

Abstract
A multiplexed peptide quantification strategy using the iTRAQ™ reagent has been described for relative measurements of peptides in digested protein mixtures. To validate the chemical specificity of the iTRAQ reaction, we have performed a detailed study of iTRAQ reactivity with two sets of synthetic peptides. The first set of peptides had sequences of Tyr-Xaa-Ser-Glu-Gly-Leu-Ser-Lys and Tyr-Xaa-Ser-Glu-Tyr-Leu-Ser-Lys where Xaa = Ala, Pro, Trp, Tyr, or Glu and was designed to study the extent of O-acylation by iTRAQ, especially hydroxyl-containing residues in different positions. The second set of peptides included Ala-Ser-Glu-His-Ala-Xaa-Tyr-Gly where Xaa = Ser, Thr, or Tyr and was selected to investigate the effect of histidyl residues separated by one amino acid residue from seryl, tyrosyl, or threonyl residues. Our findings indicated that in addition to variable levels of O-acylation of non-sequence-specific hydroxyl-containing residues, significant sequence-specific O-acylation of seryl, threonyl, and tyrosyl hydroxyls occurred when separated one residue removed from a histidyl residue; i.e., (Tyr/Ser)-Xaa-His or His-Xaa-(Tyr/Ser/Thr). This behavior was verified by a separate spiking experiment of one of the first set of peptides into E. coli protein extracts, followed by retention time targeted LC/MS/MS to demonstrate the occurrence of modifications in a complex mixture. These sequence-dependent O-acylation modifications can be confounding factors to accurate MS quantification. Reversal of peptide O-acylation by the iTRAQ reagent can be accomplished by reaction with hydroxylamine with virtually no cleavage of N-acylation and is a recommended modification of the iTRAQ protocol for many applications.
Keywords: quantitative proteomics, iTRAQ™, sequence-specific modification, peptide O-acylation, mass spectrometry, hydroxyl amino acid reactivity
Introduction
Differential proteomics depends on accurate and reproducible quantification of proteins or peptides. Because the wide variety of biological material to which this powerful approach can be applied makes a generalized quantification strategy difficult, multiple approaches have been devised and their utility depends upon the peculiarities of the samples and the goals of the experiment. In general, proteomic applications can be divided into two categories: those focusing on maintaining proteins intact during separation and quantification (“top-down”), and those analyzing either naturally occurring or enzymatically derived peptides (“bottom-up”). Because bottom-up approaches increase the complexity of the samples, chromatographic separation before mass spectrometric identification is typically important; however, losses due to chromatography are unavoidable and largely unpredictable. Such problems have dictated the need for pre-separation covalent modification of peptides using stable isotope-containing reagents to allow for normalization of the losses across experimental samples. Since quantification is ultimately relative, the assumption is that all stable isotope-labeled peptide cognates will behave similarly upon chromatographic separation and elution. Thus, the ratio of the relative intensities upon MS or MS/MS analysis will accurately reflect their abundance in their original protein state. Additionally, there are several other assumptions that further impact the validity of these claims. Two important assumptions are that the chemical modification reactions should proceed to completion and that the reactions are specific to targeted residues. Since successful quantification relies at the outset on specific, predictable, and saturating modifications of targeted residues, significant side-reactions should be avoided.
With this in mind, we have performed a study directed to address the reaction specificity of the iTRAQ chemistry1 realizing that the operative reaction should be the N-acylation of primary amines employing an NHS-ester reaction mechanism. The iTRAQ reagent has garnered interest by the proteomics community as evidenced by the many publications reporting its use since its commercial introduction. Along with its increased popularity, several papers have recently appeared evaluating its accuracy and reproducibility2-6, most notably, the work of Gan et al.2 and Quaglia et al.3, who describe reaction kinetics and the chemical/biological error rate.
In this report, we investigated the specificity of the iTRAQ reaction with particular attention to O-acylation since we had previously observed heightened reactivity of sequence-specific Tyr, Ser, and Thr hydroxyls using NHS-esters of biotin7, 8, both in model, as well as complex systems. Although Ross et al. described minimal iTRAQ reactivity with tyrosyl residue side-chains (<3%), they reported no sequence specific reactivity with tyrosyl, seryl, or threonyl residues1. In this report, we provide evidence of O-acylation of hydroxylated side-chains of amino acid residues with iTRAQ, especially in positions near histidyl residues in synthetic peptides that can significantly impact peptide quantification.
Experimental Procedures
Chemicals
All peptides used for this study were synthesized in-house using solid state Fmoc-technology with an Applied Biosystems 433 peptide synthesizer. Synthesized peptides were purified by C18 RP-HPLC and evaluated by MS for homogeneity and mass authenticity. The iTRAQ™ Reagent Methods Development Kit (consisting of the 4-plex mixture of reporters) was obtained from Applied Biosystems (Foster City, CA, USA). HPLC-grade acetonitrile was purchased from Fisher Scientific (Fair Lawn, NJ, USA). Boric acid was acquired from Mallinckrodt Laboratory Chemicals (Phillipsburg, NJ, USA). Hydroxylamine hydrochloride and TFA were purchased from Sigma-Aldrich (St. Louis, MO, USA). For MS analyses, alpha-cyano-4-hydroxycinnamic acid was purchased from Sigma-Aldrich and compressed air was purchased from Airgas (Texas City, TX, USA). Peptides used for calibrating the mass spectrometer (MS) were synthesized in-house in UTMB's Biomolecular Resource facility.
iTRAQ modification of synthetic peptides
All reactions were conducted using reagents and procedures provided by the Applied Biosystems'; kit unless otherwise indicated, with the exception that samples (synthetic and E. coli peptides) were precipitated after iTRAQ reaction. The reactions were run individually at room temperature using 50 μg of peptide in 15 μl of iTRAQ dissolution solution. At the end of the 2 h reaction time, 350 μl 0.5% TFA were added prior to RP-HPLC. As negative control, each peptide was dissolved under the same conditions as the reaction mixture except that no iTRAQ reagent was added. The molar ratio of iTRAQ reagent to reactive amino groups employed was 45:1 (personal communication, Applied Biosystems, Foster City, CA).
