the ParDE toxin antitoxin module, provides the structural basis for DNA and toxin binding. Protein Sci. 2007;16:1676–88. doi: 10.1110/ps.062680707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gotfredsen M, Gerdes K. The Escherichia coli relBE genes belong to a new toxin-antitoxin gene family. Mol Microbiol. 1998;29:1065–76. doi: 10.1046/j.1365-2958.1998.00993.x. [DOI] [PubMed] [Google Scholar]
- 140.Engelberg-Kulka H, Hazan R, Amitai S. mazEF: a chromosomal toxin-antitoxin module that triggers programmed cell death in bacteria. J Cell Sci. 2005;118:4327–32. doi: 10.1242/jcs.02619. [DOI] [PubMed] [Google Scholar]
- 141.Arcus VL, Backbro K, Roos A, Daniel EL, Baker EN. Distant structural homology leads to the functional characterization of an archaeal PIN domain as an exonuclease. J Biol Chem. 2004;279:16471–8. doi: 10.1074/jbc.M313833200. [DOI] [PubMed] [Google Scholar]
- 142.Pandey DP, Gerdes K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33:966–76. doi: 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ramage HR, Connolly LE, Cox JS. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009;5:e1000767. doi: 10.1371/journal.pgen.1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Miallau L, Faller M, Chiang J, Arbing M, Guo F, Cascio D, Eisenberg D. Structure and proposed activity of a member of the VapBC family of toxin-antitoxin systems. VapBC-5 from Mycobacterium tuberculosis. J Biol Chem. 2009;284:276–83. doi: 10.1074/jbc.M805061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hosfield DJ, Mol CD, Shen B, Tainer JA. Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: coupling DNA and PCNA binding to FEN-1 activity. Cell. 1998;95:135–46. doi: 10.1016/s0092-8674(00)81789-4. [DOI] [PubMed] [Google Scholar]
- 146.Li GY, Zhang Y, Inouye M, Ikura M. Structural mechanism of transcriptional autorepression of the Escherichia coli RelB/RelE antitoxin/toxin module. J Mol Biol. 2008;380:107–19. doi: 10.1016/j.jmb.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 147.Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, Ehrenberg M. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell. 2003;112:131–40. doi: 10.1016/s0092-8674(02)01248-5. [DOI] [PubMed] [Google Scholar]
- 148.Korch SB, Contreras H, Clark-Curtiss JE. Three Mycobacterium tuberculosis Rel toxin-antitoxin modules inhibit mycobacterial growth and are expressed in infected human macrophages. J Bacteriol. 2009;191:1618–30. doi: 10.1128/JB.01318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Singh R, Barry CE, 3rd, Boshoff HI. The three RelE homologs of Mycobacterium tuberculosis have individual, drug-specific effects on bacterial antibiotic tolerance. J Bacteriol. 2010;192:1279–91. doi: 10.1128/JB.01285-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cherny I, Rockah L, Gazit E. The YoeB toxin is a folded protein that forms a physical complex with the unfolded YefM antitoxin. Implications for a structural-based differential stability of toxin-antitoxin systems. J Biol Chem. 2005;280:30063–72. doi: 10.1074/jbc.M506220200. [DOI] [PubMed] [Google Scholar]
- 151.Kamada K, Hanaoka F. Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol Cell. 2005;19:497–509. doi: 10.1016/j.molcel.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 152.Neubauer C, Gao YG, Andersen KR, Dunham CM, Kelley AC, Hentschel J, Gerdes K, Ramakrishnan V, Brodersen DE. The structural basis for mRNA recognition and cleavage by the ribosome-dependent endonuclease RelE. Cell. 2009;139:1084–95. doi: 10.1016/j.cell.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]