

Abstract
Rhabdomyosarcoma (RMS) is a morphologically and clinically heterogeneous group of malignant tumors that resemble developing skeletal muscle and is the most common soft-tissue sarcoma in children and adolescents. The most prominent sites involve head and neck structures (~40%), genito-urinary track (~25%), and extremities (~20%). Embryonal (ERMS) and alveolar (ARMS) are the two major RMS subtypes that are distinct in their morphology and genetic make-up. The prognosis for this cancer depends strongly on tumor size, location, staging, and child’s age. In general, ERMS has a more favorable outcome, whereas the mortality rate remains high in patients with ARMS, because of its aggressive and metastatic nature. Over the past two decades, researchers have made concerted efforts to delineate genetic and epigenetic changes associated with RMS pathogenesis. These molecular signatures have presented golden opportunities to design targeted therapies for treating this aggressive cancer. This article highlights recent advances in understanding the molecular pathogenesis of RMS, and addresses promising research areas for further exploration.
Keywords: pediatric cancer, sarcoma, muscle, genetic abnormalities, oncogenesis, fusion protein
Introduction
Rhabdomyosarcoma (RMS) is the most common soft-tissue malignancy in the pediatric population. Current treatment for RMS relies on chemotherapy, with surgery and radiation as adjunct therapies. The cytotoxic actions of chemotherapeutic agents are not tumor-specific and are not effective in treating advanced and metastatic RMS. Since this cancer afflicts young patients, there are concerns of impairment in normal development and increased risks in developing secondary cancers as resulting from the long-term effects of the treatment. This article aims to (1) highlight recent advances in our understanding of RMS cell biology, which forms the critical basis for developing novel therapies that are more specific for tumor cells and less harmful to normal cells, and (2) offer prospective views on underexplored and promising research topics that could have further impact on this field.
Rhabdomyosarcoma
The two most common rhabdomyosarcoma subtypes are embryonal (ERMS) and alveolar (ARMS). Clinically, they are distinguished by differences in histopathology, genetics, and clinical presentation and outcome (Saab et al., 2011). ERMS and ARMS comprise ~60 to 70% and ~25 to 30% of US RMS cases, respectively (Ognjanovic et al., 2009). Studies have not established an association of RMS with any environmental factor. ERMS most commonly presents in the head and neck region and the genito-urinary tract of children aged 1 to 14 yrs. ARMS occurs more frequently in the extremities and trunk region and is more prevalent among older children and adolescents. In general, RMS is more common in white than black children (3:1), and the male-to-female ratio is approximately 1.5:1.
Histopathology
ERMS is composed of round or spindle-shaped cells and resembles embryonic muscle. ARMS displays a distinct alveolar architecture with aggregates of small round undifferentiated cells separated by dense hyalinized fibrous septa. Although ARMS cells express high levels of muscle differentiation markers (Dias et al., 2000), these tumors lack mature muscle characteristics. Some solid ARMS variants lack this alveolar structure and may display overlapping histology with ERMS. Recent adoption of cytogenetic and molecular screening has greatly improved diagnosis.
Genetics
Several non-random chromosome alterations have been identified in RMS (Fig. 1). Loss of heterozygosity for chromosome 11p15.5 and chromosome gains (chr. 2, 8, 12, 13) are commonly detected in ERMS (Scrable and Witte, 1989; Anderson et al., 1999). The 11p15.5 region contains several imprinted genes from which the loss of one allele could inactivate the tumor suppressor gene (e.g., H19, CDKN1C) or activate the proto-oncogene (e.g., IGF2, HRAS) leading to cancer development. By contrast, recurrent reciprocal chromosomal translocations are observed in ARMS. A t(2;13) translocation is detected in ~70% of ARMS; a less common t(1;13) variant is found in ~10% of ARMS (Turc-Carel et al., 1986; Wang-Wuu et al., 1988). These translocations fuse PAX3 (chr.2)/ PAX7 (chr.1) with FKHR (FOXO1A, chr.13) to generate chimeric PAX3/PAX7-FKHR oncogenes (Barr et al., 1993; Shapiro et al., 1993). A rare t(2;X) involving fusion between PAX3 and another FOXO family member, MLLT7 (FOXO4), has also been reported (Barr et al., 2002). ARMS tumors that do not carry these typical PAX-FOXO translocations are referred to as “fusion-negative” ARMS (ARMSn) in the literature. Williamson et al. (2010) have reported a striking similarity between ARMSn and ERMS based on their clinical behaviors (e.g., location, metastasis rate, treatment responses) and mRNA profiles. This suggests that ARMSn and ERMS arise from the same cellular lineage but undergo different developmental paths to produce distinct histological features. This is supported by recent findings that ARMSn and ERMS have distinct microRNA profiles (Gougelet et al., 2011), and that some ARMSn harbor atypical translocations (Wachtel et al., 2004; Sumegi et al., 2010). A recurrent t(2;2) or t(2;8) translocation leading to the formation of respective PAX3-NCOA1/NCOA2 fusions is detected in a subset of ARMSn tumors. The altered microRNA and gene re-arrangement profiles may explain why some ARMSn behave as aggressively as PAX-FOXO-positive ARMS. Given these new developments, it will be worthwhile to know if ERMS and ARMSn can be further clustered into subsets based on their mRNA profile, microRNA profile, translocation status, and clinical features. Such information can improve the diagnosis, prognosis, and treatment selection for the patients.
Figure 1.

Non-random chromosomal alterations found in association with RMS tumors. (A) ERMS-associated loss of heterozygosity in the p15.5 region of chromosome 11. Altered expression of some imprinted genes in ERMS is highlighted in red (down-regulation) and blue (up-regulation). (B) Reciprocal chromosomal translocations identified in ARMS tumors. The t(2;13) translocation is the most common gene re-arrangement. All translocations involve disruption of muscle-developmental PAX genes, PAX3 and PAX7. Each reciprocal translocation generates two fusion gene products, PAX-X and X-PAX, where X represents a member of the FOXO transcription factor family or a member of the nuclear co-activator NCOA family. Only the PAX-X fusion genes have been cloned and studied. FKHR-PAX3 is the only reciprocal gene whose transcript is detected in a fraction of t(2;13)-positive ARMS tumors and cell lines. Other X-PAX reciprocal genes have not been investigated.
Clinical Outcome
The overall survival rate for RMS patients has significantly improved over the past two decades. In patients with low-grade, localized ERMS, the five-year survival rate has reached >85%. This rate drops dramatically with increased age and more advanced tumor stages at diagnosis. There is also a striking difference in mortality between patients with metastatic PAX3-FKHR and PAX7-FKHR ARMS; the estimated five-year survival rate is 8% (PAX3-FKHR) as compared with 75% (PAX7-FKHR) (Sorensen et al., 2002; Kazanowska et al., 2007).