Separation by RP-HPLC
Each reaction mixture, including negative controls, were separated by RP-HPLC on an Agilent 1100 HPLC system (San Jose, CA, USA) using a Vydac C18 RP column (2.1 × 250 mm, 5 μm, 300 Å). The reaction mixture was loaded onto the column and washed for 10 min at 0.2 ml/min using buffer A (0.1 % TFA in Milli-Q water). The peptides were eluted with a linear gradient of 0-35% buffer B (0.1 % TFA in ACN) for 105 min at a flow rate of 0.2 ml/min with samples collected at 1 min intervals. Buffer B was then changed to 95% for 3 min and held at 95% for another 12 min. The column was re-equilibrated by decreasing the gradient to 0% B over 3 min and held at 0% for another 30 min. The samples exhibiting absorbance peaks at 215 nm were collected and analyzed by MALDI tandem time-of-flight and ESI/LC/MS/MS.
O-deacylation reaction with hydroxylamine-HCl
The reaction mixtures of peptides with iTRAQ reagent or collected fractions of HPLC-purified reaction mixtures were dried by vacuum centrifugation (Jouan Technology, Herbain, France). The samples were then dissolved in 10 μl of 0.1% TFA and reacted at 25°C for 5 h with 50 μl of hydroxylamine-HCl/boric acid buffer, pH 9.2 (0.8 M hydroxylamine-HCl in 0.1 M boric acid adjusted to pH 9.2 with NaOH).
MS analysis of selected peptides with iTRAQ tag(s) by MALDI-TOF/TOF
One μl of sample solution was spotted directly onto a MALDI target plate and allowed to dry, followed by 1 μl of matrix solution applied on the sample spot and allowed to dry. The matrix solution consisted of alpha-cyano-4-hydroxycinnamic acid dissolved in 50:50 ACN/water at 5 mg/ml. Data were acquired with an Applied Biosystems 4800 MALDI-TOF/TOF MS. The Applied Biosystem's software package included the 4000 Series Explorer (v. 3.6 RC1) with Oracle Database Schema Version (v. 3.19.0), Data Version (3.80.0) to acquire both MS and MS/MS spectral data. The instrument was operated in positive ion reflectron mode with mass range set at 850 – 3000 Da and the focus mass manually set at 1500 Da. For MS data, 2000 laser shots were acquired and averaged from several sites on each sample spot. Automatic external calibration was performed using a peptide mixture with reference masses 904.46 1296.68, 1570.67, and 2465.19 Da. MALDI MS was performed on ions indicative of peptides with one to four iTRAQ tags. A 1 kV positive ion MS/MS method was used to acquire data under post-source decay and/or collision-induced dissociation conditions. The instrument precursor selection window was +/− 3 Da. For MS/MS data 4000 laser shots were acquired and averaged from numerous sites on each sample spot. Automatic external calibration was performed using reference fragment masses 175.12, 480.25, 684.34, 1056.47, and 1441.63 Da from a precursor mass of 1570.70 Da.
Peptide spiking into E. coli extract
E. coli BL21 (DE3) cells were grown in LB media at 37° C to log phase. The harvested cell pellet was washed with PBS buffer once and was then lysed with a French press using 40 mM Tris and stored at −80°C. The extract was then centrifuged (100,000 × g for 4 h) and the protein supernatant precipitated with acetone and extracted with 8 M urea, 1% CHAPS and stored at −80°C until immediately needed. In preparation for iTRAQ labeling, the extract was re-precipitated in acetone and resuspended in iTRAQ reaction buffer and used for the study at a concentration of 10 mg/mL.
Reaction of E. coli proteins and synthetic peptide 619 with iTRAQ reagent
E. coli proteins (100 μg) were mixed with peptide 619 (4.5 μg) in 10 μL iTRAQ dissolution buffer. Tryptic digestion of the mixture and iTRAQ reaction was performed according to the manufacturer's recommendations, and the reaction mixture treated with the ReadyPrep 2-D Cleanup Kit from Bio-Rad (Hercules, CA, USA) according to the manufacturer's recommendations. O-deacylation was also performed separately as described above.
Retention time targeted LC/MS/MS analysis (Peptide 619)
E. coli extract (5 μl) containing peptide 619 (YASEGLSK) digested with trypsin and subsequently labeled with iTRAQ was analyzed by nano-LC/MS/MS with an Eksigent NanoLC Plus (Foster City, CA) and Thermo Finnigan LTQ Orbitrap Velos (San Jose, CA) MS and RP C18 capillary chromatography (FTMS [Fourier transform MS] performed on MS1, and ITMS [ion trap MS] performed on MS/MS acquisitions). Flow rate was 400 nL/min for a linear 60 min LC gradient, where mobile phase is A (5% ACN, 0.1% FA) and B (100% ACN, 0.1% FA).
MS-specific parameters included the following: tip voltage at +2.0 kV, FTMS mode for MS acquisition of precursor ions (60,000 resolution setting); ITMS mode for subsequent MS/MS of top 6 precursors selected; fragmentation accomplished via collision-induced dissociation (CID).
XCalibur raw data was analyzed by Protein Pilot. Protein match probabilities were determined using expectation values and/or MASCOT protein scores. For protein identification, the appropriate taxonomy (E. coli) was searched in NCBI and/or SwissProtein databases. Other parameters included maximum missed cleavages = 1; variable modifications include iTRAQ 4-plex (N-terminus, K, and Y); precursor tolerance set at 0.02 Da; MS/MS fragment tolerance set at 0.3 Da; and peptide charges considered were +2 and +3. The significance of a protein match was based on expectation values; each protein match was accompanied by an expectation value. The expectation value was the number of matches with equal or better scores that were expected to occur by chance alone. The default significance threshold was p < 0.05, so an expectation value of 0.05 was considered to be on this threshold. We used a more stringent threshold of 10−3 for protein identification—the lower the expectation value, the more significant the score.
