

Abstract
We have previously shown that the inhibition of histone deacetylases (HDACs) protects the heart against acute myocardial ischemia and reperfusion injury. We also demonstrated that HDAC inhibition stimulates myogenesis and angiogenesis in a cultured embryonic stem cell model. We investigate whether in vivo inhibition of HDAC preserves cardiac performance and prevents cardiac remodeling in mouse myocardial infarction (MI) through the stimulation of endogenous regeneration. MI was created by ligation of the left descending artery. Animals were divided into three groups: 1) sham group, animals that underwent thoracotomy without MI; 2) MI, animals that underwent MI; and 3) MI + trichostatin A (TSA), MI animals that received a daily intraperitoneal injection of TSA. In addition, infarcted mice received a daily intraperitoneal injection of TSA (0.1 mg/kg), a selective HDAC inhibitor. 5-Bromo-2-deoxyuridine (50 mg/kg) was delivered every other day to pulse-chase label in vivo endogenous cardiac replication. Eight weeks later, the MI hearts showed a reduction in ventricular contractility. HDAC inhibition increased the improvement of myocardial functional recovery after MI, which was associated with the prevention of myocardial remodeling and reduction of myocardial and serum tumor necrosis factor α. HDAC inhibition enhanced the formation of new myocytes and microvessels, which was consistent with the robust increase in proliferation and cytokinesis in the MI hearts. An increase in angiogenic response was demonstrated in MI hearts receiving TSA treatment. It is noteworthy that TSA treatment significantly inhibited HDAC activity and increased phosphorylation of Akt-1, but decreased active caspase 3. Taken together, our results indicate that HDAC inhibition preserves cardiac performance and mitigates myocardial remodeling through stimulating cardiac endogenous regeneration.
Introduction
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) have recently emerged as important enzymes in the regulation of a variety of cellular responses. Histone acetylation is mediated by histone acetyl transferase. The resulting modification in the structure of chromatin leads to nucleosomal relaxation and altered transcriptional activation. The reverse reaction is mediated by HDAC, which induces deacetylation, chromatin condensation, and transcriptional repression (Luger et al., 1997; Hansen et al., 1998; Cheung et al., 2000; Strahl and Allis, 2000; Turner, 2000). In addition to histones, growing evidence suggests that HAT and HDAC target nonhistone proteins, including transcriptional factors (Chen et al., 2001), which may represent general regulatory mechanisms in biological signaling.
Since the identification of HDAC1 (named HD1) (Hassig et al., 1998), 18 HDACs have been described in mammals and are divided into three distinct classes on the basis of secondary structure (Verdin et al., 2003). Class I HDACs consist of HDAC1, HDAC2, HDAC3, and HDAC8, which are predominantly nuclear proteins and ubiquitously expressed. Class II HDACs include HDAC4, HDAC5, HDAC7, and HDAC9. In contrast to class I, class II HDACs exhibit a tissue-specific pattern of expression. HDAC4 and HDAC5 are found at high levels in the heart, brain, and skeletal muscles (Fischle et al., 1999; Grozinger et al., 1999; Wang et al., 1999). Class III HDACs were identified on the basis of sequence similarity with Sir, a yeast transcriptional repressor that requires the cofactor NDA+ for its deacetylase activity. HDAC inhibitors have shown efficacy as anticancer reagents in human and animal models and are emerging as an exciting clinical treatment targeting solid and hematological malignancies (Vigushi and Coombes, 2004).
Inhibition of class II HDACs silences fetal gene activation, renders myocytes insensitive to hypertrophic agonists, blocks cardiac hypertrophy, and prevents cardiac remodeling (Antos et al., 2003; Kee et al., 2006; Kong et al., 2006; Granger et al., 2008). Furthermore, HDAC inhibition has previously been shown to markedly decrease infarct size in the focal cerebral ischemia model of rats (Ren et al., 2004). We and others have demonstrated that inhibition of HDAC with trichostatin A (TSA) protects the heart against ischemic injury (Lee et al., 2007; Zhao et al., 2007, 2010; Granger et al., 2008; Zhang et al., 2010), which is associated with activation transcriptional factor κB, stimulation of gp91, and activation of p38 mitogen-activated protein kinase. Recently, we demonstrated that HDAC inhibition increased the resistance of embryonic stem cells (ESCs) to oxidant stress, which is associated with the reduction of cell death, an increase in cell viability, and a decrease in apoptosis. It is noteworthy that we also found that HDAC inhibition stimulates the growth of embryoid bodies, which is linked to the cardiac lineage commitment and an increased up-regulation of cardiac-specific transcriptional factors, suggesting that HDAC inhibition constitutes a major cascade in controlling cardiogenesis and promoting the survival of ESCs (Chen et al., 2011).