Pax3-Fkhr Fusion
Since the discovery of the RMS-associated chromosomal re-arrangements, major progress has been made in the characterization of the fusion products. The prototype, PAX3-FKHR, has been the main research focus for the past two decades. These studies highlight the critical role of PAX3-FKHR in ARMS pathology and provide blueprints for investigating the newly emerged fusion gene products PAX3-NCOA1/2. Because NCOA1/2 are transcriptional co-activators for nuclear hormone receptors instead of transcription factors like FKHR, future studies are needed to compare the mechanisms and gene targets of PAX3-NCOA1/2 with those of PAX-FOXO to see if different mechanisms are responsible for the heterogeneity of the ARMS subtype.
Transcription Properties
PAX3 is a paired-box transcription factor with roles in developmental and adult regenerative myogenesis (Buckingham and Relaix, 2007; Lagha et al., 2008). FKHR is a wing-helix transcription factor with roles in cell growth, survival, and metabolic regulation (Birkenkamp and Coffer, 2003). The translocation breaks within intron-7 of the PAX3 gene and intron-1 of the FKHR gene, leading an in-frame PAX3-FKHR chimera (Fig. 2). Although the sequence-specific DNA-binding domains (paired- and homeo-) of PAX3 are conserved in PAX3-FKHR, comparative expression profiles of ERMS and ARMS tumors, and of PAX3/PAX3-FKHR-expressing culture cells, suggest that these two proteins do not function equally (Khan et al., 1999; Begum et al., 2005; De Pitta et al., 2006; Mercado et al., 2008). Several key differences setting PAX3-FKHR apart from PAX3 have been identified: PAX3-FKHR activates PAX3-dependent genes at a higher level as the result of the stronger activation domain of FKHR (Fredericks et al., 1995), activates a unique set of genes not targeted by PAX3 by uncoupling the interdependence of the two DNA-binding domains necessary for PAX3 function (Epstein et al., 1998; Zhang and Wang, 2006, 2011), and is not inactivated by several PAX3 interacting inhibitory proteins (Wiggan et al., 1998; Hollenbach et al., 1999). The cumulative effects of these novel properties in PAX3-FKHR are further magnified through altered responses toward post-translational regulation. Phosphorylation destabilizes PAX3 and controls the nuclear-cytoplasm localization of FKHR. While PAX3-FKHR retains many of the active phosphorylation sites, it is more resistant to degradation (Miller and Hollenbach, 2007) and resides constitutively in the nucleus (del Peso et al., 1999). Together, these studies have identified novel mechanisms by which PAX3-FKHR transforms cells.
Figure 2.
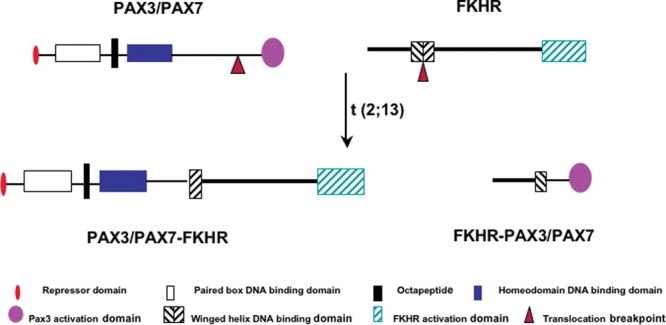
Diagrammatic illustration of PAX3, FKHR, PAX3-FKHR, and FKHR-PAX3 protein structures. The translocation breaks the PAX3 gene between the DNA-binding domains and activation domain, and breaks the FKHR gene within the FKHR DNA-binding domain. The repressor (R) domain represents the first 10-amino-acid sequence of the PAX3 protein, which inhibits PAX3 but not PAX3-FKHR transcription activity. The octapeptide sequence is thought to be involved in dimerization of PAX3, but whether it plays a similar role in PAX3-FKHR is not known. The balanced translocation results in expression of a 97-kDa PAX3-FKHR fusion protein and a putative 35-kDa FKHR-PAX3 fusion protein.
Oncogenic Properties
Several lines of evidence support an oncogenic role of PAX3-FKHR in ARMS development. Ectopic expression of PAX3-FKHR in fibroblasts and myoblasts causes accelerated proliferation and anchorage-independent growth (Scheidler et al., 1996; Lam et al., 1999), and blocks terminal differentiation into multinucleated myotubes (Epstein et al., 1995). Interestingly, expression of a PAX3-FKHR mutant with an impaired paired-DNA-binding domain in fibroblasts and myoblasts can induce cellular transformation and high-grade tumor formation similar to the wild-type PAX3-FKHR. Although myoblasts are not transformed by a PAX3-FKHR homeo-domain mutant, these cells form metastatic tumors in nude mice (Zhang et al., 2009). These mutational studies highlight the biological significance of the gain-of-transcription target specificity in the oncogenic activity of the fusion protein. When introduced into ERMS cells, PAX3-FKHR produces morphological and proliferation rate changes associated with a higher transformation grade (Anderson et al., 2001). Antisense knockdown of PAX3-FKHR expression reduces ARMS cell growth followed by apoptosis (Bernasconi et al., 1996). However, establishing a direct role for PAX3-FKHR in the initiation of ARMS tumorigenesis in genetically engineered animals has not been as straightforward. Transgenic or knock-in mice expressing PAX3-FKHR under the control of PAX3 promoter show developmental lethality without signs of muscle tumor formation (Anderson et al., 2001; Lagutina et al., 2002). The first PAX3-FKHR-driven ARMS animal model uses a conditional knock-in strategy, targeting the fusion gene expression in differentiated muscle cells (Keller et al., 2004a). In this model, tumor incidence is extremely low (1/218) but is increased with additional mutation in the p53 or Ink4A/ARF gene. This observation suggests that ARMS formation depends on cooperation between PAX3-FKHR and other genetic mutations (Fig. 3). It remains to be determined whether the cooperating event occurs before or after translocation during ARMS establishment. Collectively, the cell culture and animal studies indicate that persistent PAX3-FKHR expression is essential for the initiation and maintenance of ARMS.
Figure 3.