Results
Reaction of synthetic peptides with iTRAQ
Ideally, the use of the iTRAQ reagent for peptide quantification should satisfy the following conditions: 1) the reagent should react specifically with N-terminal α-amino and lysyl ε-amino groups in a peptide; 2) the reaction conditions should ensure saturation; i.e., all reactive amino groups should be 100% modified; 3) since fractionation is required before MS quantification, all forms of the peptide should behave similarly during LC and the first MS; 4) the percentage of reporter groups released during MS/MS should be equal for all isotopic versions of the reagent; and 5) significant side reactions should be avoided. Failure to satisfy these conditions would likely negatively impact accuracy and reproducibility. To investigate whether the iTRAQ reagent and labeling protocol satisfied these conditions, we embarked on a directed study to evaluate the specificity of the reaction chemistry with synthetic peptides containing sequences known to be reactive with NHS-esters. As shown in Table 1, peptide Set 1 represents the peptides used to investigate the reactivity of tyrosyl and seryl residues adjacent to random residues and in multiple positions within the sequence of peptides. Peptide Set 2 tested the hypothesis, based on our previous observations, that a histidyl residue in proximity to a Ser, Thr, or Tyr residue increased the reactivity of their side-chain hydroxyls to NHS-esters. Since the NHS-group is the basis of iTRAQ modification chemistry, this could significantly reduce its quantification accuracy and reproducibility.
Table 1. Synthetic test peptides reacted with iTRAQa,b,c.
| Set 1 | Set 2 |
|---|---|
| 619 - YASEGLSK | 679 - ASEHASYG |
| 620 - YPSEGLSK | 680 - ASEHAYYG |
| 621 - YWSEGLSK | 681 - ASEHATYG |
| 622 - YYSEGLSK | |
| 623 - YESEYLSK |
Variable residues in bold.
Number before peptide sequence is an internal reference number referred to in the text
All peptides were synthesized as amides
The following HPLC, MS, and MS/MS results demonstrate that various peptides, whether in pure form or spiked into a complex mixture of proteins, reacted not only with the iTRAQ reagent through their α- and ε-amino groups but also with the side-chain hydroxyls of certain Tyr, Ser, and Thr residues. HPLC of pure synthetic peptides modified with the iTRAQ reagent according to the manufacturer's protocol showed multiple peaks (Figs. 1-3), each of which contained a mixture of peptides exhibiting multiple iTRAQ groups as observed by MALDI TOF/TOF analyses. Examples below identify the amino acid sites that reacted with the iTRAQ. Additionally, the reaction enhancement ability of a histidyl residue one residue removed from Tyr, Ser, or Thr (Tyr/Ser-Xaa-His or His-Xaa-Tyr/Ser/Thr) was demonstrated. Moreover, regardless of the sequence, some side-chain hydroxyl group reactivity was observed even when histidine was not present in the peptide. Importantly, however, we show that O-acylation reactions can be reversed by reaction with hydroxylamine-HCl (Fig. 3). Reaction results for several synthetic peptides are summarized in Tables 2 and 3.
Figure 1. RP-HPLC of peptide 623 (YESEYLSK).
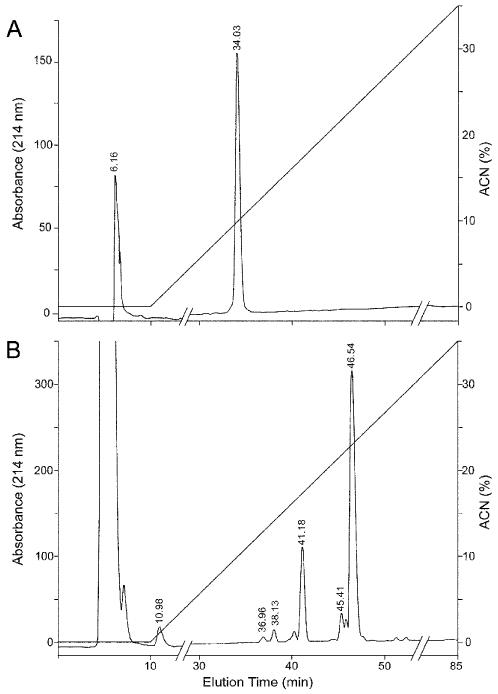
(A) The unmodified peptide. (B) iTRAQ-treated peptide analyzed by RP-HPLC as described in Experimental Procedures. Each of the major peaks observed at times 41.1, 45.4, and 46.5 were collected and analyzed by MALDI. The peak at 41.4 consisted of primarily peptide 623 with 1 iTRAQ label (m/z 1161.5) with a minor intensity peak representing two iTRAQ labels (m/z 1305.6), and the peak at 45.4 contained 1, 2, and to a lesser degree, 3 iTRAQ tags (m/z 1449.7) on the peptide (data not shown). The peptide fraction eluting at 46.5 min was further analyzed for iTRAQ modification(s) by MS/MS (Fig. 4B).
Figure 3. RP-HPLC of hydroxylamine-treated peptide 681 (ASEHATYG).
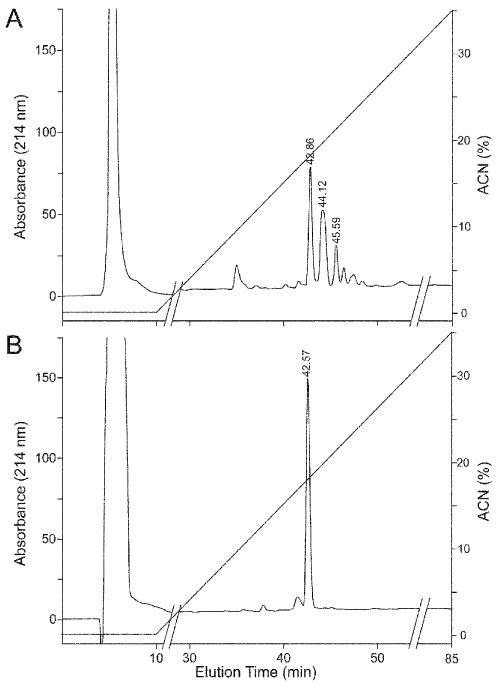
(A) iTRAQ-modified peptide. This HPLC run was performed at a different time than Fig. 2B, thus the peak RTs are somewhat different. (B) Modified peptide treated with hydroxylamine. Analysis of the large peak eluting at 42.6 min for remaining iTRAQ modification(s) by MS showed only singly modified peptide.
