

The catalytic domain of human Dus2-like enzyme was purified and crystallized, and data were collected to 1.9 Å resolution.
Keywords: dihydrouridine synthases, tRNA
Abstract
Dihydrouridine synthases catalyse the reduction of uridine to dihydrouridine in the D-loop and variable loop of tRNA. The human dihydrouridine synthase HsDus2L has been implicated in the development of pulmonary carcinogenesis. Here, the purification, crystallization and preliminary X-ray characterization of the HsDus2L catalytic domain are reported. The crystals belonged to space group P21 and contained a single molecule of HsDus2L in the asymmetric unit. A complete data set was collected to 1.9 Å resolution using synchrotron radiation.
1. Introduction
Post-transcriptional modifications influence the three-dimensional structure of tRNA, rendering regions either more flexible or more rigid in order to modulate and reinforce the structure of the core (Motorin & Helm, 2010 ▶). Dihydrouridine synthase (Dus) enzymes principally target the D-loop of tRNA, named for incorporation of dihydrouridine (D), which is understood to increase regional flexibility (Fig. 1 ▶ a). Residues within the D-loop are involved in conserved tertiary base pairs which stabilize the fold of tRNA (Shi & Moore, 2000 ▶). Psychrophilic bacteria living at temperatures of 268–285 K possess 40–70% more D than the mesophilic Escherichia coli. This modification is almost absent from hyperthermophilic bacterial tRNAs (Dalluge et al., 1997 ▶).
Figure 1.

Secondary structure of tRNA, domain topology of HsDus2L, and SDS–PAGE and SEC–MALLS of H6-HsDus2L 1–340. (a) Secondary structure of tRNA with D target sites (U16, 17, 20, 20a, 20b and 47) shaded. (b) Domain topology of HsDus2L. Lines representing expression constructs are annotated with the results of expression tests. (c) SDS–PAGE and (d) SEC–MALLS analysis of SEC-purified H6-HsDus2L 1–340.
Little is known of the function of the human Dus2-like (HsDus2L) enzyme. Increased incorporation of D in tRNAPhe has been identified in malignant tissues (Kuchino & Borek, 1978 ▶). HsDus2L expression is upregulated in non-small-cell lung carcinoma (NSCLC), potentiates hyperproliferation and correlates with a poorer prognosis (Kato et al., 2005 ▶). HsDus2L is known to interact with the apoptotic regulator dsRNA-activated protein kinase PKR (Mittelstadt et al., 2008 ▶).
In Saccharomyces cerevisiae Dus2p (ScDus2p), D-loop modification has been characterized at position 20 and other sites (Fig. 1 ▶ a; Xing et al., 2004 ▶). HsDus2L has moderate sequence identity to ScDus2p and low identity to two structurally characterized enzymes (Table 1 ▶). The first crystal structure of a Dus enzyme, that of TM0096 from Thermotoga maritima (TmDUS; PDB entry 1vhn; Park et al., 2004 ▶), identified a flavin mononucleotide (FMN) cofactor binding in the TIM barrel. FMN is used as a cofactor to perform an NADPH-dependent redox cycle in which the oxidative half-reaction reduces target uracil (Rider et al., 2009 ▶). The crystal structure of Thermus thermophilus Dus–tRNA (TthDus; PDB entry 3b0p) identified the mechanism of specific modification (U20) (PDB entries 3b0u and 3b0v; Yu et al., 2011 ▶). Binding involves recognition at the nexus of D-loops and T-loops of folded tRNA. Given that D is more important for tRNA flexibility in psychrophiles and mesophiles, a complete understanding of the panoply of Dus enzymes awaits structural characterization of these groups.