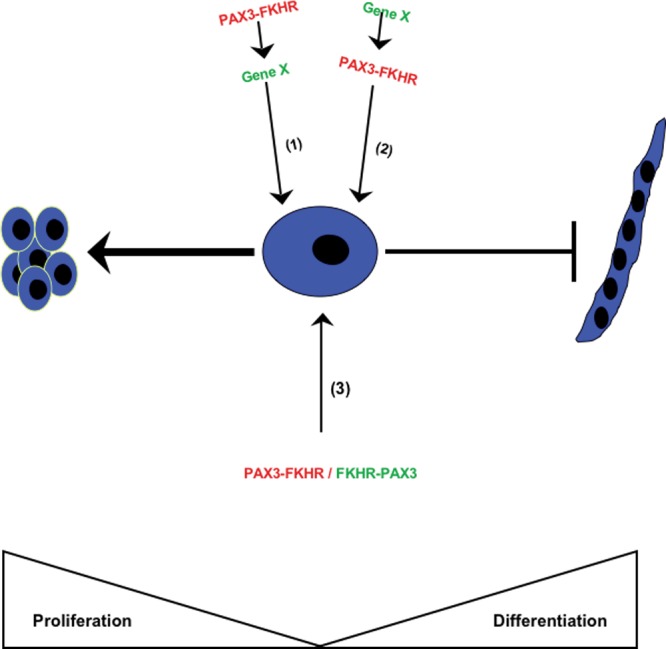
Three putative mechanisms of chromosomal translocation initiating events in ARMS development. (1) ARMS forms as a result of de novo translocation leading to the formation of PAX3-FKHR oncoprotein that convert cells into a permissible state for acquiring additional mutations. (2) ARMS forms from cells with existing genetic mutation that predisposes to chromosomal translocation. (3)De novo translocation leading to the co-expression of PAX3-FKHR and FKHR-PAX3 is sufficient to initiate the ARMS phenotype. The non-translocation mutations shown in the first two models may be variable, contributing to the heterogeneity of ARMS.
FKHR-PAX3 Fusion Gene: Just a Byproduct?
Reciprocal translocations can lead to the formation of two fusion genes. One fusion product is considered to be oncogenic, whereas the reciprocal product is often not detected or is detected in only a percentage of the tumors. All t(2;13)-positive ARMS express the oncogenic PAX3-FKHR protein. The reciprocal FKHR-PAX3 transcript is detected in ~40 to 60% of ARMS tumors tested, but the status of protein expression is unknown (Barr et al., 1998; Frascella et al., 1998). The inconsistent FKHR-PAX3 mRNA expression suggests that it may not have a role in ARMS. This could be a misconception, since primary tumors have already accumulated numerous secondary mutations at presentation. Analysis of recent data shows that some leukemia-associated reciprocal products are themselves not oncogenic but work in concert with the oncogenic partner in determining tumor pathology and lineage specificity, thus renewing interest in the role of other reciprocal products in human cancers (Rego and Pandolfi, 2002; Zheng et al., 2009). In the absence of animal models that faithfully recapitulate the entire spectrum of human ARMS, a role for FKHR-PAX3 during tumor initiation cannot be ruled out (Fig. 3).
Disrupted Balance Between Growth And Differentiation
Proliferation and differentiation are mutually exclusive events mediated by opposing cellular signals. In muscle, this balance is largely controlled through the actions of cell-cycle regulators and myogenic regulatory factors (MRFs). While cell-cycle and myogenesis pathways are mostly independently regulated, a coordinated interaction between proliferation and myogenic pathways is critical for normal development of mature muscle fibers. RMS cells exhibit aberrant regulation of several of these pathways, which could underlie the mechanistic basis for uncontrolled growth and failed differentiation.
Growth Defects
RMS cells display many defects in cell-cycle checkpoints and growth factor signaling pathways, leading to accelerated proliferation, typically at the G1-S transition. Notable changes include elevated expression of positive cell-cycle regulators such as cyclin D types (Zhang et al., 2004), cdk4 (Ragazzini et al., 2004), and Skp2 (Zhang and Wang, 2007), as well as repression of negative cell-cycle regulators such as cyclin inhibitors p16Ink4a (Iolascon et al., 1996; Obana et al., 2003), p21 (Moretti et al., 2002), p27 (Zhang and Wang, 2003), and p57 (Roeb et al., 2007). Ink4A and cdk4 mutations occur more frequently in ARMS. Several mechanisms explain the direct effects of PAX3-FKHR on these cell-cycle regulators: PAX3-FKHR silences p16Ink4a expression by increasing methylation of the Ink4A promoter (Linardic et al., 2007), up-regulates Skp2 transcription through activating a novel E2F complex binding to the Skp2 promoter (Zhang and Wang, 2007), and enhances degradation of ERG1 transcription factor involved in p57 gene activation (Roeb et al., 2007). Several dysregulated growth factor signaling pathways further augment the cell-cycle regulator-mediated growth effects in RMS. The most frequently affected pathways include signaling controlled by IGF, FGF, HGF, and PDGF. Dysregulated FGF and HGF pathways are linked to increased invasion and metastasis (Taulli et al., 2006; Taylor et al., 2009). PAX3-FKHR is known to activate these pathways by transcriptional activation of the corresponding receptor genes, IGFR1, FGFR4, c-met, and PDGFαR (Epstein et al., 1996, 1998; Ayalon et al., 2001; Cao et al., 2010). The ability of PAX3-FKHR to target both cell-cycle checkpoints and mitogenic signaling pathways contributes to ARMS aggressiveness.
Differentiation Defects
Myogenesis occurs through an orderly cascade of events involving myogenic regulatory factor (MRF) family and the myocyte enhancer factor-2 (MEF2) family (Perry and Rudnick, 2000; Pownall et al., 2002). The onset of myogenesis is controlled through a sequential activation of the MRFs, beginning with MyoD/Myf5 (muscle specification factors) followed by myogenin (muscle differentiation factor), and finally by Myf6 (muscle maturation factor). MEF2 factors work in concert with MRFs to stimulate transcription of muscle-specific structural genes. Although RMS cells express MyoD, myogenin, and MEF2, they fail to complete myogenesis under differentiation conditions. RMS-expressed MRFs and MEF2 bind their cognate DNA sequences in in vitro binding assays, but they cannot activate reporter genes containing the same binding sites (Tapscott et al., 1993). This implies that these factors are kept inactive. Recent studies suggest that increased FGF and TGFβ-signaling inhibits MRF and MEF2 function. FGF induces MEF2 phosphorylation that blocks MEF2 DNA-binding activity (Li et al., 1992). Although FGF level is not affected in RMS, over-expression of FGFR4 may lead to increased FGF signaling and consequent inactivation of MEF2 in RMS cells. Myostatin and TGFβ1, members of the TGFβ superfamily, are over-expressed in RMS cells. Since HGF can induce myostatin expression in normal muscle cells (Yamada et al., 2010), elevated myostatin in RMS may result from amplified signaling through the HGF receptor (c-met) that is frequently over-expressed in these cells. Myostatin and TGFβ1 inhibit myogenesis by repressing MyoD and MEF2 transcription function, respectively (Liu et al., 2004; Yamada et al., 2010). One proposed mechanism for these inhibitory effects of TGFβ members involves down-regulation of pro-myogenic microRNAs, including miR-1, miR-206, and miR-29. These microRNAs negatively regulate HDAC4, a repressor of MyoD and MEF2 transcription activities (Winbanks et al., 2011).