Table 2. Multiple sites of attachment of peptide amides reacted with iTRAQ reagent.
| Peptide | Theoretical [M+H]+ | Observed [M+H]+ | Observed iTRAQ tagsα | % Relative Intensity |
|---|---|---|---|---|
| 619-YASEGLSK (853)b | 1141.6 | 1141.5 | 2 | 100.0 |
| 1285.7 | 1285.6 | 3 | 42.9 | |
| 620-YPSEGLSK (879) | 1167.6 | 1167.7 | 2 | 94.9 |
| 1311.7 | 1311.8 | 3 | 100.0 | |
| 1455.8 | 1455.9 | 4 | 16.2 | |
| 621-YWSEGLSK (968) | 1256.6 | 1256.5 | 2 | 100.0 |
| 622-YYSEGLSK (946) | 1233.6 | 1233.7 | 2 | 100.0 |
| 1377.7 | 1377.8 | 3 | 80.7 | |
| 1521.8 | 1521.9 | 4 | 3.0 | |
| 623-YESEYLSK (1017) | 1161.5 | 1161.5 | 1 | 4.8 |
| 1305.6 | 1305.6 | 2 | 28.1 | |
| 1449.7 | 1449.7 | 3 | 100.0 | |
| 1593.8 | 1593.8 | 4 | 2.9 | |
| 623 after hydroxylamine | 1161.5 | 1161.5 | 1 | 2.6 |
| 1305.6 | 1305.6 | 2 | 100.0 | |
| 679-ASEHASYG (820) | 964.4 | 964.4 | 1 | 1.8 |
| 1108.5 | 1108.5 | 2 | 35.8 | |
| 1252.6 | 1252.6 | 3 | 100.0 | |
| 1396.7 | 1396.7 | 4 | 0.89 | |
| 680-ASEHAYYG (896) | 1040.4 | 1040.4 | 1 | 3.4 |
| 1184.5 | 1184.5 | 2 | 35.6 | |
| 1328.6 | 1328.6 | 3 | 100.0 | |
| 1472.7 | 1472.7 | 4 | 40.2 | |
| 681-ASEHATYG (834) | 1122.5 | 1122.5 | 2 | 17.1 |
| 1266.6 | 1266.6 | 3 | 100.0 | |
| 1410.7 | 1410.8 | 4 | 0.47 | |
| 681 after hydroxylamine | 978.4 | 978.5 | 1 | 100.0 |
| 1122.5 | 1122.5 | 2 | 2.1 |
Determined using MALDI TOF/TOF.
Number before peptide sequence is an in-house reference number referred to in the text; number in parenthesis is [M+H]+ of unmodified peptide amide.
Table 3. RP-HPLC fractionated iTRAQ modified Set-2 peptides.
| Peptide | RP-HPLC RT (min) |
Theoretical [M+H]+ (m/z) |
Observed [M+H]+ (m/z) |
iTRAQ tags (Observed) |
|---|---|---|---|---|
| 679-ASEHASYG (820) a | 47.4 | 1396.7 | 1396.7 | 4 |
| 1252.6 | 1252.6 | 3 | ||
| 1108.5 | 1108.5 | 2 | ||
| 680-ASEHAYYG (896) | 55.9 | 1472.7 | 1472.7 | 4 |
| 1328.6 | 1328.6 | 3 | ||
| 1184.5 | 1184.5 | 2 | ||
| 681-ASEHATYG (834) | 45.1 | 1410.7 | 1410.8 | 4 |
| 1266.6 | 1266.6 | 3 | ||
| 1122.5 | 1122.5 | 2 | ||
| ASEHATYG O-deacylated | 42.6 | 978.4 | 978.5 | 1 |
Number before peptide sequence is an internal reference number referenced in the text; number in parenthesis is [M+H]+ of unmodified peptide amide.
YESEYLSK (623)
This peptide contains six potential iTRAQ modification sites: α-amine on N-terminal Tyr, ε-amine on C-terminal Lys, and four possible O-acylation sites on the hydroxyl groups of the two Ser and two Tyr residues. Fig. 1 is an HPLC chromatogram showing the RT of intact peptide before (1A) and after (1B) iTRAQ labeling. Each of the major peaks observed at times 41.1, 45.4, and 46.5 were collected and analyzed by MALDI. The peak at 41.4 consisted of primarily peptide 623 with 1 iTRAQ label (m/z 1161.5) with a minor intensity peak representing two iTRAQ labels (m/z 1305.6), and the peak at 45.4 contained 1, 2, and to a lesser degree, 3 iTRAQ tags (m/z 1449.7) on the peptide (data not shown). The MALDI analysis of the major peak at 46.5 min is described in Fig. 4. Fig. 4A is a MALDI mass spectrum of a specific HPLC fraction (t = 46 min) showing modified peptide with m/z 1161.5 (1 iTRAQ), 1305.6 (2 iTRAQ), 1449.7 (3 iTRAQ), and 1593.8 (4 iTRAQ). Clearly, α- and ε-amines were not the only residues modified. MS analysis of the other HPLC fraction eluting at t = 41 min showed three peaks attributed to multiple (1-3) iTRAQ tags bound to the peptide. The b- and y-ion series of the MS/MS analysis of the singly tagged peptide (m/z 1161.5) showed that the iTRAQ tag attachment was primarily on the amino-terminus of Tyr. The precise iTRAQ attachment on the tyrosyl residue, either α-amine or hydroxyl, could not be definitively established solely based on the MS/MS results, but was clearly identified as an α-amine attachment by the subsequent hydroxylamine treatment (Fig. 4C). MS/MS analysis of the doubly tagged peptide showed that the modified sites were located primarily on the amino terminal tyrosyl residue and the ε-amine of the C-terminal Lys residue (results not shown).
Figure 4. MALDI MS Spectrum of iTRAQ modified peptide YESEYLSK.