The discovery of cardiac stem cells (CSCs) has generated considerable enthusiasm toward the idea of the regenerating myocardium by using resident stem cells (Beltrami et al., 2003; Oh et al., 2003; Laugwitz et al., 2005; Loffredo et al., 2011). It is noteworthy that epigenetic intervention and/or HDAC inhibition have been identified as the most critical determination for cell programming in vitro studies (Huangfu et al., 2008; Ware et al., 2009). However, it is not clear whether HDAC inhibition can trigger an endogenous regeneration that is sufficient to repair infarcted tissue. In this study, we used the chronic mouse myocardial infarction (MI) model and systematically delivered HDAC inhibitor to examine the effect of HDAC inhibition on cardiac performance and myocardial remodeling. We used 5-bromo-2-deoxyuridine (BrdU) to pulse-chase label the newly formed myocardial tissue and vascular structure after HDAC inhibition in MI hearts. We also detected whether HDAC inhibition-induced myocardial regeneration resulted in the improvement of functional recovery. Our results demonstrate that HDAC inhibition improves myocardial function and promotes endogenous regeneration and angiogenic response in the infarcted heart.
Materials and Methods
Animals.
Adult male ICR mice (10–12 weeks old) were supplied by Charles River Laboratories, Inc. (Wilmington, MA). All animal experiments were conducted under a protocol approved by the Institutional Animal Care and Use Committee of Roger Williams Medical Center, which conforms to the National Institutes of Health's Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996).
In Vivo Myocardial Infarction.
The mouse myocardial infarction model was created after thoracotomy by applying permanent ligation of the left anterior descending artery as described previously (Zhao et al., 2008; Tseng et al., 2010). In brief, mice were anesthetized with an intraperitoneal injection of sodium pentobarbital at a dose of 50 mg/kg; additional doses of pentobarbital were given as needed during the procedure to maintain an anesthetized state. Mice were placed in a supine position and intubated with an endotracheal tube. Ventilation was achieved with a mini rodent ventilator (Harvard Apparatus Inc., Holliston, MA). Thoracotomy was performed with tenotomy scissors. A 7-0 nylon suture was passed with a tapered needle under the left anterior descending coronary artery. The suture was tied to create coronary occlusion. Upon completion of ligation, the chest was closed in a layered fashion, and air was evacuated to prevent pneumothorax. Mice in the sham group were anesthetized and underwent thoracotomy without coronary ligation. Animals were divided into three groups: 1) sham, animals that underwent thoracotomy without MI; 2) MI, animals that underwent MI; and 3) MI + TSA, MI animals that received a daily intraperitoneal injection of TSA (0.1 mg/kg; Sigma, St. Louis, MO) for 8 consecutive weeks after MI. MI control animals received a daily intraperitoneal injection of saline (0.1 ml). To evaluate the effect of HDAC inhibition on newly formed angiomyogenesis, BrdU (50 mg/kg; Sigma) was delivered every other day to pulse-chase label in vivo endogenous cardiac replication. Eight weeks later, cardiac ventricular function and immunohistochemistry were determined.
Langendorff Isolated Heart Perfusion.
The methodology of Langendorff's perfused heart preparation and measurement of left ventricular (LV) function has been described previously in detail (Zhao and Kukreja, 2002; Zhao et al., 2006, 2008; Tseng et al., 2010). In brief, mice were anesthetized with a lethal intraperitoneal injection of sodium pentobarbital (120 mg/kg). Hearts were rapidly excised and arrested in ice-cold Krebs-Henseleit buffer. They were then cannulated via the ascending aorta for retrograde perfusion by the Langendorff method using Krebs-Henseleit buffer containing 110 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4 · 7H2O, 2.5 mM CaCl2 · 2H2O, 11 mM glucose, 1.2 mM KH2PO4, 25 mM NaHCO3, and 0.5 mM EDTA. The buffer, aerated with 95% O2:5% CO2 to give a pH of 7.4 at 37°C, was perfused at a constant pressure of 55 mm Hg. A water-filled latex balloon, inserted into the left ventricle and attached to the tip of polyethylene tubing, was then inflated sufficiently to provide a left ventricular end-diastolic pressure (LVEDP) of approximately 10 mm Hg measured by means of a disposable Gould pressure transducer (Gould Instrument Systems Inc., Cleveland, OH). Left ventricular functional analysis was recorded by using software and a computer-based recording system (BIOPAC, Goleta, CA). These parameters included left ventricular systolic pressure, LVEDP, heart rate (HR), and LV developed pressure (LVDP), where LVDP is LV systolic pressure minus LVEDP. LV dP/dtmax and LV dP/dtmin were continuously recorded. At the end of the experiments, myocardial samples were collected for histological analysis.
Determination of Microvascular Density.
To identify microvascular density, isolated hearts were perfused with the modified Krebs-Henseleit buffer containing fluorescent-isothiocyanate-conjugated lycopersicon esculentum lection (Sigma), which uniformly binds to the surface of the endothelium. The hearts were then fixed in 4% paraformaldehyde, and paraffin-embedded cross-sections were prepared. Myocytes were identified by α-sarcomeric actinin by using a red fluorescent secondary antibody. Microvascular density was determined on 18 different, randomly selected fields of cross-sectioned fibers from each heart (Frederick et al., 2010).
Tissue and Cellular Immunoctyochemistries.
