

A purified blue-light-absorbing proteorhodopsin D97N mutant protein (BPR_D97N) has been crystallized using the vapour-diffusion method.
Keywords: proteorhodopsins, BPR
Abstract
Proteorhodopsins (PRs), seven-transmembrane chromoproteins with retinal as a chromophore, are light-driven proton pumps. To elucidate the light-driven proton-pumping mechanism of PRs, a pET28a vector containing the blue-light-absorbing proteorhodopsin (BPR) gene was constructed and the protein was overexpressed in Escherichia coli. The protein was purified by immobilized metal-ion affinity chromatography (IMAC). The purified BPR D97N mutant protein (BPR_D97N) was crystallized using the vapour-diffusion method. Preliminary X-ray diffraction data analysis showed that the crystal belonged to the orthorhombic space group P21212, with unit-cell parameters a = 161.6, b = 168.6, c = 64.7 Å. A complete data set was collected to 3.3 Å resolution using synchrotron radiation on beamline X06 of the Swiss Light Source (SLS). Molecular replacement was unsuccessful. To solve the structure of BPR_D97N by experimental phasing, selenomethionine-substituted protein crystals were prepared. These crystals diffracted to 3.0 Å resolution and a complete data set was collected on beamline BL17U of the Shanghai Synchrotron Radiation Facility (SSRF). Heavy-atom substructure determination and phasing by SAD clearly showed that the crystal contained five molecules in the asymmetric unit, with a V M of 3.26 Å3 Da−1 and a solvent content of 62.3%.
1. Introduction
Proteorhodopsins (PRs) are seven-transmembrane proteins which use retinal as a chromophore. This microbial rhodopsin family was first reported ten years ago (Béjà et al., 2000 ▶). Studies have shown that these microbial rhodopsins are widely distributed in oceanic environments (Béjà et al., 2001 ▶; Venter et al., 2004 ▶; Sabehi et al., 2004 ▶; Man-Aharonovich et al., 2004 ▶; Torre et al., 2003 ▶), including the Mediterranean and Red Seas (Man et al., 2003 ▶; Sabehi et al., 2007 ▶; Man-Aharonovich et al., 2004 ▶), China Seas (Zhao et al., 2009 ▶) and Antarctic Ocean (Koh et al., 2010 ▶), and fresh water (Atamna-Ismaeel et al., 2008 ▶) around the world. PR-bearing bacteria are some of the numerically richest microorganisms on Earth, accounting for around 13% of the total bacteria in sea surface water and with an average of 2.5 × 104 PR molecules per cell (Sabehi et al., 2005 ▶). Amino-acid sequence analysis has shown that PRs share less than 30% sequence identity with other microbial rhodopsins such as bacteriorhodopsin (bR), sensory rhodopsin (SR) and halorhodopsin (HR) (Béjà et al., 2000 ▶). PRs have been demonstrated to be light-driven proton pumps which generate proton motive force by translocating protons across the cell inner membrane in the same way as bR (Friedrich et al., 2002 ▶; Dioumaev et al., 2003 ▶; Wang et al., 2003 ▶). The primary proton acceptor Asp97 and donor Glu108 play crucial roles in the light-driven proton-translocation process (Dioumaev et al., 2002 ▶). A recent study has shown that proteorhodopsin confers a fitness advantage to marine bacteria during periods of resource deprivation at the ocean’s surface. Marine Vibrio cells which express PR were able to survive for longer times during starvation in light than in darkness (Gómez-Consarnau et al., 2010 ▶).
In order to understand the global ecological implications of PR and to solve the puzzle of its light harvesting and proton translocation, it is very important to resolve its three-dimensional structure. Various methods such as EM and NMR have been used to attempt to solve the structure of PR (Shastri et al., 2007 ▶; Liang et al., 2007 ▶; Schäfer et al., 2009 ▶; Pfleger et al., 2008 ▶; Shi et al., 2009 ▶), but no three-dimensional structure has been reported to date. To solve the crystal structure of a proteorhodopsin, we attempted to crystallize wild-type blue-light-absorbing proteorhodopsin (BPR) and green-light-absorbing proteorhodopsin (GPR; Man et al., 2003 ▶) and various single and multiple mutants of these and other PRs. Here, we report the crystallization and preliminary X-ray crystallographic analysis of the D97N mutant of BPR (BPR_D97N). The BPR gene was originally isolated from uncultured marine plankton collected at the Hawaiian Ocean Time Station.