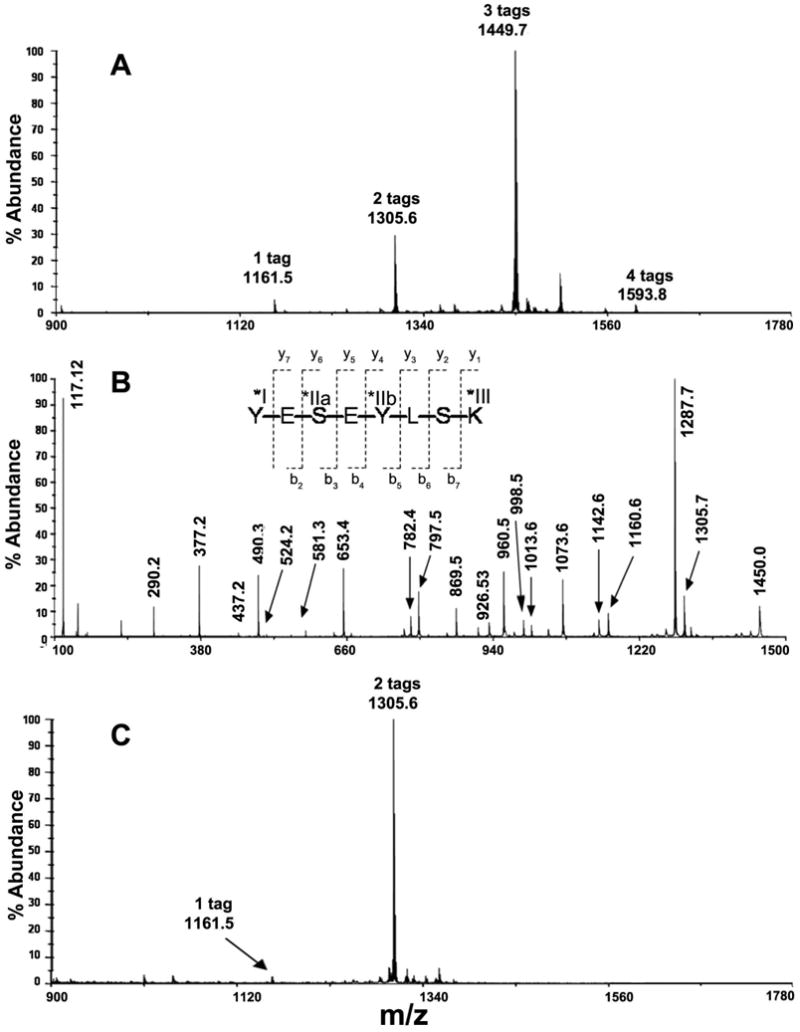
(A) The MS spectrum of the HPLC fraction eluting at t = 46.5 min (Fig. 1B) showing peaks at m/z 1161.5 (1 × iTRAQ), 1305.6 (2 × iTRAQ), 1449.7 (3 × iTRAQ), and 1593.8 (4 × iTRAQ). Four out of six possible sites, labeled with Roman numerals, reacted with the iTRAQ reagent—α-amine on N-terminal Tyr (*I), ε-amine on C-terminal Lys (*III), and four hydroxyls on all Ser and Tyr residues were possible iTRAQ reactive sites in this peptide, however Ser-3 (IIa) and Tyr-5 (IIb) were likely variable modification sites. (B) MS/MS spectrum of peptide YESEYLSK with three bound iTRAQ groups. The b-ion and y-ion series revealed an iTRAQ derivative at the N-terminal Tyr and the C-terminal Lys, as well as a mixture of iTRAQ modifications at internal residues Tyr (as indicated by b-ion series) and Ser (as indicated by y-ion series). (C) MALDI MS mass spectrum of unfractionated modified peptide YESEYLSK after reaction with hydroxylamine. This result indicated the presence of only two iTRAQ tags on the peptide without any O-acylation evident. MS/MS data (not shown) indicated that the two iTRAQ groups primarily modified the peptide at the N-terminal tyrosine and the ε-amine of the C-terminal lysine.
Figure 4B shows the MS/MS spectrum of the triply tagged peptide (m/z 1449.7). The spectrum was comprised of typical b- and y-ions, some of which suggested the possible iTRAQ attachment sites. The iTRAQ reporter ion was observed at m/z 117.1. Analysis of the b-ion series showed residual intact precursor ion that had not undergone fragmentation at m/z 1450.0 (3 iTRAQ + intact ion). Peaks are also observed at m/z 1160.6 (2 iTRAQ + b7-ion), m/z 1073.6 (2 iTRAQ + b6-ion), m/z 960.5 (2 iTRAQ + b5-ion), and at m/z 653.4 and 797.5, which were indistinguishable from two specific y-ions (see below) indicating either (1 iTRAQ + b4-ion) or (2 iTRAQ + b4-ion), respectively. Small peaks at m/z 581.2 (2 iTRAQ + b2-ion), m/z 524.2 (1 iTRAQ + b3-ion), and 437.2 (1 iTRAQ + b2-ion) were also observed, indicating the likely site of attachment at the N-terminal Tyr.
The y-ion series shown in Fig. 4B confirmed the occurrence of iTRAQ tags on N-terminal Tyr, the C-terminal Lys ε-amino group, and perhaps a heterogeneous mixture of internal Ser-3 and internal Tyr-5 residues. Furthermore, beginning with the precursor at m/z 1450.0 (3 iTRAQ + intact ion), the peak at m/z 1305.7 indicated the ion after cleavage of an iTRAQ residue, while the peak at m/z 1287.7 can be attributed to the ion after cleavage of an iTRAQ residue and cleavage of one water molecule. Peaks were also observed at m/z 1142.6 (2 iTRAQ + y7-ion), m/z 998.5 (1 iTRAQ + y7-ion), m/z 1013.6 (2 iTRAQ + y6-ion), m/z 926.5 (2 iTRAQ + y5-ion), m/z 869.5(1 iTRAQ + y6-ion), and at m/z 782.4 (1 iTRAQ + y5-ion). Additional peaks were observed at m/z 653.4 (1 iTRAQ + y4-ion), which was isobaric with a b-ion discussed above, at m/z 490.3 (1 iTRAQ + y3-ion), m/z 377.2 (1 iTRAQ + y2-ion), and at m/z 290.2 (1 iTRAQ + y1-ion) or more specifically iTRAQ plus Lys. Thus, these results indicated that there were iTRAQ modifications not only at the N-terminal Tyr and the C-terminal Lys residues, but also at either internal residues (e.g., Ser-3 and Tyr-5) as indicated by the y-ion series. All of the described modified species containing one to four tags were found within the same HPLC fraction (Fig. 1B, t = 46 min).