Cardiac tissues and sections were prepared from the paraffin-embedded hearts as described previously (Tseng et al., 2010). Tissue sections were deparaffinized for 30 min at 70°C and subsequently immersed in xylene and ethanol at decreasing concentrations. Slides were then washed in distilled water. Myocytes were identified by α-sarcomeric actinin; smooth muscle cells were identified by anti-α-smooth muscle actin (α-SMA) monoclonal antibodies, and endothelial cells were identified by von Willebrand factor (vWF) polyclonal rabbit antibody (Sigma). BrdU was used to detect the proliferative cells; Ki67 was used to detect the cycling cells; and phosphorylated histone 3 (pH3) was used to detect mitosis. The total number of vessels from each group was calculated and normalized to the tissue area. CSCs were labeled with c-kit antibodies. The stained numbers of each section were counted in approximately 20 randomized fields of the tissue sections, which were taken in the middle plane of each heart and contained infarct and border regions.
Histological Analysis.
Sections (10 μm) were prepared from paraffin-embedded tissues, and sections from apex, mid-left ventricle, and base were stained with Masson's trichrome according to the manufacturer's protocol (Sigma). Images of the three sections, from the base to the apex of the left ventricle, were taken by using an Olympus BX51 microscope (Olympus, Tokyo, Japan) with SPOT Advanced software (SPOT Imaging Solutions, Sterling Heights, MI). Infarct scar area and total area of LV were traced manually and measured by using Image J software (National Institutes of Health, Bethesda, MD). Wall thickness of left ventricle, viable myocardium, and scar size were measured as described previously (Cheng et al., 2010). To quantitate the degree of LV dilation, the LV expansion index was calculated by using a modification of the method: expansion index = (LV cavity area/total area) × (noninfarcted region wall thickness/risk region wall thickness) (Cheng et al., 2010).
HDAC Activity Assay in the Myocardium.
Measurement of HDAC activity in cardiac tissue was conducted by using a colorimetric HDAC activity assay kit (BioVision, Mountain View, CA).
Western Blot Analysis.
The cardiac tissues were lysed with radioimmunoprecipitation assay buffer (150 mM sodium chloride, 1.0% nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0), supplemented with protease inhibitor cocktail. The proteins detected included antiphosphorylated Akt-1, Akt-1 (Cell Signaling Technology, Danvers, MA), and active caspase 3 (Abcam plc, Cambridge, UK) and β-actin rabbit polyclonal antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA). Proteins (30 μg/lane) were separated by 10% SDS-polyacrylamide gel electrophoresis. The proteins were then transferred onto a nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk in phosphate-buffered saline containing 0.5% Tween 20 for 1 h. The blots were incubated with primary antibodies (1:1000) overnight at 4 h and visualized by incubation with anti-rabbit or anti-mouse horseradish peroxidase-conjugated antibodies (1:5000 dilution) for 1 h. The immunoblots were developed with ECL Chemiluminescence Detection Reagent (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). The densitometric results were normalized to the control group and expressed as percentages of control values.
Measurements of Myocardial TNF-α.
TNF-α contents were measured by using an enzyme-linked immunosorbent assay kit (BD Biosciences, San Jose, CA). In brief, a standard curve was generated for samples assayed by standards of various concentrations. Assays were performed in duplicate. Development of a colored reaction caused by the conversion of chromogenic substrate (tetramethylbenzidine) that was directly proportional to the amount of the analyte assayed followed for 10 min with analysis via an enzyme-linked immunosorbent assay plate reader at 450 nm. Results were expressed in picograms per milligram.
Statistics.
All measurements are expressed as means ± S.E. Differences among the groups were analyzed by one-way analysis of variance, followed by post hoc Bonferroni correction or Student's unpaired t test for two groups. Statistical differences were considered significant with a value of p < 0.05.
Results
Ventricular Function of Postinfarction Mouse Heart In Vitro.
Left ventricular functional parameters were measured in isovolumetric perfused hearts. Ventricular function is illustrated in Fig. 1. Significant reductions in left ventricular systolic pressure and developed pressure were observed in infarcted hearts compared with sham control heart. Left ventricular developed pressure in MI vehicle-injected hearts decreased to 50% of that of sham- operated mice. There was a greater improvement in LV dP/dtmax and LV dP/dtmin restoration in the TSA-treated groups. There was no significant difference in heart rate between the groups. The assessment of effluent was used as a measure of the coronary flow. TSA treatment increased the content of coronary effluent (CF) compared with the control group. Taken together, the administration of TSA in MI mice improved cardiac functional recovery in MI hearts.
Fig. 1.

HDAC inhibition augmented the restoration of ventricular function in MI mice. LV function was assessed in isovolumetric hearts 8 weeks after MI. Measured parameters include left ventricular systolic pressure, HR (beats/min), and LVDP [mm Hg, where LVDP is systolic pressure minus LVEDP (mm Hg)], and rate pressure product (RPP; mm Hg · beats/min). RPP is expressed as the product of LVDP and HR, where developed pressure is systolic pressure minus LVEDP. LV dP/dtmax (mm Hg/s) and LV dP/dtmin (mm Hg/s) were continuously recorded. CF is ml/min. Values represent mean ± S.E. (n = 5/group). *, p < 0.05 compared with MI group.
Evidence of Newly Formed Myocardial Components in Postinfarction Heart.