Table 1. Sequence identity of ScDus2p, TthDus and TmDus to HsDus2L (PSIBLAST; Altschul et al., 1997 ▶).
| Protein | Region | HsDus2L | Identity (%) |
|---|---|---|---|
| ScDus2p | 2–301 | 8–289 | 39 |
| TthDus | 5–237 | 13–254 | 22 |
| TmDus | 4–248 | 13–270 | 23 |
The low sequence identity of the HsDus2L Dus domain to TmDus and TthDus suggests that while the overall fold may be moderately conserved, the C-terminal α-helical domain (253–340) could vary substantially (Table 1 ▶). Hence, the structure of HsDus2L should generate novel mechanistic details for eukaryotic Dus enzymes. The structure of the enzymatic domain will be crucial to address the structure-based design of specific inhibitors of D incorporation as potential NSCLC therapeutics. Here, we describe the purification and crystallization of the Dus domain of Dus2L from Homo sapiens.
2. Methods and materials
2.1. Cloning
Bioinformatic analysis with JPred was utilized to predict secondary structure (Cole et al., 2008 ▶). Three constructs were designed based on predicted domain boundaries (Fig. 1 ▶ b). The cDNA of HsDus2L was accessed through the DNASU plasmid repository (clone HsCD00303989; Cormier et al., 2011 ▶). The coding sequences (accession code AAH06527) were PCR-amplified, digested and ligated into the NheI/XhoI sites of the pET28a vector, generating an N-terminal fusion with a His6 tag/thrombin cleavage site.
2.2. Overexpression and purification
Clones were transformed into Escherichia coli BL21 (DE3) pLysS competent cells. An LB culture containing 30 µg ml−1 kanamycin and 40 µg ml−1 chloramphenicol was inoculated and incubated at 310 K to an OD600 of 0.6. Expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside followed by incubation at 290 K for 20 h. The cells were harvested and resuspended in 20 mM Tris pH 8, 500 mM NaCl, 20 mM imidazole, 2 mM DTT, sonicated for 3 min and the lysate was cleared by centrifugation at 40 000g for 1 h at 277 K. The lysate was loaded onto a charged HisTrap HP column (GE Healthcare) and protein was eluted using an imidazole gradient (20–500 mM, 20 column volumes). The protein was concentrated using an Amicon centrifugal filter (Millipore). The protein concentrate was diluted in Tris/DTT to an NaCl concentration of 50 mM, loaded onto a pre-equilibrated MonoQ 5/50 GL column (GE Healthcare) and eluted with an NaCl gradient (50 mM–1 M, 20 column volumes). H6-HsDus2L 1–340 (molar mass 40 326 Da) was purified by size-exclusion chromatography (SEC) as described below. The protein was digested using thrombin and incubated at 277 K for 16 h. Untagged HsDus2L 1–340 (molar mass 38 605 Da) was separated from uncleaved material by passage through a charged HisTrap HP column. Cleaved and uncleaved HsDus2L 1–340 were concentrated and purified by SEC using a Superdex 75 HR 10/30 column (GE Healthcare) equilibrated in 20 mM Tris pH 8, 100 mM NaCl, 5 mM DTT (Fig. 1 ▶ c). The protein concentration was determined from the absorbance at 280 nm using molar extinction coefficients of 21 890 M −1 cm−1 (untagged) and 22 515 M −1 cm−1 (hexahistidine tagged). The tagged and untagged proteins were concentrated to 10 mg ml−1. H6-HsDus2L 1–340 (2.5 mg ml−1) was analysed by SEC multi-angle laser light scattering (SEC–MALLS) using a Biosep-SEC-s3000 column (Phenomenex), a Dawn HELEOS-II 18-angle light-scattering detector and an Optilab rEX refractive-index monitor (Wyatt). Purified tagged protein was characterized using matrix-assisted laser desorption/ionization mass spectrometry (MALDI–MS).