2. Materials and methods
2.1. Expression of BPR_D97N
The BPR D97N mutant, originally obtained from Hot75m4 (Swiss-Prot Q9AFF7.2), was constructed as described previously (Wang et al., 2003 ▶). The PCR product was digested with NcoI and XhoI and ligated into digested pET28a vector to create the expression vector. The mutation was confirmed by DNA sequencing. The recombinant plasmid encodes BPR_D97N with a C-terminal hexahistidine tag (KLLEHHHHHH). The recombinant plasmid was then transformed into Escherichia coli C43 (DE3) (Miroux & Walker, 1996 ▶). Cells with the recombinant plasmid were grown in LB medium with 30 mg ml−1 kanamycin to an OD600 of 0.9 at 310 K. The culture was then induced by adding 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). All-trans retinal was added to a final concentration of 5 µM at the same time. The cells were harvested by centrifugation (5000 rev min−1 for 10 min; Sorvall rotor F10S 6X500Y) after induction at 303 K for 4 h. Selenomethionine-substituted (SeMet) BPR was produced (Van Duyne et al., 2003 ▶) and purified as above.
2.2. Purification of BPR_D97N
The BPR_D97N protein was purified by IMAC. The cell pellets containing the target protein from 6 l culture were resuspended in buffer A (50 mM potassium phosphate buffer pH 7.6, 300 mM NaCl, 5 mM imidazole) supplemented with 2% Triton X-100 and 1 mM phenylmethylsulfonyl fluoride. The cells were disrupted by three cycles of sonication at 273 K. The suspension was stirred by shaking at 277 K and then subjected to centrifugation (12 000 rev min−1, 30 min, 277 K; Sorvall rotor SA300) to remove insoluble material. The supernatant was applied onto 2 ml Ni–NTA resin (GE Healthcare) at 277 K. The column was sequentially washed with buffer A, buffer B [buffer A supplemented with 20 mM imidazole and 0.1% n-dodecyl-β-d-maltoside (DDM)] and buffer C (buffer A supplemented with 50 mM imidazole and 0.1% DDM), and then eluted with buffer D and buffer E (buffer A supplemented with 100 and 250 mM imidazole, respectively, and 0.01% DDM). The protein purity was judged by SDS–PAGE. The purified protein was then concentrated and applied at room temperature onto a Sephadex G-25 column (GE Healthcare) which had been pre-equilibrated with 10 mM Tris–HCl pH 8.0, 300 mM KCl and 0.01% DDM. BPR_D97N was eluted, collected and concentrated to 20 mg ml−1 using an Amicon Ultra-15 filter (Millipore).
2.3. Crystallization of BPR_D97N
Initial crystallization screens were performed using six different screening kits from Hampton Research (Index, MembFac, Crystal Screen, Crystal Screen 2, Crystal Screen Lite and Crystal Screen Cryo) employing the sitting-drop vapour-diffusion method. All of the trials were stored at 295 K. After around two weeks, small crystals were obtained from several different conditions [0.1 M sodium citrate tribasic dehydrate pH 5.6, 2.5 M 1,6-hexanediol (Crystal Screen 2, condition No. 19), 12% PEG 4000, 0.1 M sodium citrate pH 5.6, 0.1 M sodium chloride (MembFac, condition No. 17), 12% PEG 4000, 2% isopropanol, 0.1 M ADA pH 6.5, 0.1 M lithium sulfate (MembFac, condition No. 23), 0.2 M magnesium chloride hexahydrate, 0.1 M Na HEPES pH 7.5, 30%(v/v) polyethylene glycol 400 (Crystal Screen, condition No. 23) and 0.2 M ammonium acetate, 0.1 M bis-tris pH 6.5, 45%(v/v) 2-methyl-2,4-pentanediol (Index, condition No. 51)]. The condition Index No. 51 was chosen for further optimization. This condition was optimized by grid screening of the MPD concentration and the pH of the crystallization condition and the use of additive screens and detergent screens. Crystals suitable for diffraction were grown by mixing 1 µl 20 mg ml−1 protein solution with the same amount of reservoir solution plus 1–2% Tween 80 and equilibrating against 200 µl reservoir solution [0.2 M ammonium acetate, 0.1 M bis-tris pH 6.5, 44–49%(w/v) MPD] using the hanging-drop method in 24-well plates. The crystals reached maximum dimensions after 4–6 weeks (Fig. 1 ▶). The SeMet protein was crystallized using the same conditions as for the native protein.