Also, within the same HPLC fraction described above (Fig. 1B, t = 46 min), there was a peak at m/z 1593.8 (4 iTRAQ + peptide) in the first MS (Fig. 4A). Analysis of the y-ions in the MS/MS spectrum (results not shown) showed that the N-terminal Tyr residue had two iTRAQ tags, the internal Tyr residue had one iTRAQ tag, and the C-terminal Lys also had one iTRAQ tag. The b-ion series revealed that two iTRAQ tags were attached to the b4-ion (Tyr-Glu-Ser-Glu); hence, one tag could be on Ser and one tag on Tyr, or both tags could be on Tyr; however, the y-ion results indicated that the N-terminal Tyr residue contained the two iTRAQ tags. Thus, overall the HPLC fraction at t = 46 min contained peptide YESEYLSK that had reacted with one to four iTRAQ groups.
YESEYLSK (623) reacted with hydroxylamine
The reaction of iTRAQ with the hydroxyl groups of Tyr and Ser in this peptide could be reversed by reaction with hydroxylamine. Fig. 4C shows a mass spectrum of iTRAQ-modified peptide YESEYLSK, initially modified with 1-4 tags, after reaction with hydroxylamine-HCl. The major MS peak at m/z 1305.6 indicated the attachment of two iTRAQ tags on the peptide. A considerably less abundant peak at m/z 1161.5 indicated the same peptide with only one attached iTRAQ group. Compared with Fig. 4A, the spectrum was substantially different due to the removal of two iTRAQ groups from the peptide. MS/MS analysis revealed that the two iTRAQ groups were still attached to the peptide and gave peaks at m/z 290.2 (iTRAQ + Lys) and m/z 308.2 (iTRAQ + Tyr) corresponding to an iTRAQ on the α-amine of the N-terminus and an iTRAQ on the ε-amine of the C-terminus Lys residue.
ASEHATYG (681)
We reported previously that histidine could enhance the reaction of hydroxyl side-chains of amino acid residues that are one residue removed7, 8. We thus synthesized several peptides that included a histidine residue near residues with hydroxyl side-chains; i.e., Ser, Thr, and Tyr. Fig. 5A is a MALDI MS spectrum of peptide ASEHATYG as obtained from a single HPLC peak. This fraction contained peptides with two, three, and four iTRAQ groups at m/z 1122.5, 1266.6, and 1410.8, respectively. The four possible attachment sites included the α-amine at N-terminal Ala and the hydroxyl groups on residues Ser, Tyr, and Thr. Upon reaction with hydroxylamine, the mass spectrum was reduced to a single peak observed at m/z 978.5, indicating the presence of only one iTRAQ primarily on the N-terminal Ala (results not shown) and confirming the hydroxyl reactivity of the reagent. The peptide mass at m/z 261.1 (iTRAQ + Ala) was followed by a b-ion series that contained one iTRAQ on N-terminal alanine at m/z 303.1, 432.2, 569.3, 640.3, 741.4, 904.4, and 978.5 corresponding to fragment ions from m/z 978.5.
Figure 5. Super reactivity of Ser and Thr in peptide ASEHATYG when near to His.
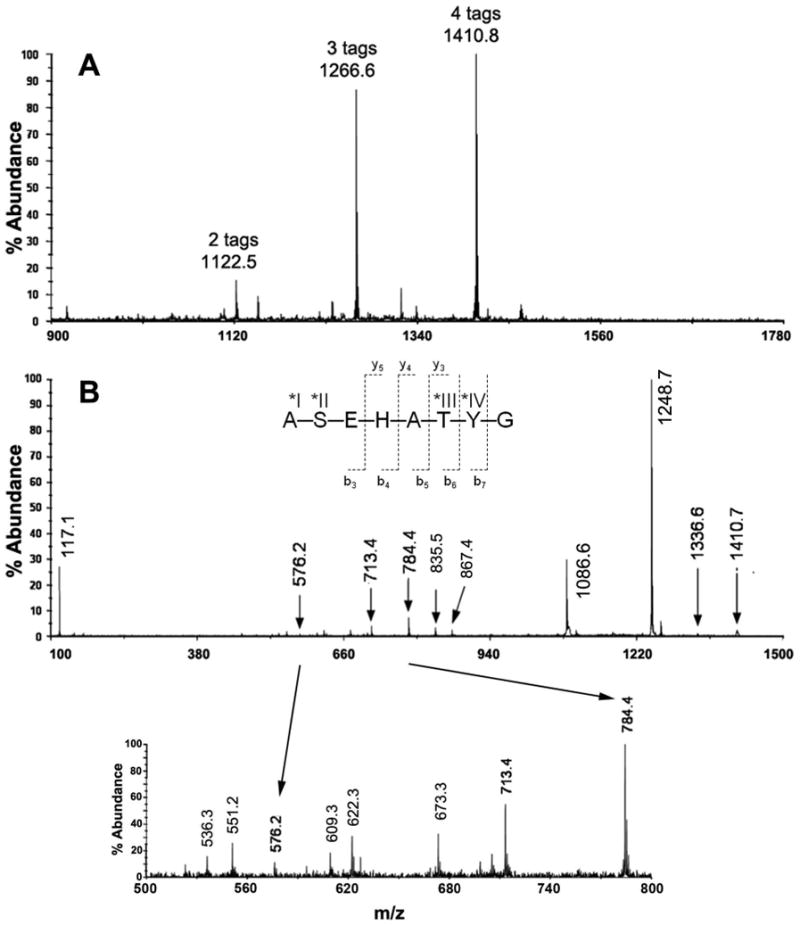
(A) MALDI MS spectrum of ASEHATYG from a single HPLC fraction, containing a mixture of peptide modified with two, three, and four iTRAQ groups at m/z 1122.5, 1266.6, and 1410.8, respectively. All four possible reactive sites—α-amine at N-terminal Ala (*I), and the hydroxyls on Ser (*II), Thr (*III), and Tyr (*IV) reacted with the iTRAQ reagent. (B) MS/MS spectrum of ASEHATYG with four iTRAQ tags. The insert is a zoom region from m/z 500-900. According to this MS/MS result, the remaining two iTRAQ tags were located primarily on the N-terminal α-amine Ala, and on the hydroxyl-group of Ser. Hydroxylamine reversed the O-acyl iTRAQ modification (results not shown).