To test whether HDAC inhibition serves as a major determinant to activating c-kit+ CSCs to regenerate myocardium, mouse MI was induced by ligation of the left descending coronary artery. TSA treatment led to a dramatic increase in c-kit+ CSCs, with increases that were approximately 2-fold (Fig. 2C). In addition, TSA treatments markedly increased BrdU incorporation within the infarcted area (Fig. 2, A and B). Ki67 was included to provide a quantitative estimation of the fraction of cells in the cell cycle at the time of observation. An increase in Ki67-positive components was evident in TSA-treated MI hearts. Mitosis was assessed by using pH3 (Fig. 2D). The pH3-positive nuclei and cardiomyocytes that were elevated in the hearts received TSA (Fig. 2E). The representative images of BruU, c-kit, Ki67, and pH3 in sham and the infarcted area of MI and MI+TSA-treated hearts are shown in Fig. 3. Taken together, these results indicate that TSA stimulates the self-renewal of c-kit+ CSCs and enhances endogenous myocardial proliferation and cytokinesis in vivo in MI hearts.
Fig. 2.

HDAC inhibition stimulates endogenous cardiac regeneration after myocardial infarction. ICR mice were subjected to MI by ligation of the left descending coronary artery for 8 weeks. Trichostatin A and 5-bromo-2-deoxyuridine were administrated. Detailed methods of producing myocardial infarction and delivery of HDAC inhibitor and BrdU are under Materials and Methods. Quantitative analyses of BrdU (A and B), c-kit+ CSC (C), Ki67 (D), and pH3 (E) staining in paraffin-embedded sections are shown. Hearts were fixed and sectioned 8 weeks after MI. Values represent mean ± S.E. (n = 3–4/per group). *, p < 0.05.
Fig. 3.

Representative images from sham (left), MI (center), and TSA-treated (right) MI hearts. A, BrdU. B, c-kit+ CSC. C, Ki67. D, pH3. Nuclei are stained in blue (DAPI), and cardiomyocytes are in green (α-sarcomeric actinin). BrdU, Ki67, pH3, and c-kit are in red. Scale bars, 50 μm.
HDAC Inhibition, Angiogenesis, and Vascular Structure.
Angiogenic responses were examined by immunofluorescent staining for vascular smooth muscle actin and vWF. TSA treatment substantially increased the densities of microvessels in the infarcted zone, as indicated by an increase in α-SMA, lectin, and vWF (Fig. 4). In the border and center regions of the infarct zone, α-SMA staining was observed. As shown in Fig. 4, A and D, administration of TSA significantly increased α-SMA-positive vascular density in the border zones of infarcted hearts compared with left anterior descending artery ligation alone. Furthermore, to identify the microvascular density, isolated hearts were perfused with fluorescent isothiocyanate-conjugated lycopersicon esculentum lection, which uniformly binds to the surface of the endothelium. As shown in Fig. 4, B and E, TSA treatment caused a marked increase in lectin-stained vascular structure density in MI hearts compared with the control hearts. Likewise, we also observed a significant increase in vWF-positive staining in infarcted and border zones after administration of TSA (Fig. 4, C and F).
Fig. 4.

Quantitative analysis of microvessel formation in sham, MI, and TSA-treated MI hearts after HDAC inhibition. A to C, densities of α-SMA (A), lectin (B), and vWF (C). Values represent mean ± S.E. (n = 5/group). *, p < 0.05 versus sham and MI. D to F, representative images of α-SMA-positive (D), lectin-positive (E), and vWF-postive (F) microvascular densities. Scale bars, 50 μm.
Prevention of Myocardial Hypertrophy and Remodeling.
Administration of TSA reduced the content of fibrosis of left ventricle in response to myocardial infarction and myocardial infarction size (Fig. 5, A and B). Compared with the sham control group, morphometric analysis of infarcted hearts showed severe LV chamber dilatation and infarct wall thinning. MI hearts that received HDAC inhibition illustrated attenuated LV remodeling, which demonstrated a more viable myocardium and thicker infarcted wall but smaller scar size and less LV expansion (Fig. 5C). MI heart resulted in the increase of infarction. Heart/body ratio, an index of hypertrophy, increased after myocardial infarction. However, the hypertrophic response to MI was reduced in the TSA-treated mouse MI hearts (Fig. 6A). The Kaplan-Meier survival curve was used to assess animal survival. As shown in Fig. 6B, TSA treatment significantly improved the survival rate of animals exposed to myocardial infarction.
Fig. 5.

HDAC inhibition mitigates myocardial remodeling in MI hearts. A and B, representative images of hematoxylin and eosin (H&E) staining (A) and Masson-tranchrome staining (B). Scale bars, 50 μm. C, viable myocardium, wall thickness, scar sizes, and the expansion index are shown. Values represent mean ± S.E. (n = 3/per group). *, P < 0.05 versus MI group.
Fig. 6.

HDAC inhibition prevented hypertrophic response and increased the animal survival of postoperated animals. A, heart/body ratio in infarcted myocardium. *, p < 0.05 versus sham group and MI. B, survival rate of animals in MI hearts.