2.3. Crystallization and data collection
Initial screening for crystallization conditions for tagged and untagged proteins was performed by the sitting-drop vapour-diffusion method using the PACT (Newman et al., 2005 ▶; Qiagen), Ammonium Sulfate (Qiagen), MPD (Qiagen), Morpheus (Gorrec, 2009 ▶; Molecular Dimensions) and Index (Hampton Research) screens. 0.30 µl sample solution and 0.15 µl reservoir solution were aliquoted using a Mosquito robot into an MRC/Wilden 96-well plate with 54 µl reservoir solution and incubated at 292 K. Crystals were cryoprotected in protein buffer and reservoir supplemented with PEG 1500 to 32%(w/v) and flash-cooled in liquid N2. X-ray data for untagged HsDus2L 1–340 crystals were collected on beamline I04 at Diamond Light Source, Didcot, England using an ADSC Q315r CCD detector at 100 K over 180° (crystal-to-detector distance of 274 mm, 0.5° oscillation, 0.5 s exposure for high-resolution data and crystal-to-detector distance of 424 mm, 1° oscillation and 0.5 s exposure for low-resolution data). Indexing and integration was performed with XDS (Kabsch, 2010 ▶). The Laue group was confirmed by POINTLESS and scaling and merging of the data were performed by SCALA (Evans, 2006 ▶). Data-collection statistics are summarized in Table 2 ▶.
Table 2. Data-collection statistics for HsDus2L.
Values in parentheses are for the highest resolution shell.
| X-ray source | Beamline I04, Diamond |
| Wavelength (Å) | 0.9795 |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 46.3, b = 49.6, c = 69.6, β = 95.7 |
| Resolution limits (Å) | 49.6–1.9 (2.0–1.9) |
| Unique reflections | 24244 (3515) |
| Completeness (%) | 97.6 (97.8) |
| Multiplicity | 3.0 (2.8) |
| 〈I/σ(I)〉 | 10.4 (1.7) |
| Rmerge† (%) | 6.3 (58.5) |
| Rp.i.m.‡ (%) | 4.0 (41.4) |
| CC1/2§ | 0.997 (0.716) |
| No. of molecules in asymmetric unit | 1 |
| VM (Å3 Da−1) | 2.1 |
| Solvent content (%) | 40.2 |
R
merge = 
 , where I(hkl) is the integrated intensity of a given reflection.
, where I(hkl) is the integrated intensity of a given reflection.
R
p.i.m. = 
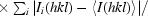 .
.
CC1/2 is the half-data-set correlation coefficient.
3. Results
Expression trials were performed for three constructs in small-scale cultures (Fig. 1 ▶ b). Purification of the full-length enzyme resulted in the isolation of a degraded product. HsDus2L 1–340 was expressed in E. coli and purified by nickel-affinity, anion-exchange and size-exclusion chromatography to >95% purity as estimated by SDS–PAGE analysis (Fig. 1 ▶ c). Purified HsDus2L 1–340 had an absorption peak at 450 nm and was yellow-coloured, which was suggestive of the presence of oxidized FMN, a cofactor that is critical for enzymatic reduction of uracil and that has also been identified in TthDus and TmDus (Park et al., 2004 ▶; Yu et al., 2011 ▶). MALDI–MS of H6-HsDus2L 1–340 identified a single peak of 40 370 Da consistent with the predicted mass. The molar mass in solution estimated using SEC–MALLS was 40 700 ± 2850 Da, indicating a monomeric species (Fig. 1 ▶ d).
Crystals of H6-HsDus2L 1–340 grew in 2 d in PACT screen conditions B11, C11 and D11 [0.2 M CaCl2, 20%(w/v) PEG 6000 and 0.1 M MES pH 6/HEPES pH 7/Tris pH 8, respectively]. These crystals grew in clumps, proved impossible to separate and were difficult to reproduce in optimization screens. Crystallization screening with untagged enzyme resulted in the growth of rod-like clusters that could be mechanically separated. These crystals grew in 2 d from PACT screen condition D1 in a reservoir consisting of 0.1 M MES/malic/Tris buffer pH 4 (Newman, 2004 ▶) and 25%(w/v) PEG 1500. Crystals of untagged HsDus2L 1–340 (Fig. 2 ▶ a) diffracted to 1.9 Å resolution (Fig. 2 ▶ b) and belonged to space group P21, with unit-cell parameters a = 46.2, b = 49.6, c = 69.6 Å, β = 95.7°. The Matthews coefficient estimated that the asymmetric unit comprised a single molecule of HsDus2L (Matthews, 1968 ▶).