Figure 1.

Single crystals of BPR_D97N grown using MPD as a precipitant.
2.4. Data collection and processing
The crystals were directly picked up in nylon loops and flash-cooled in liquid nitrogen. A complete X-ray diffraction data set was collected from a native crystal on beamline X06 of the Swiss Light Source (SLS) at 100 K. Each frame was exposed for 3.2 s with a rotation range of 1.0° (Fig. 2 ▶). For the SeMet crystal, a complete data set was collected on beamline BL17U of the Shanghai Synchrotron Radiation Facility (SSRF). The data were processed using the XDS package (Kabsch, 2010a ▶,b ▶). The data-collection and processing statistics are summarized in Table 1 ▶. Initial experimental phasing was attempted using HKL2MAP (Pape & Schneider, 2004 ▶) from the SHELX program suite (Sheldrick, 2008 ▶).
Figure 2.

Diffraction image of a BPR_D97N crystal.
Table 1. Summary of X-ray data for BPR_D97N.
Values in parentheses are for the highest resolution shell.
| Native | SeMet | |
|---|---|---|
| Wavelength (Å) | 1.0 | 0.9792 |
| Temperature (K) | 100 | 100 |
| Space group | P21212 | P21212 |
| Unit-cell parameters (Å) | a = 161.6, b = 168.6, c = 64.7 | a = 161.4, b = 169.0, c = 65.3 |
| Resolution range (Å) | 20.0–3.3 (3.4–3.3) | 20.0–3.0 (3.1–3.0) |
| Total observations | 98622 (8449) | 449797 (40668) |
| Unique observations | 26814 (2282) | 68739 (6400) |
| Completeness (%) | 97.8 (98.9) | 99.0 (99.5) |
| Multiplicity | 3.7 (3.7) | 6.54 (6.35) |
| Rmerge† (%) | 16.3 (95.9) | 24.7 (92.7) |
| Mean I/σ(I) | 14.36 (2.07) | 8.59 (2.30) |
| Crystal-to-detector distance (mm) | 300 | 350 |
| Detector | MAR 225 | ADSC Q315r |
R
merge = 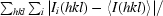
 , where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple measurements. Calculated using reflections with I > −3σ(I).
, where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple measurements. Calculated using reflections with I > −3σ(I).
3. Results and discussion
The best diffraction data for native BPR_D97N were collected to 3.3 Å resolution using synchrotron X-rays. The crystals belonged to space group P21212, with unit-cell parameters a = 161.6, b = 168.6, c = 64.7 Å (Fig. 2 ▶). Attempts at crystal structure determination of BPR_D97N by molecular replacement using the crystal structures of bR, sensory rhodopsin II and xanthorhodopsin as search models failed. We therefore prepared SeMet BPR_D97N crystals and collected a complete 3.0 Å resolution data set in order to solve the crystal structure. Experimental phasing in SHELXD/SHELXE using HKL2MAP showed that the asymmetric unit contained five molecules of BPR. The corresponding Matthews coefficient V M (Matthews, 1968 ▶) is 3.26 Å3 Da−1, with a solvent content of 62.3%. Examination of the solution revealed good crystal packing and no clashes between symmetry-related molecules. Model building and structure refinement are under way.
Acknowledgments
We are grateful to the staff members of SLS and SSRF for help with data collection. We thank Dr Jianping Ding of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, for testing the initial diffraction in his laboratory. We thank Dr Kwang-Hwan Jung of Sogang University, Republic of Korea for supplying the PR gene. This work was supported by grants from the National Natural Science Foundation of China (30700135 and 31170686).