It is known that TNF-α in myocardium was markedly up-regulated in response to myocardial ischemia. The myocardial and serum TNF-α levels were suppressed in MI hearts from mice receiving TSA treatment compared with the control MI group (Fig. 7).
Fig. 7.

The myocardial and serum TNF-α levels in sham and MI hearts among groups. A, serum TNF-α. B, myocardial TNF-α. Values represent mean ± S.E. (n = 6/per group). *, p < 0.001 versus sham and MI.
HDAC Inhibition Activates Mitogen-Activated Protein Kinase Signaling.
We have previously demonstrated that Akt-1 is involved in the protection of hearts against ischemic injury (Tseng et al., 2010). Akit-1 is pivotal in the regulation of cardiac development, cardiac stress, and protective effects. We next attempted to examine whether HDAC inhibition would activate these specific signaling pathways in MI hearts. HDAC activity in MI hearts increased compared with the sham group, but was abrogated by TSA treatment (Fig. 8A). It is noteworthy that relative to the control MI TSA treatment showed an increase in phosphorylated Akt-1 (Fig. 8B). Furthermore, HDAC inhibition significantly reduced the content of active caspase 3 in MI hearts (Fig. 8C), suggesting that stimulation of Akt-1 signaling pathway and inhibition of apoptosis also involves the beneficial effects of TSA.
Fig. 8.
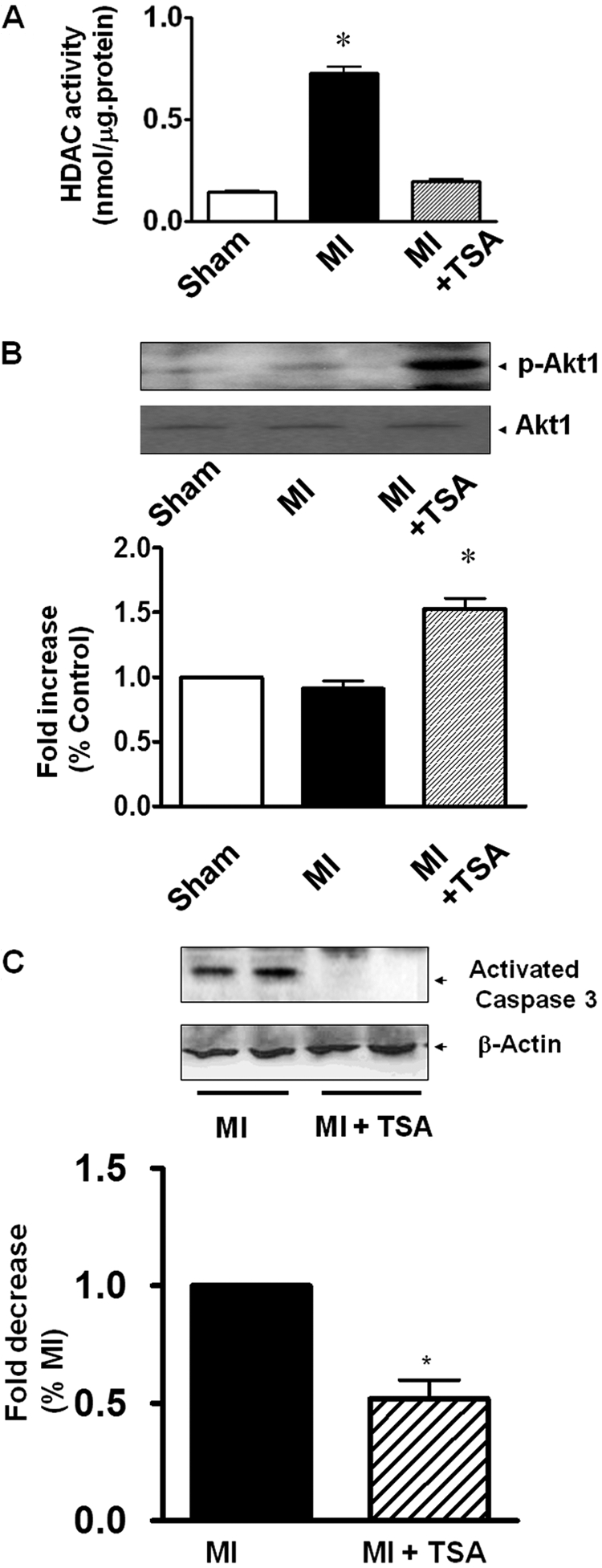
Measurements of HDAC activity, activated Akt-1, and apoptosis in myocardium. A, HDAC activity was inhibited in TSA-treated MI hearts. B, HDAC inhibition resulted in increases in phosphorylated Akt-1(p-Akt1). C, activated caspase-3. Values represent mean ± S.E. (n = 3–5/group). *, p < 0.05 versus MI.