Etiology of Rhabdomyosarcoma
The issue of RMS cellular origin remains controversial. Because RMS tumors express markers of developing skeletal muscle, it is thought that they arise from myogenic cells. Some tumors, however, develop in organs that lack a skeletal muscle component, thus raising the possibility that RMS could develop from non-muscle cells. Given the heterogeneity of RMS tumors and their variable clinical presentation, identification of cell-of-origin is important to understanding RMS biology as well as for improving patient prognosis and treatment. Current knowledge on RMS cellular origin comes from two general approaches: xenograft mouse models with genetically modified cells of defined lineages, and genetically engineered mice. Analysis of these data suggests that multiple cell origins are involved in RMS tumorigenesis (Fig. 4), and points to the important contributions of both cell type and specific genetic alteration in defining the RMS subtype development.
Figure 4.

Model of multiple cells-of-origin of RMS development based on xenograft and genetically engineered animal studies. The diagram depicts three general stages in muscle development: specification, where activation of MRFs commits cells to myogenic lineage (e.g., satellites); determination, where committed muscle cells lose self-renewing capacity (e.g., myoblasts); and differentiation, where cells acquire muscle-specific phenotype (e.g., myotubes) and muscle-specific gene expression (e.g., myogenin, Myf6, myosin). RMS heterogeneity may result from different muscle development stages when the initial oncogenic event occurs as well as from different cooperating primary and secondary genetic events involved in tumor initiation.
Xenograft Models
Studies have shown that genetically manipulated satellite and MSC cultured cells can induce RMS tumor formation when implanted into immunodeficient mice. Linardic et al. (2005) have shown that concomitant expression of T-antigen/Ras/hTERT genes in post-natal human satellite cells gives rise to ERMS-like tumors. ARMS-like tumors can develop from PAX3-FKHR expressing MSC and satellite cells. Ren et al. (2008) have shown that MSC expressing PAX3-FKHR in the presence of T-antigen, H-Ras activation, and/or p53 mutation gives rise to ARMS. The finding that ARMS tumors can develop from non-myogenic MSC cells is significant, due to their widespread distribution throughout the body, and could account, in part, for tumor development at sites that lack skeletal muscle. Murine satellite C2 cells expressing PAX3-FKHR induce ARMS-like tumors in nude mice without bringing in additional genetic mutation (Zhang et al., 2009). It is worth noting that C2 is a spontaneously immortalized line that likely has acquired unknown genetic modifications that cooperate with PAX3-FKHR for promoting ARMS tumors; however, they do not carry mutations known to cooperate with PAX3-FKHR in other experimental models (Keller et al., 2004a; Ren et al., 2008).
Genetically Engineered Models
The major drawback of xenograft tumor models is the reliance on cell lines with unknown genetic modifications. Additionally, cell-line-induced tumors do not reflect the architectural complexity of in vivo tumors. Genetically engineered models with germ-line or conditional mutations are more likely to mirror the natural history of RMS development (see De Giovanni et al., 2009, for review). This article focuses only on models that provide direct information on the cellular origin of RMS.
Using a conditional knock-in approach to control temporal-and lineage-specific PAX3-FKHR expression from the PAX3 locus, Keller et al. (2004a,b) have shown that ARMS develops in mice only when PAX3-FKHR expression is activated in Myf6+ cells. Activation of PAX3-FKHR in PAX7+ cells that represent embryonic muscle stem cells and the majority of satellite cells in adult muscle reduces satellite cell number but does not induce ARMS. These findings are intriguing from two aspects:
(1) ARMS develop from Myf6+ cells that are thought to represent mature muscle fibers, yet these tumors lack muscle characteristics that are typically observed in human ARMS. The authors suggest that the ARMS tumors in the Myf6+ model could also arise from a sub-population of myogenic progenitor cells that transiently express Myf6+ but do not contribute to muscle development during embryogenesis. This conditional model cannot distinguish these two cell populations. Nevertheless, the concept of mature muscle as the origin of ARMS is supported by a Drosophila model, which has a much better characterized genetic program for myogenic lineage specification. In this model, the authors show that ARMS develops when PAX7-FKHR is conditionally induced in syncytial muscle fibers (Galindo et al., 2006). An alternative explanation for the lack of muscular characteristics in the Myf6+-driven ARMS tumors is that PAX3-FKHR has the potential to de-differentiate mature muscle cells.
(2) The lack of ARMS in the PAX7+ cell lineage challenges the findings in the C2 xenograft model, because C2 cells are thought to resemble satellite cells. Keller’s model produces ARMS at a low frequency unless p53 or INK4A/ARF is mutated. One possible explanation is that other types of cooperating events are needed for PAX3-FKHR-induced ARMS in satellite cells, as previously mentioned. Another possibility is that only satellite cells with low PAX7 or capable of losing PAX7 expression can be coaxed into rhabdomyosarcomagenesis. It has been reported that satellite progenitor cells prepared from human muscle fibers contain both PAX7− and PAX7+ populations (Reimann et al., 2004). Furthermore, PAX3-FKHR represses PAX7 expression in ERMS (Tomescu et al., 2004) and C2 cells (C. Wang, unpublished observations); however, down-regulation of PAX7 by PAX3-FKHR is not observed in Keller’s model. Although it is not clear why PAX7 expression is resistant to repression by PAX3-FKHR in Keller’s model, the high level of PAX7 might offset the oncogenic potency of the fusion protein. More research is needed to clarify the identity and role of secondary mutations and cell lineage in ARMS initiation and development.