References
- Atamna-Ismaeel, N., Sabehi, G., Sharon, I., Witzel, K. P., Labrenz, M., Jürgens, K., Barkay, T., Stomp, M., Huisman, J. & Béjà, O. (2008). ISME J. 2, 656–662. [DOI] [PubMed]
- Béjà, O., Aravind, L., Koonin, E. V., Suzuki, M. T., Hadd, A., Nguyen, L. P., Jovanovich, S. B., Gates, C. M., Feldman, R. A., Spudich, J. L., Spudich, E. N. & DeLong, E. F. (2000). Science, 289, 1902–1906. [DOI] [PubMed]
- Béjà, O., Spudich, E. N., Spudich, J. L., Leclerc, M. & DeLong, E. F. (2001). Nature (London), 411, 786–789. [DOI] [PubMed]
- Dioumaev, A. K., Brown, L. S., Shih, J., Spudich, E. N., Spudich, J. L. & Lanyi, J. K. (2002). Biochemistry, 41, 5348–5358. [DOI] [PubMed]
- Dioumaev, A. K., Wang, J. M., Bálint, Z., Váró, G. & Lanyi, J. K. (2003). Biochemistry, 42, 6582–6587. [DOI] [PubMed]
- Friedrich, T., Geibel, S., Kalmbach, R., Chizhov, I., Ataka, K., Heberle, J., Engelhard, M. & Bamberg, E. (2002). J. Mol. Biol. 321, 821–838. [DOI] [PubMed]
- Gómez-Consarnau, L., Akram, N., Lindell, K., Pedersen, A., Neutze, R., Milton, D. L., González, J. M. & Pinhassi, J. (2010). PLoS Biol. 8, e1000358. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010a). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010b). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Koh, E. Y., Atamna-Ismaeel, N., Martin, A., Cowie, R. O., Beja, O., Davy, S. K., Maas, E. W. & Ryan, K. G. (2010). Appl. Environ. Microbiol. 76, 5918–5925. [DOI] [PMC free article] [PubMed]
- Liang, H., Whited, G., Nguyen, C. & Stucky, G. D. (2007). Proc. Natl Acad. Sci. USA, 104, 8212–8217. [DOI] [PMC free article] [PubMed]
- Man, D., Wang, W., Sabehi, G., Aravind, L., Post, A. F., Massana, R., Spudich, E. N., Spudich, J. L. & Béjà, O. (2003). EMBO J. 22, 1725–1731. [DOI] [PMC free article] [PubMed]
- Man-Aharonovich, D., Sabehi, G., Sineshchekov, O. A., Spudich, E. N., Spudich, J. L. & Béjà, O. (2004). Photochem. Photobiol. Sci. 3, 459–462. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Miroux, B. & Walker, J. E. (1996). J. Mol. Biol. 260, 289–298. [DOI] [PubMed]
- Pape, T. & Schneider, T. R. (2004). J. Appl. Cryst. 37, 843–844.
- Pfleger, N., Lorch, M., Woerner, A. C., Shastri, S. & Glaubitz, C. (2008). J. Biomol. NMR, 40, 15–21. [DOI] [PubMed]
- Sabehi, G., Béjà, O., Suzuki, M. T., Preston, C. M. & DeLong, E. F. (2004). Environ. Microbiol. 6, 903–910. [DOI] [PubMed]
- Sabehi, G., Kirkup, B. C., Rozenberg, M., Stambler, N., Polz, M. F. & Béjà, O. (2007). ISME J. 1, 48–55. [DOI] [PubMed]
- Sabehi, G., Loy, A., Jung, K. H., Partha, R., Spudich, J. L., Isaacson, T., Hirschberg, J., Wagner, M. & Béjà, O. (2005). PLoS Biol. 3, 1409–1417. [DOI] [PMC free article] [PubMed]
- Schäfer, G., Shastri, S., Verhoefen, M. K., Vogel, V., Glaubitz, C., Wachtveitl, J. & Mäntele, W. (2009). Photochem. Photobiol. 85, 529–534. [DOI] [PubMed]
- Shastri, S., Vonck, J., Pfleger, N., Haase, W., Kuehlbrandt, W. & Glaubitz, C. (2007). Biochim. Biophys. Acta, 1768, 3012–3019. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Shi, L., Ahmed, M. A., Zhang, W., Whited, G., Brown, L. S. & Ladizhansky, V. (2009). J. Mol. Biol. 386, 1078–1093. [DOI] [PubMed]
- Torre, J. R., Christianson, L. M., Béjà, O., Suzuki, M. T., Karl, D. M., Heidelberg, J., DeLong, E. F., Spudich, E. N., Spudich, J. L., Leclerc, M. & DeLong, E. F. (2003). Nature (London), 411, 786–789.
- Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L. & Clardy, J. (1993). J. Mol. Biol. 229, 105–124. [DOI] [PubMed]
- Venter, J. C. et al. (2004). Science, 304, 66–74. [DOI] [PubMed]
- Wang, W.-W., Sineshchekov, O. A., Spudich, E. N. & Spudich, J. L. (2003). J. Biol. Chem. 278, 33985–33991. [DOI] [PubMed]
- Zhao, M., Chen, F. & Jiao, N. (2009). Appl. Environ. Microbiol. 75, 529–533. [DOI] [PMC free article] [PubMed]