Discussion
HDAC inhibition has been demonstrated to play an essential role in the regulation of cardiac growth, prevention of cardiac hypertrophy, and protection against ischemia injury (Kee et al., 2006; Kong et al., 2006; Zhao et al., 2007, 2010; Granger et al., 2008; Haberland et al., 2009). The availability of well characterized heart progenitor cells will allow for a direct examination of their biological function and specific pathway that drives cardiogenesis in the developmental stage and regeneration of infarcted myocardium (Matsuura et al., 2009; Tang et al., 2010). In this study, we have demonstrated: 1) administration of TSA improved myocardial functional recovery in the infarcted heart; 2) TSA stimulated endogenous myocardial regeneration, which involves c-kit+ CSC proliferation in MI hearts; 3) TSA augmented cardiac proliferation and mitosis as indicated by increases in Ki67 and phosphorylated histone 3, which are closely associated with an increase of newly formed cardiac components; the angiogenesis and newly formed vascular structure were up-regulated in MI hearts after HDAC inhibition; and 4) TSA administration prevented myocardial remodeling and cardiac hypertrophy and mitigated the proinflammatory response.
HATs and HDAC govern gene expression patterns by being recruited to target genes through association with specific transcription factors (Yuan et al., 2005; Haberland et al., 2009). We and others have demonstrated that inhibition of HDACs with a selective inhibitor shows cardioprotection and blocks cardiac hypertrophy (Antos et al., 2003; Kee et al., 2006; Kong et al., 2006; Lee et al., 2007; Zhao et al., 2007, 2010; Granger et al., 2008; Zhang et al., 2010). Consistent with our previous findings, chronic MI hearts with HDAC inhibition manifested an antihypertrophic effect and prevented cardiac modeling, which is associated with the improvement of myocardial functional recovery.
It is known that development and cell fate determination require genetic and epigenetic coordination (Surani et al., 2007). The most striking example shown here is that HDAC inhibitors increase reprogramming efficiency of fibroblasts into induced pluripotent stem cells and ESC self-renewal across species (Huangfu et al., 2008; Ware et al., 2009), which support the possibility of reprogramming through HDAC inhibition, which may make the therapeutic use of reprogrammed cells safer and more practical. However, it remains unknown whether HDAC inhibition could stimulate endogenous angiomyogenesis and repair the infarcted myocardium in vivo. Our evidence shows that the administration of TSA stimulates the self-renewal of the c-kit CSCs, which is consistent with the marked increase in newly formed myocytes in the infarcted hearts, indicating that in vivo HDAC inhibition effectively activated cardiac progenitor cells to facilitate myocardial regeneration. In addition, c-kit+ CSCs are characteristically positively stained with c-kit and cardiac actinin, a specific cardiac marker, which lacks hematopoietic stem cell marker CD34, indicating that peripheral c-kit+ stem cells might not play a major role in myocardial regeneration induced by HDAC inhibition. It will be interesting to see whether HDAC inhibition promotes myocardial regeneration through the mobilization of peripheral progenitor cells. Our observation is supported by our previous findings that trichostatin A and sodium butyrate stimulate the growth of embryoid bodies, which are closely associated with a faster spontaneous rhythmic contraction, the cardiac lineage commitment, and an increased up-regulation of cardiac actin and cardiac-specific transcriptional factors (Chen et al., 2011). Therefore, HDAC inhibition plays an important role in mediating endogenous myocardial regeneration.
Angiogenesis is a complex physiologic process consisting of local degradation of the surrounding capillaries and migration of ECs to angiogenic stimuli after proliferation, leading to the formation of a three-dimensional capillary network. We have shown that angiogenesis after TSA treatment is associated with the restoration/preservation of myocardial function after myocardial infarction. Coronary effluent was increased by the administration of TSA compared with that of control MI, suggesting that an increase in coronary effluent might be associated with a more robust angiogenic response after administration of TSA. In addition, an increase in CF might be related to vascular dilation, which is worthwhile for the future investigation. It is known that pathological cardiac hypertrophy with reduced contractility is accompanied by impaired coronary angiogenesis. It is likely that the increase in angiogenic response in TSA treatment contributes to the improvement in contractile performance in our studies. This emphasizes the importance of TSA in regulation of the angiogenic response in myocardial infarction. It has recently been reported that both cardiac α- and β-myosin heavy chain isoforms were found to be reversibly acetylated and lysine acetylation increased the actin sliding velocity of α-myosin and β-myosin compared with their respective nonacetylated isoforms (Samant et al., 2011). It is likely that TSA treatment also contributes to the improvement of myocardial functional recovery in our study, which is a subject that merits further investigation. Akt is pivotal in the regulation of cardiac development (Mangi et al., 2003; Tseng et al., 2010). Our observation shows that relative to the control MI TSA treatments promote an increase in phosphorylated Akt-1. It is likely that HDAC inhibition stimulates Akt-1 to facilitate myocardial regeneration and prevents cardiac modeling under in vivo conditions.