Rubin et al. (2011) have reported that ERMS tumors are formed in a series of Ptch (+/−) and/or p53 (−/−) engineered mice with conditionally targeted expression in PAX3+ cells (pre- and post-natal myogenic progenitor), PAX7+ cells (satellite), or Myf6+ cells (mature muscle). All lineages in Ptch (+/−)/p53 (−/−) mice develop mesenchymal tumors, of which various percentages are of the ERMS type. Strikingly, mice with the p53 mutation in Myf6+ mature muscle cells are 100% ERMS. The authors suggest that the precise pairing of cell lineage with specific mutations is key to determining the type of sarcoma that develops. The degree of differentiation observed in these models seems to correlate with the targeted cell lineages, with PAX7+ satellite cell-derived tumors displaying the least myogenic differentiation and Myf6+ cell-derived tumors showing the most myogenic differentiation. The various extents of myogenesis in the mouse ERMS correlate well with the differentiation spectrum observed in human ERMS. The high degree of myogenesis in Myf6+-derived ERMS is in stark contrast to the lack of myogenesis in Myf6+-derived ARMS. The difference might reflect the different Myf6+ cell populations being targeted for ERMS vs. ARMS. Alternatively, the degree of myogenesis in the RMS may depend on the specific combination of genetic mutations.
Targeted Cancer Therapy
Several target-specific compounds have been identified that are effective in suppressing the growth and tumorigenesis of RMS cells. These compounds include inhibitors for receptor tyrosine kinases (Crose and Linardic, 2011), intracellular signaling molecules (Martins et al., 2011), and angiogenic factors (Bid and Houghton, 2011). Many of these drugs and analogs are in clinical trials. Because PAX3-FKHR is specific for ARMS, investigators have focused on developing reagents that directly alter its expression or function. These reagents include siRNAs (Bernasconi et al., 1996), transcription repressors (Fredericks et al., 2001), vaccines against peptide spanning the fusion region (Rodeberg et al., 2005), and kinase inhibitors (Zeng et al., 2010). Readers are referred to a comprehensive review on these topics for more information (Wachtel and Schafer, 2010).
This article focuses on the therapeutic value of “differentiation therapy” for treating RMS. Differentiation therapy aims to suppress tumor behavior by forcing cancer cells to terminally differentiate with a concomitant reduction in proliferation. There is evidence that RMS cells are susceptible to such therapeutic interventions. For example, some RMS cells differentiate upon treatment with retinoic acid (Crouch and Helman, 1991) or TPA (Aguanno et al., 1990; Bouche et al., 1993). Introduction of a constitutively active MKK6 induces myogenesis in RMS cells by reactivating the p38 MAPK that is normally required for muscle differentiation but is impaired in RMS cells (Puri et al., 2000). However, these approaches work only on selected RMS cell lines, suggesting that the myogenic defects in RMS are heterogeneous, involving multiple, most likely parallel, pathways. Since RMS cells can be coaxed into differentiation, there must be common downstream effectors in the various defective pathways, which are susceptible for reactivation in all RMS cell types. Indeed, introduction of late-stage effectors such as Myf6 (Sirri et al., 2003) and microRNAs (Wang et al., 2008; Taulli et al., 2009; Yan et al., 2009; Rao et al., 2010) restores myogenesis in many RMS cell lines. Re-expression of miR-29 and miR-1/206 in RMS cells also blocks tumor formation in mice. Additionally, Taulli et al. (2009) showed that activation of miR1/206 in xenograft tumors attenuates further tumor expansion.
One of the challenges of chemical-based therapies is the risk that cancer cells will develop drug resistance or activate alternative pathways to overcome the drug effects. The high frequency of overactive IGF/IGFR signaling in RMS has sparked interest in testing IGFR inhibitors in clinical trials. However, analysis of recent data shows that RMS cells treated with IGFR inhibitors become resistant through up-regulation of EGFR/Her1 or Her2/Neu receptor (Huang et al., 2009; Abraham et al., 2011). Since microRNAs occur naturally in cells and target multiple genes and pathways, they may represent unique opportunities for therapeutic intervention that requires lower dosage with minimal side effects. MicroRNA-related research in RMS is still in its infancy. Recent profiling studies comparing microRNA expression patterns in RMS subtypes and normal skeletal muscle have expanded the number of microRNA candidates that can be further explored (Wei et al., 2009; Sarver et al., 2010).
Concluding Remarks
There has been tremendous progress made in the understanding of RMS biology since the identification of associated genetic defects. Considering the high degree of heterogeneity in RMS, there remain many aspects of RMS pathogenesis in need of clarification, such as (1) developing additional genetically relevant pre-clinical animal models that reflect the diverse RMS groups, (2) establishing the origin and evolution of different RMS phenotypes, (3) defining the spectrum of cooperating genetic and molecular events that drive RMS initiation and progression, and (4) determining if chromosomal translocation is the first step in the transformation of MSC or myogenic cells to cancerous ARMS cells. This information is critical for assessing the heterogeneity of clinical responses, and for designing the most effective molecular targeted therapies for RMS treatment.
Acknowledgments
I thank Dr. Reed Graves for his valuable suggestions in the preparation of this manuscript. I apologize for the omission of topics and references due to space limitations.
Footnotes
This work is supported by an NIH grant (CA074904) to C.W.