Fig. 5B is the MS/MS spectrum of ASEHATYG with four iTRAQ tags, where b-ions and some y-ions (additional cleavage of a water molecule in some cases) dominated the spectrum. A peak can be seen at m/z 1410.7 for the intact ion (peptide with four iTRAQ tags). The iTRAQ reporter group ion was indicated by the peak at m/z 117.1. Upon cleavage of Gly, a peak at m/z 1336.6 was observed (4 iTRAQ + b7-ion). The intense peak at m/z 1248.7 was attributed to the intact ion losing 1 iTRAQ and a water molecule. Also, a peak at m/z 1086.6 can be attributed to the ion after subsequent cleavage of an additional iTRAQ and a water molecule. A peak at m/z 867.5 (2 iTRAQ + b6-ion with cleavage of one water molecule) was observed. The next identifiable peak at m/z 784.4 was attributed to the ion after cleavage of the Thr-Tyr fragment ion with 2 iTRAQ groups, confirming two iTRAQ modifications plus the b5-ion, and a peak at m/z 622.3 (1 iTRAQ + b5-ion with cleavage of one water molecule) was observed. A peak was also observed at m/z 713.4 with the cleavage of Ala (2 iTRAQ + b4-ion), at m/z 551.2 (1 iTRAQ + b4-ion with cleavage of one water molecule), and at m/z 576.3 upon cleavage of His (2 iTRAQ + b3-ion). The cleavage of His at m/z 576.3 resulted in two iTRAQ tags plus the Ala-Ser-Glu fragment ion. Therefore, these MS/MS results indicated that the remaining two iTRAQ tags were located on the N-terminal α-amine of Ala and on the hydroxyl group of Ser.
The y-ion series included the cleavage of 2 or 3 iTRAQ molecules as well as water molecules, including peaks at m/z 835.5 (2 iTRAQ + y5-ion), m/z 609.3 (2 iTRAQ + y3-ion with cleavage of one water molecule), m/z 673.3 (1 iTRAQ + y5-ion with cleavage of one water molecule), and m/z 536.3 (1 iTRAQ + y4-ion with cleavage of one water molecule). We can infer from the MS/MS results of the y-ion series that, for the 4-iTRAQ peptide, one iTRAQ tag was located on the N-terminal α-amine of Ala, and one iTRAQ tag was on each of the hydroxyl groups of Ser, Thr, and Tyr (based on the MS/MS peaks observed and the availability of attachment for iTRAQ molecules).
MS/MS analysis of the MS peak at 1266.67 of peptide ASEHATYG (Fig. 5A) showed three tags (MS/MS results not shown). Combining the b-ion and y-ion series results, we concluded that the iTRAQ tags were located primarily on the N-terminal α-amine, on the reactive hydroxyl group of Ser and on the hydroxyl group of either Thr or Tyr. Peptide ASEHATYG also had a two-iTRAQ tagged MS peak shown in Fig. 5A. Since this peak was much less abundant when compared with the peaks for the three-and four-labeled peptides (Fig. 5A), obtaining useful MS/MS data was difficult.
iTRAQ peptide 619 spiked in E. coli extract confirmed by retention time targeted nano-LC/MS/MS
To confirm iTRAQ variable reactivity and peptide side-chain O-acylation, we spiked peptide 619 (YASEGLSK-amide) into E. coli lysate and processed the mixture as described in Experimental Procedures. The peptide exhibited up to 4 iTRAQ tags (out of the potential 5) following the iTRAQ labeling chemistry based on LC/MS/MS data (ions detected included both 2+ and 3+ charge states, and possible sites include N-terminal Tyr, C-terminal Lys, and two internal Ser residues). However, manual MS/MS sequencing confirmed the heterogeneous presence of (only) 1, 2, and 3 iTRAQ tags. The complete sequence of b- and y-ions was obtained for all 1, 2, and 3 iTRAQ forms of the peptide. We were unable to sequence the peptide with 4 iTRAQ tags for confirmation; however, we were able to confirm at least one iTRAQ O-acylation on the tyrosyl or an internal seryl residue by sequencing the triply charged, triply iTRAQ-tagged peptide (Fig. 6).
Figure 6. Retention time targeted nano-LC/MS/MS and sequence of iTRAQ-labeled E. coli sample with spiked peptide 619.
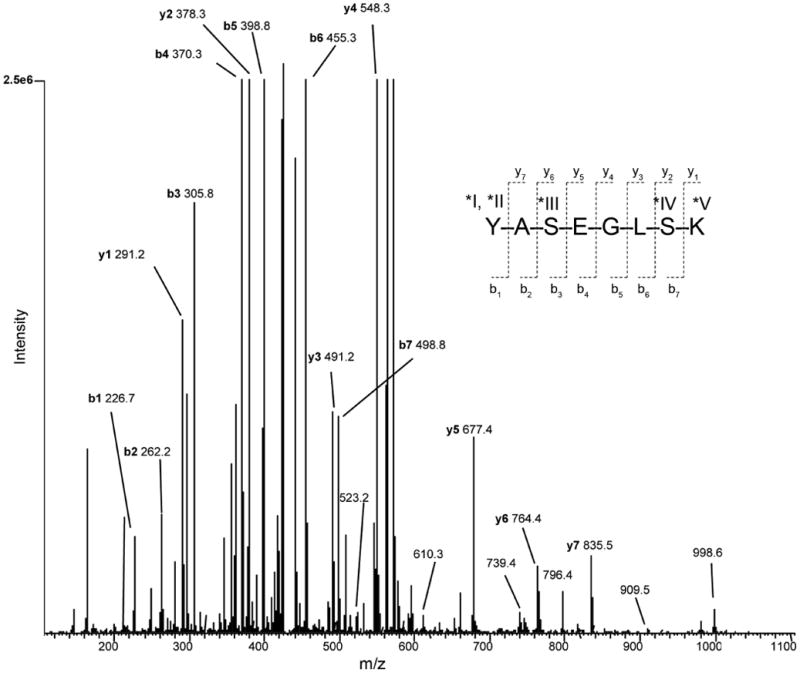
MS/MS spectrum of precursor synthetic peptide 619 (YASEGLSK) with 3 iTRAQ tags in a +3 charge state (m/z 429.5, peak window: 29.0-31.0 min, maximum peak at 30.6 min) generated by CID and by nano-LC/MS/MS. The peptide was spiked into an E. coli lysate, followed by trypsin digestion and iTRAQ labeling of the entire mixture. Complete y- and b-ion series are observed for fragment ions in the +1 and +2 charge states, respectively. Additional labeled peaks are observed for b ion series in the +1 charge state as well. Manual MS/MS sequencing of this peptide demonstrates that O-acylation has occurred on the synthetic peptide spiked into the complex sample.