In conclusion, although we have previously shown that TSA induces a pharmacological preconditioning effect against acute myocardial ischemia and reperfusion injury, it is not clear whether TSA promotes endogenous angiomyogenesis in infarcted mouse hearts. The present study demonstrates new evidence that in vivo inhibition of HDAC improved cardiac functional recovery and antagonized myocardial remodeling in chronic MI. It is noteworthy that HDAC inhibition significantly improved animal survival rate of post-MI, which is associated with the mitigation of both myocardial and serum TNF-α levels in MI heart. Furthermore, HDAC inhibition stimulated the self-renewal of CSCs and resulted in robust increases in proliferation and cytokinesis in the MI hearts, which are associated with the enhancement of the newly formed myocytes and angiogenic responses. In addition, HDAC inhibition-induced cardioprotection involves the activation of Akt-1 and inhibition of apoptosis. Our study provides novel evidence that a new therapeutic strategy could be developed based on HDAC inhibition that elicits the stimulation of cardiac endogenous regeneration and angiogenesis in infarcted heart.
The work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grant R01-HL089405] and the American Heart Association National Center [Grant 0735458N] (to T.C.Z.).
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- HAT
- histone acetyltransferase
- HDAC
- histone deacetylase
- MI
- myocardial infarction
- TSA
- trichostatin A
- BrdU
- 5-bromo-2-deoxyuridine
- TNF-α
- tumor necrosis factor α
- ESC
- embryonic stem cell
- CSC
- cardiac stem cell
- LV
- left ventricular
- LVEDP
- LV end-diastolic pressure
- LVDP
- LV developed pressure
- LV dP/dtmax
- LV dP/dt maximum
- LV dP/dtmin
- LV dP/dt minimum
- RPP
- rate pressure product
- HR
- heart rate
- CF
- coronary effluent
- α-SMA
- α-smooth muscle actin
- vWF
- von Willebrand factor
- pH3
- phosphorylated histone 3.
Authorship Contributions
Participated in research design: Zhang, Qin, Liu, Cheng, and T. C. Zhao.
Conducted experiments: Zhang, Qin, Y. Zhao, Fast, and Cheng.
Performed data analysis: Zhang, Qin, Y. Zhao, and Cheng.
Wrote or contributed to the writing of the manuscript: Zhang, Qin, Zhuang, Liu, and T. C. Zhao.
References
- Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, Rindt H, Gorczynski RJ, Olson EN. (2003) Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem 278:28930–28937 [DOI] [PubMed] [Google Scholar]
- Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, et al. (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114:763–776 [DOI] [PubMed] [Google Scholar]
- Chen HP, Denicola M, Qin X, Zhao Y, Zhang L, Long XL, Zhuang S, Liu PY, Zhao TC. (2011) HDAC inhibition promotes cardiogenesis and the survival of embryonic stem cells through proteasome-dependent pathway. J Cell Biochem 112:3246–3255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LF, Fischle W, Verdin E, Greene WC. (2001) Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293:1653–1657 [DOI] [PubMed] [Google Scholar]
- Cheng K, Li TS, Malliaras K, Davis DR, Zhang Y, Marbán E. (2010) Magnetic targeting enhances engraftment and functional benefit of iron-labeled cardiosphere-derived cells in myocardial infarction. Circ Res 106:1570–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung P, Allis CD, Sassone-Corsi P. (2000) Signaling to chromatin through histone modifications. Cell 103:263–271 [DOI] [PubMed] [Google Scholar]
- Fischle W, Emiliani S, Hendzel MJ, Nagase T, Nomura N, Voelter W, Verdin E. (1999) A new family of human histone deacetylases related to Saccharomyces cerevisiae HDA1p. J Biol Chem 274:11713–11720 [DOI] [PubMed] [Google Scholar]
- Frederick JR, Fitzpatrick JR, 3rd, McCormick RC, Harris DA, Kim AY, Muenzer JR, Marotta N, Smith MJ, Cohen JE, Hiesinger W, et al. (2010) Stromal cell-derived factor-1α activation of tissue-engineered endothelial progenitor cell matrix enhances ventricular function after myocardial infarction by inducing neovasculogenesis. Circulation 122 (11 Suppl): S107–S117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger A, Abdullah I, Huebner F, Stout A, Wang T, Huebner T, Epstein JA, Gruber PJ. (2008) Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J 22:3549–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozinger CM, Hassig CA, Schreiber SL. (1999) Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci U S A 96:4868–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10:32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JC, Tse C, Wolffe AP. (1998) Structure and function of the core histone N-termini: more than meets the eye. Biochemistry 37:17637–17641 [DOI] [PubMed] [Google Scholar]
- Hassig CA, Tong JK, Fleischer TC, Owa T, Grable PG, Ayer DE, Schreiber SL. (1998) A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc Natl Acad Sci U S A 95:3519–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, Muhlestein W, Melton DA. (2008) Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol 26:1269–1275 [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources (1996) Guide for the Care and Use of Laboratory Animals 7th ed Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington, DC [Google Scholar]
- Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, et al. (2006) Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 113:51–59 [DOI] [PubMed] [Google Scholar]
- Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN, Hill JA. (2006) Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 113:2579–2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugwitz KL, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, Lin LZ, Cai CL, Lu MM, Reth M, et al. (2005) Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 433:647–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TM, Lin MS, Chang NC. (2007) Inhibition of histone deacetylase on ventricular remodeling in infarcted rats. Am J Physiol Heart Circ Physiol 293:H968–H977 [DOI] [PubMed] [Google Scholar]
- Loffredo FS, Steinhauser ML, Gannon J, Lee RT. (2011) Bone marrow-derived cell therapy stimulates endogenous cardiomyocyte progenitors and promotes cardiac repair. Cell Stem Cell 8:389–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389:251–260 [DOI] [PubMed] [Google Scholar]
- Mangi AA, Noiseux N, Kong D, He H, Rezvani M, Ingwall JS, Dzau VJ. (2003) Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat Med 9:1195–1201 [DOI] [PubMed] [Google Scholar]
- Matsuura K, Honda A, Nagai T, Fukushima N, Iwanaga K, Tokunaga M, Shimizu T, Okano T, Kasanuki H, Hagiwara N, et al. (2009) Transplantation of cardiac progenitor cells ameliorates cardiac dysfunction after myocardial infarction in mice. J Clin Invest 119:2204–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, et al. (2003) Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci U S A 100:12313–12318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. (2004) Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem 89:1358–1367 [DOI] [PubMed] [Google Scholar]
- Samant SA, Courson DS, Sundaresan NR, Pillai VB, Tan M, Zhao Y, Shroff SG, Rock RS, Gupta MP. (2011) HDAC3-dependent reversible lysine acetylation of cardiac myosin heavy chain isoforms modulates their enzymatic and motor activity. J Biol Chem 286:5567–5577 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Surani MA, Hayashi K, Hajkova P. (2007) Genetic and epigenetic regulators of pluripotency. Cell 128:747–762 [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. (2000) The language of covalent histone modifications. Nature 403:41–45 [DOI] [PubMed] [Google Scholar]
- Tang XL, Rokosh G, Sanganalmath SK, Yuan F, Sato H, Mu J, Dai S, Li C, Chen N, Peng Y, et al. (2010) Intracoronary administration of cardiac progenitor cells alleviates left ventricular dysfunction in rats with a 30-day-old infarction. Circulation 121:293–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng A, Stabila J, McGonnigal B, Yano N, Yang MJ, Tseng YT, Davol PA, Lum LG, Padbury JF, Zhao TC. (2010) Effect of disruption of Akt-1 of lin(−)c-kit(+) stem cells on myocardial performance in infarcted heart. Cardiovasc Res 87:704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BM. (2000) Histone acetylation and an epigenetic code. Bioessays 22:836–845 [DOI] [PubMed] [Google Scholar]
- Verdin E, Dequiedt F, Kasler HG. (2003) Class II histone deacetylases: versatile regulators. Trends Genet 19:286–293 [DOI] [PubMed] [Google Scholar]
- Vigushin DM, Coombes RC. (2004) Targeted histone deacetylase inhibition for cancer therapy. Curr Cancer Drug Targets 4:205–218 [DOI] [PubMed] [Google Scholar]
- Wang AH, Bertos NR, Vezmar M, Pelletier N, Crosato M, Heng HH, Th'ng J, Han J, Yang XJ. (1999) HDAC4, a human histone deacetylase related to yeast HDA1, is a transcriptional corepressor. Mol Cell Biol 19:7816–7827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware CB, Wang L, Mecham BH, Shen L, Nelson AM, Bar M, Lamba DA, Dauphin DS, Buckingham B, Askari B, et al. (2009) Histone deacetylase inhibition elicits an evolutionarily conserved self-renewal program in embryonic stem cells. Cell Stem Cell 4:359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZL, Guan YJ, Chatterjee D, Chin YE. (2005) Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307:269–273 [DOI] [PubMed] [Google Scholar]
- Zhang LX, Zhao Y, Cheng G, Guo TL, Chin YE, Liu PY, Zhao TC. (2010) Targeted deletion of NF-κB p50 diminishes the cardioprotection of histone deacetylase inhibition. Am J Physiol Heart Circ Physiol 298:H2154–H2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Parikh P, Bhashyam S, Bolukoglu H, Poornima I, Shen YT, Shannon RP. (2006) Direct effects of glucagon-like peptide-1 on myocardial contractility and glucose uptake in normal and postischemic isolated rat hearts. J Pharmacol Exp Ther 317:1106–1113 [DOI] [PubMed] [Google Scholar]
- Zhao TC, Kukreja RC. (2002) Late preconditioning elicited by activation of adenosine A3 receptor in heart: role of NF-κB, iNOS and mitochondrial K(ATP) channel. J Mol Cell Cardiol 34:263–277 [DOI] [PubMed] [Google Scholar]
- Zhao TC, Cheng G, Zhang LX, Tseng YT, Padbury JF. (2007) Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc Res 76:473–481 [DOI] [PubMed] [Google Scholar]
- Zhao TC, Tseng A, Yano N, Tseng Y, Davol PA, Lee RJ, Lum LG, Padbury JF. (2008) Targeting human CD34+ hematopoietic stem cells with anti-CD45 x anti-myosin light-chain bispecific antibody preserves cardiac function in myocardial infarction. J Appl Physiol 104:1793–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao TC, Zhang LX, Cheng G, Liu JT. (2010) gp-91 mediates histone deacetylase inhibition-induced cardioprotection. Biochim Biophys Acta 1803:872–880 [DOI] [PMC free article] [PubMed] [Google Scholar]