The author declares no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Abraham J, Prajapati SI, Nishijo K, Schaffer BS, Taniguchi E, Kilcoyne A, et al. (2011). Evasion mechanisms to igf1r inhibition in rhabdomyosarcoma. Mol Cancer Ther 10:697-707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguanno S, Bouchè M, Adamo S, Molinaro M. (1990). 12-O-tetradecanoyl phorbol-13-acetate-induced differentiation of a human rhabdomyosarcoma cell line. Cancer Res 50:3377-3382 [PubMed] [Google Scholar]
- Anderson J, Gordon A, Pritchard-Jones K, Shipley J. (1999). Genes, chromosomes, and rhabdomyosarcoma. Genes Chromosomes Cancer 26: 275-285 [PubMed] [Google Scholar]
- Anderson MJ, Shelton GD, Cavenee WK, Arden KC. (2001). Embryonic expression of the tumor-associated PAX3-FKHR fusion protein interferes with the developmental functions of Pax3. Proc Natl Acad Sci USA 98:1589-1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayalon D, Glaser T, Werner H. (2001). Transcriptional regulation of IGF-I receptor gene expression by the PAX3-FKHR oncoprotein. Growth Horm IGF Res 11:289-297 [DOI] [PubMed] [Google Scholar]
- Barr FG, Galili N, Holick J, Biegel JA, Rovera G, Emanuel BS. (1993). Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nature Genetics 3:113-117 [DOI] [PubMed] [Google Scholar]
- Barr FG, Nauta EL, Hollows CJ. (1998). Structural analysis of PAX3 genomic rearrangements in alveolar rhabdomyosarcoma. Cancer Genet Cytogenet 102:32-39 [DOI] [PubMed] [Google Scholar]
- Barr FG, Qualman SJ, Macris MH, Melnyk N, Lawlor ER, Strzelecki DM, et al. (2002). Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res 62:4704-4710 [PubMed] [Google Scholar]
- Begum S, Emami N, Cheung A, Wilkins O, Der S, Hamel PA. (2005). Cell-type-specific regulation of distinct sets of gene targets by Pax3 and Pax3/FKHR. Oncogene 24:1860-1872 [DOI] [PubMed] [Google Scholar]
- Bernasconi M, Remppis A, Fredericks WJ, Rauscher FJ, 3rd, Schäfer BW. (1996). Induction of apoptosis in rhabdomyosarcoma cells through down-regulation of PAX proteins. Proc Natl Acad Sci USA 93:13164-13169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bid HK, Houghton PJ. (2011). Targeting angiogenesis in childhood sarcomas. Sarcoma 10.1155/2011/601514 10.1155/2011/601514 [Epub ahead of print, Dec 10, 2010]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenkamp KU, Coffer PJ. (2003). Regulation of cell survival and proliferation by the FOXO (Forkhead box, class O) subfamily of Forkhead transcription factors. Biochem Soc Trans 31(Pt 1):292-297 [DOI] [PubMed] [Google Scholar]
- Bouche M, Senni MI, Grossi AM, Zappelli F, Polimeni M, Arnold HH, et al. (1993). TPA-induced differentiation of human rhabdomyosarcoma cells: expression of the myogenic regulatory factors. Exp Cell Res 208:209-217 [DOI] [PubMed] [Google Scholar]
- Buckingham M, Relaix F. (2007). The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu Rev Cell Dev Biol 23:645-673 [DOI] [PubMed] [Google Scholar]
- Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, et al. (2010). Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res 70:6497-6508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crose LE, Linardic CM. (2011). Receptor tyrosine kinases as therapeutic targets in rhabdomyosarcoma. Sarcoma 10.1155/2011/756982 10.1155/2011/756982 [Epub ahead of print, Jan 2, 2011]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch GD, Helman LJ. (1991). All-trans-retinoic acid inhibits the growth of human rhabdomyosarcoma cell lines. Cancer Res 51:4882-4887 [PubMed] [Google Scholar]
- De Pitta C, Tombolan L, Albiero G, Sartori F, Romualdi C, Jurman G, et al. (2006). Gene expression profiling identifies potential relevant genes in alveolar rhabdomyosarcoma pathogenesis and discriminates PAX3-FKHR positive and negative tumors. Int J Cancer 118:2772-2781 [DOI] [PubMed] [Google Scholar]
- De Giovanni C, Landuzzi L, Nicoletti G, Lollini PL, Nanni P. (2009). Molecular and cellular biology of rhabdomyosarcoma. Future Oncol 5:1449-1475 [DOI] [PubMed] [Google Scholar]
- del Peso L, Gonzalez VM, Hernandez R, Barr FG, Nunez G. (1999). Regulation of the forkhead transcription factor FKHR, but not the PAX3-FKHR fusion protein, by the serine/threonine kinase Akt. Oncogene 18:7328-7333 [DOI] [PubMed] [Google Scholar]
- Dias P, Chen B, Dilday B, Palmer H, Hosoi H, Singh S, et al. (2000). Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol 156:399-408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein JA, Lam P, Jepeal L, Maas RL, Shapiro DN. (1995). Pax3 inhibits myogenic differentiation of cultured myoblast cells. J Biol Chem 270:11719-11722 [DOI] [PubMed] [Google Scholar]
- Epstein JA, Shapiro DN, Cheng J, Lam PY, Maas RL. (1996). Pax3 modulates expression of the c-Met receptor during limb muscle development. Proc Natl Acad Sci USA 93:4213-4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein JA, Song B, Lakkis M, Wang C. (1998). Tumor-specific PAX3-FKHR transcription factor, but not PAX3, activates the platelet-derived growth factor alpha receptor. Mol Cell Biol 18:4118-4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frascella E, Toffolatti L, Rosolen A. (1998). Normal and rearranged PAX3 expression in human rhabdomyosarcoma. Cancer Genet Cytogenet 102:104-109 [DOI] [PubMed] [Google Scholar]
- Fredericks WJ, Galili N, Mukhopadhyay S, Rovera G, Bennicelli J, Barr FG, et al. (1995). The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol 15:1522-1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericks WJ, Ayyanathan K, Rauscher FJ., 3rd (2001). Regulating the neoplastic phenotype using engineered transcriptional repressors. Cancer Lett 162(Suppl):23S-32S [DOI] [PubMed] [Google Scholar]
- Galindo RL, Allport JA, Olson EN. (2006). A Drosophila model of the rhabdomyosarcoma initiator PAX7-FKHR. Proc Natl Acad Sci USA 103:13439-13444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gougelet A, Perez J, Pissaloux D, Besse A, Duc A, Decouvelaere AV, et al. (2011). miRNA profiling: how to bypass the current difficulties in the diagnosis and treatment of sarcomas. Sarcoma 10.1155/2011/460650 10.1155/2011/460650 [Epub ahead of print, Feb 22, 2011]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbach AD, Sublett JE, McPherson CJ, Grosveld G. (1999). The Pax3-FKHR oncoprotein is unresponsive to the Pax3-associated repressor hDaxx. EMBO J 18:3702-3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Greer A, Hurlburt W, Han X, Hafezi R, Wittenberg GM, et al. (2009). The mechanisms of differential sensitivity to an insulin-like growth factor-1 receptor inhibitor (BMS-536924) and rationale for combining with EGFR/HER2 inhibitors. Cancer Res 69:161-170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iolascon A, Faienza MF, Coppola B, Rosolen A, Basso G, Della Ragione F, et al. (1996). Analysis of cyclin-dependent kinase inhibitor genes (CDKN2A, CDKN2B, and CDKN2C) in childhood rhabdomyosarcoma. Genes Chromosomes Cancer 15:217-222 [DOI] [PubMed] [Google Scholar]