Figure 2.

Crystals and diffraction of untagged HsDus2L 1–340. (a) Rod-like crystals from PACT screen (condition D1). (b) Diffraction recorded using synchrotron radiation with the crystal rotated by 0.5°. The resolution at the edge is 1.9 Å.
We attempted to phase the data by molecular replacement using the structures of TthDus and TmDus. MR experiments were performed using MOLREP, Phaser and BALBES (Vagin & Teplyakov, 2010 ▶; McCoy et al., 2007 ▶; Long et al., 2008 ▶); however, no solutions were identified. We are performing selenomethionine labelling to attempt to determine the structure of the HsDus2L Dus domain by multiwavelength anomalous dispersion phasing.
Acknowledgments
We would like to thank Dr Johan Turkenburg and Sam Hart for their assistance with data collection. We acknowledge the support of beamline I04 at Diamond Light Source, Didcot, England for crystallographic data collection.
References
- Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997). Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed]
- Cole, C., Barber, J. D. & Barton, G. J. (2008). Nucleic Acids Res. 36, W197–W201. [DOI] [PMC free article] [PubMed]
- Cormier, C. Y., Park, J. G., Fiacco, M., Steel, J., Hunter, P., Kramer, J., Singla, R. & LaBaer, J. (2011). J. Struct. Funct. Genomics, 12, 55–62. [DOI] [PMC free article] [PubMed]
- Dalluge, J. J., Hamamoto, T., Horikoshi, K., Morita, R. Y., Stetter, K. O. & McCloskey, J. A. (1997). J. Bacteriol. 179, 1918–1923. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Gorrec, F. (2009). J. Appl. Cryst. 42, 1035–1042. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kato, T., Daigo, Y., Hayama, S., Ishikawa, N., Yamabuki, T., Ito, T., Miyamoto, M., Kondo, S. & Nakamura, Y. (2005). Cancer Res. 65, 5638–5646. [DOI] [PubMed]
- Kuchino, Y. & Borek, E. (1978). Nature (London), 271, 126–129. [DOI] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Mittelstadt, M., Frump, A., Khuu, T., Fowlkes, V., Handy, I., Patel, C. V. & Patel, R. C. (2008). Nucleic Acids Res. 36, 998–1008. [DOI] [PMC free article] [PubMed]
- Motorin, Y. & Helm, M. (2010). Biochemistry, 49, 4934–4944. [DOI] [PubMed]
- Newman, J. (2004). Acta Cryst. D60, 610–612. [DOI] [PubMed]
- Newman, J., Egan, D., Walter, T. S., Meged, R., Berry, I., Ben Jelloul, M., Sussman, J. L., Stuart, D. I. & Perrakis, A. (2005). Acta Cryst. D61, 1426–1431. [DOI] [PubMed]
- Park, F., Gajiwala, K., Noland, B., Wu, L., He, D., Molinari, J., Loomis, K., Pagarigan, B., Kearins, P., Christopher, J., Peat, T., Badger, J., Hendle, J., Lin, J. & Buchanan, S. (2004). Proteins, 55, 772–774. [DOI] [PubMed]
- Rider, L. W., Ottosen, M. B., Gattis, S. G. & Palfey, B. A. (2009). J. Biol. Chem. 284, 10324–10333. [DOI] [PMC free article] [PubMed]
- Shi, H. & Moore, P. B. (2000). RNA, 6, 1091–1105. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Xing, F., Hiley, S. L., Hughes, T. R. & Phizicky, E. M. (2004). J. Biol. Chem. 279, 17850–17860. [DOI] [PubMed]
- Yu, F., Tanaka, Y., Yamashita, K., Suzuki, T., Nakamura, A., Hirano, N., Suzuki, T., Yao, M. & Tanaka, I. (2011). Proc. Natl Acad. Sci. USA, 108, 19593–19598. [DOI] [PMC free article] [PubMed]