- Kazanowska B, Reich A, Stegmaier S, Bekassy AN, Leuschner I, Chybicka A, et al. (2007). Pax3-fkhr and pax7-fkhr fusion genes impact outcome of alveolar rhabdomyosarcoma in children. Fetal Pediatr Pathol 26:17-31 [DOI] [PubMed] [Google Scholar]
- Keller C, Hansen MS, Coffin CM, Capecchi MR. (2004a). Pax3-Fkhr interferes with embryonic Pax3 and Pax7 function: implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev 18:2608-2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, Capecchi MR. (2004b). Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev 18:2614-2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J, Bittner ML, Saal LH, Teichmann U, Azorsa DO, Gooden GC, et al. (1999). cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc Natl Acad Sci USA 96:13264-13269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagha M, Sato T, Bajard L, Daubas P, Esner M, Montarras D, et al. (2008). Regulation of skeletal muscle stem cell behavior by Pax3 and Pax7. Cold Spring Harb Symp Quant Biol 73:307-315 [DOI] [PubMed] [Google Scholar]
- Lagutina I, Conway SJ, Sublett J, Grosveld GC. (2002). Pax3-FKHR knock-in mice show developmental aberrations but do not develop tumors. Mol Cell Biol 22:7204-7216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam PY, Sublett JE, Hollenbach AD, Roussel MF. (1999). The oncogenic potential of the Pax3-FKHR fusion protein requires the Pax3 homeodomain recognition helix but not the Pax3 paired-box DNA binding domain. Mol Cell Biol 19:594-601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhou J, James G, Heller-Harrison R, Czech MP, Olson EN. (1992). FGF inactivates myogenic helix-loop-helix proteins through phosphorylation of a conserved protein kinase C site in their DNA-binding domains. Cell 71:1181-1194 [DOI] [PubMed] [Google Scholar]
- Linardic CM, Downie DL, Qualman S, Bentley RC, Counter CM. (2005). Genetic modeling of human rhabdomyosarcoma. Cancer Res 65:4490-4495 [DOI] [PubMed] [Google Scholar]
- Linardic CM, Naini S, Herndon JE, 2nd, Kesserwan C, Qualman SJ, Counter CM. (2007). The PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescence. Cancer Res 67:6691-6699 [DOI] [PubMed] [Google Scholar]
- Liu D, Kang JS, Derynck R. (2004). TGF-beta-activated Smad3 represses MEF2-dependent transcription in myogenic differentiation. EMBO J 23:1557-1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins AS, Olmos D, Missiaglia E, Shipley J. (2011). Targeting the insulin-like growth factor pathway in rhabdomyosarcomas: rationale and future perspectives. Sarcoma 10.1155/2011/209736 10.1155/2011/209736 [Epub ahead of print, Mar 3, 2011]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado GE, Xia SJ, Zhang C, Ahn EH, Gustafson DM, Lae M, et al. (2008). Identification of PAX3-FKHR-regulated genes differentially expressed between alveolar and embryonal rhabdomyosarcoma: focus on MYCN as a biologically relevant target. Genes Chromosomes Cancer 47:510-520 [DOI] [PubMed] [Google Scholar]
- Miller PJ, Hollenbach AD. (2007). The oncogenic fusion protein Pax3-FKHR has a greater post-translational stability relative to Pax3 during early myogenesis. Biochim Biophys Acta 1770:1450-1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti A, Borriello A, Monno F, Criscuolo M, Rosolen A, Esposito G, et al. (2002). Cell division cycle control in embryonal and alveolar rhabdomyosarcomas. Eur J Cancer 38:2290-2299 [DOI] [PubMed] [Google Scholar]
- Obana K, Yang HW, Piao HY, Taki T, Hashizume K, Hanada R, et al. (2003). Aberrations of p16INK4A, p14ARF and p15INK4B genes in pediatric solid tumors. Int J Oncol 23:1151-1157 [PubMed] [Google Scholar]
- Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. (2009). Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer 115:4218-4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RL, Rudnick MA. (2000). Molecular mechanisms regulating myogenic determination and differentiation. Front Biosci 5:D750-D767 [DOI] [PubMed] [Google Scholar]
- Pownall ME, Gustafsson MK, Emerson CP., Jr (2002). Myogenic regulatory factors and the specification of muscle progenitors in vertebrate embryos. Annu Rev Cell Dev Biol 18:747-783 [DOI] [PubMed] [Google Scholar]
- Puri PL, Wu Z, Zhang P, Wood LD, Bhakta KS, Han J, et al. (2000). Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev 14:574-584 [PMC free article] [PubMed] [Google Scholar]
- Ragazzini P, Gamberi G, Pazzaglia L, Serra M, Magagnoli G, Ponticelli F, et al. (2004). Amplification of CDK4, MDM2, SAS and GLI genes in leiomyosarcoma, alveolar and embryonal rhabdomyosarcoma. Histol Histopathol 19:401-411 [DOI] [PubMed] [Google Scholar]
- Rao PK, Missiaglia E, Shields L, Hyde G, Yuan B, Shepherd CJ, et al. (2010). Distinct roles for miR-1 and miR-133a in the proliferation and differentiation of rhabdomyosarcoma cells. FASEB J 24:3427-3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego EM, Pandolfi PP. (2002). Reciprocal products of chromosomal translocations in human cancer pathogenesis: key players or innocent bystanders? Trends Mol Med 8:396-405 [DOI] [PubMed] [Google Scholar]
- Reimann J, Brimah K, Schroder R, Wernig A, Beauchamp JR, Partridge TA. (2004). Pax7 distribution in human skeletal muscle biopsies and myogenic tissue cultures. Cell Tissue Res 315:233-242 [DOI] [PubMed] [Google Scholar]
- Ren YX, Finckenstein FG, Abdueva DA, Shahbazian V, Chung B, Weinberg KI, et al. (2008). Mouse mesenchymal stem cells expressing PAX-FKHR form alveolar rhabdomyosarcomas by cooperating with secondary mutations. Cancer Res 68:6587-6597 [DOI] [PubMed] [Google Scholar]
- Rodeberg DA, Nuss RA, Heppelmann CJ, Celis E. (2005). Lack of effective T-lymphocyte response to the PAX3/FKHR translocation area in alveolar rhabdomyosarcoma. Cancer Immunol Immunother 54:526-534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeb W, Boyer A, Cavenee WK, Arden KC. (2007). PAX3-FOXO1 controls expression of the p57Kip2 cell-cycle regulator through degradation of EGR1. Proc Natl Acad Sci USA 104:18085-18090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin BP, Nishijo K, Chen HI, Yi X, Schuetze DP, Pal R, et al. (2011). Evidence for an unanticipated relationship between undifferentiated pleomorphic sarcoma and embryonal rhabdomyosarcoma. Cancer Cell 19:177-191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab R, Spunt SL, Skapek SX. (2011). Myogenesis and rhabdomyosarcoma: the Jekyll and Hyde of skeletal muscle. Curr Top Dev Biol 94:197-234 [DOI] [PubMed] [Google Scholar]
- Sarver AL, Phalak R, Thayanithy V, Subramanian S. (2010). S-MED: Sarcoma microRNA expression database. Lab Invest 90:753-761 [DOI] [PubMed] [Google Scholar]
- Scheidler S, Fredericks WJ, Rauscher FJ, 3rd, Barr FG, Vogt PK. (1996). The hybrid PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma transforms fibroblasts in culture. Proc Natl Acad Sci USA 93:9805-9809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrable H, Witte D. (1989). Molecular differential pathology of rhabdomyosarcoma. Genes Chromosome Cancer 1:23-35 [DOI] [PubMed] [Google Scholar]
- Shapiro DN, Sublett JE, Li B, Downing JR, Naeve CW. (1993). Fusion of PAX3 to a member of the Forkhead family of transcripton factors in human alveolar rhabdomyosarcoma. Cancer Research 53:5108-5112 [PubMed] [Google Scholar]
- Sirri V, Leibovitch MP, Leibovitch SA. (2003). Muscle regulatory factor MRF4 activates differentiation in rhabdomyosarcoma RD cells through a positive-acting C-terminal protein domain. Oncogene 22:5658-5666 [DOI] [PubMed] [Google Scholar]
- Sorensen PH, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, Maurer HM, et al. (2002). PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol 20:2672-2679 [DOI] [PubMed] [Google Scholar]
- Sumegi J, Streblow R, Frayer RW, Dal Cin P, Rosenberg A, Meloni-Ehrig A, et al. (2010). Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes Chromosomes Cancer 49:224-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott SJ, Thayer MJ, Weintraub H. (1993). Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science 259:1450-1453 [DOI] [PubMed] [Google Scholar]
- Taulli R, Scuoppo C, Bersani F, Accornero P, Forni PE, Miretti S, et al. (2006). Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res 66:4742-4749 [DOI] [PubMed] [Google Scholar]
- Taulli R, Bersani F, Foglizzo V, Linari A, Vigna E, Ladanyi M, et al. (2009). The muscle-specific microRNA miR-206 blocks human rhabdomyosarcoma growth in xenotransplanted mice by promoting myogenic differentiation. J Clin Invest 119:2366-2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JG, 6th, Cheuk AT, Tsang PS, Chung JY, Song YK, Desai K, et al. (2009). Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest 119:3395-3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomescu O, Xia SJ, Strezlecki D, Bennicelli JL, Ginsberg J, Pawel B, et al. (2004). Inducible short-term and stable long-term cell culture systems reveal that the PAX3-FKHR fusion oncoprotein regulates CXCR4, PAX3, and PAX7 expression. Lab Invest 84:1060-1070 [DOI] [PubMed] [Google Scholar]
- Turc-Carel C, Lizard-Nacol S, Justrabo E, Favrot M, Tabone E. (1986). Consistent chromosomal translocation in alveolar rhabdomyosarcoma. Cancer Genetics Cytogenet 19:361-362 [DOI] [PubMed] [Google Scholar]
- Wachtel M, Schafer BW. (2010). Targets for cancer therapy in childhood sarcomas. Cancer Treat Rev 36:318-327 [DOI] [PubMed] [Google Scholar]
- Wachtel M, Dettling M, Koscielniak E, Stegmaier S, Treuner J, Simon-Klingenstein K, et al. (2004). Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res 64:5539-5545 [DOI] [PubMed] [Google Scholar]
- Wang H, Garzon R, Sun H, Ladner KJ, Singh R, Dahlman J, et al. (2008). NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 14:369-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang-Wuu S, Soukup S, Ballard E, Gotwals B, Lampkin B. (1988). Chromosomal analysis of sixteen human rhabdomyosarcomas. Cancer Res 48:983-987 [PubMed] [Google Scholar]
- Wei JS, Johansson P, Chen QR, Song YK, Durinck S, Wen X, et al. (2009). MicroRNA profiling identifies cancer specific and prognostic signatures in pediatric malignancies. Clin Cancer Res 15:5560-5568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggan O, Taniguchi-Sidle A, Hamel AP. (1998). Interaction of the pRB-family proteins with factors containing paired-like homeodomains. Oncogene 16:227-236 [DOI] [PubMed] [Google Scholar]
- Williamson D, Missiaglia E, de Reyniès A, Pierron G, Thuille B, Palenzuela G, et al. (2010). Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol 28:2151-2158 [DOI] [PubMed] [Google Scholar]
- Winbanks CE, Wang B, Beyer C, Koh P, White L, Kantharidis P, et al. (2011). TGF-beta regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. J Biol Chem 286:13805-13814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Tatsumi R, Yamanouchi K, Hosoyama T, Shiratsuchi S, Sato A, et al. (2010). High concentrations of HGF inhibit skeletal muscle satellite cell proliferation in vitro by inducing expression of myostatin: a possible mechanism for reestablishing satellite cell quiescence in vivo. Am J Physiol Cell Physiol 298:C465-C476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Dong Xda E, Chen X, Wang L, Lu C, Wang J, et al. (2009). MicroRNA-1/206 targets c-Met and inhibits rhabdomyosarcoma development. J Biol Chem 284:29596-29604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng FY, Dong H, Cui J, Liu L, Chen T. (2010). Glycogen synthase kinase 3 regulates PAX3-FKHR-mediated cell proliferation in human alveolar rhabdomyosarcoma cells. Biochem Biophys Res Commun 391: 1049-1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hu S, Schofield DE, Sorensen PH, Triche TJ. (2004). Selective usage of D-Type cyclins by Ewing’s tumors and rhabdomyosarcomas. Cancer Res 64:6026-6034 [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang C. (2003). PAX3-FKHR transformation increases 26 S proteasome-dependent degradation of p27Kip1, a potential role for elevated Skp2 expression. J Biol Chem 278:27-36 [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang C. (2006). F-box protein Skp2: a novel transcriptional target of E2F. Oncogene 25:2615-2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang C. (2007). Identification of a new class of PAX3-FKHR target promoters: a role of the Pax3 paired box DNA binding domain. Oncogene 26:1595-1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang C. (2011). Nephroblastoma overexpressed (NOV/CCN3) gene: a paired-domain-specific PAX3-FKHR transcription target that promotes survival and motility in alveolar rhabdomyosarcoma cells. Oncogene 10.1038/onc.2011.69 10.1038/onc.2011.69 [Epub ahead of print, Mar 21, 2011]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Schwartz J, Wang C. (2009). Comparative analysis of paired- and homeodomain-specific roles in PAX3-FKHR oncogenesis. Int J Clin Exp Pathol 2:370-383 [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Oancea C, Henschler R, Moore MA, Ruthardt M. (2009). Reciprocal t(9;22) ABL/BCR fusion proteins: leukemogenic potential and effects on B cell commitment. PLoS One 4:e7661. [DOI] [PMC free article] [PubMed] [Google Scholar]