Figure 6 shows the complete b- and y-ion sequence of peptide 619 with 3 attached iTRAQ tags at m/z 429.5 and targeted in the RT window of 29.0 - 31.0 min (peak maximum at 30.6 min). The precursor ion was a 3+ charge state at m/z 429.5 (second isotope showed better MS/MS sequencing than the first isotope at 429.2, so this data was analyzed in more detail). Upon CID of the selected precursor ion with 3 iTRAQ tags and in a 3+ charge state, the MS/MS spectrum indicates a combination of fragment ions of 1+ and 2+ charge states, with both 1 and 2 attached iTRAQ tags (upon removal of iTRAQ tag(s) following CID), respectively. Complete b-ion sequence was obtained for the peptide which, upon CID, lost one iTRAQ tag, while manual sequencing confirmed the presence of the 2 remaining iTRAQ tags with the fragment ions in the 2+ charge state. Also observed were other (less intense) b-ions in the 1+charge state at m/z 523.2, 610.3, 739.4, 796.4, and 909.5 (b2, b3, b4, b5, and b6, respectively).
Complete y-ion sequence was obtained for the peptide, from which two iTRAQ tags were cleaved upon CID, with manual sequencing confirming the presence of the 1 remaining iTRAQ tag with the fragment ions in the 1+ charge state. The remaining peak at m/z 998.6 is the peptide with only 1 iTRAQ tag, indicating 2 tags have already been cleaved from the (3+ charge state) precursor ion (manual validation of the isotopic envelope of m/z 429.5 was facilitated by the FTMS acquisition mode of 60,000 mass resolution in this experiment).
Numerous MS/MS spectra and manual sequencing of peptide 619 spiked into E. coli (followed by trypsin digestion and labeling iTRAQ chemistry) with 1, 2, and 3 iTRAQ tags matched the spectrum obtained from the positive control of peptide 619 (iTRAQ peptide only). In addition, E. coli iTRAQ peptides isobaric with peptide 619 were not observed in the pure E. coli digestion negative control (no peptide 619). All possible peptide 619 forms with iTRAQ tags were compared to the extensive peptide list matching numerous E. coli proteins via a database search. Not only was there no mass overlap, but also manual validation by MS/MS of the synthetic peptide was performed, and confirmed the presence of the O-acylated and N-alkylated forms in the spiked mixture (Fig. 6).
Discussion
We have demonstrated that the iTRAQ reagent is capable of significant O-acylation reactions involving the hydroxylated amino acid residues Ser, Thr, and Tyr, especially in certain histidine containing sequences, and even in complex protein mixtures. Miller and Kurosky7 and Miller et al.8 reported that acylation of hydroxyl amino acid residues by NHS-esters of biotin was dependent on the presence of histidine one residue removed on either side of the reactive hydroxyl residue. Furthermore, a plot of O-acyl reactivity vs pH gave a sigmoidal curve with a pKapp of 6.65 consistent with histidyl imidazole ionization5. These results indicated that the increased nucleophilicity of the oxygen of the side-chain hydroxyl group was due to the intramolecular interaction (hydrogen bonding) between the side-chain hydroxyl group and the histidyl imidazole group reminiscent, in part, to the serine protease active site. In every case studied involving some 15 peptides, enhanced reactivity of the hydroxylated residues with NHS-esters of biotin was observed when near a histidine7. It is therefore likely that the observed enhanced reactivity of the iTRAQ reagent with hydroxyl amino acid residues near histidyl residues parallels that reported for O-acylation with NHS-esters of biotin. Interestingly, however, the hydroxyl amino acid reactivity of the iTRAQ reagent seemed to be greater when compared with the reactivity observed for the NHS-esters of biotin. Typically, in previous studies, NHS-esters of biotin were only reactive with side-chain hydroxyls near histidyl residues. In our hands, however, the iTRAQ reagent showed appreciable reactivity with some side-chain hydroxyls in peptides that did not contain histidine residues; e.g., peptide 623. Thus, while iTRAQ O-acylation exhibits sequence dependence, likely due to near neighbor side-chain interactions, additional reactivity may occur with other side chain hydroxyls.
An important consideration relates to the matrix in which the reactivity is measured, and the relative ratio of hydroxylated and amine residues in the peptide vs the relative ratios of these residues in the natural extract. We therefore performed a peptide spiking experiment into a complex protein mixture from an E. coli lysate. Since the reactions were performed under denaturing conditions, all peptides were expected to be devoid of structure, and therefore all reactive residues should be equally available. To compare the impact of the peptide on the iTRAQ reactivity in the spiked and unspiked E. coli lysate, we estimated the total hydroxyl and amine ratio of the spiked extract compared to the unspiked extract. Based upon percentage compositions, Ser, Arg, Thr, Tyr, and Lys range from 2.67 – 5.58 in E. coli total protein, and 2.73 – 6.85 in mammalian cells 9. From these data and the average mass of an E. coli tryptic peptide evidenced from its genome (and ORFs) in a 100 μg tryptic sample, the molar ratio of hydroxyl to amine groups is about 1.3, while the ratio in the peptide-spiked E. coli lysate is also about 1.3 (<0.1% difference). Thus the spiked peptide O-acylations we observed could not be due to an unnaturally higher abundance of hydroxyls in the spiked peptide vs those present in an unspiked E. coli lysate.
Our results demonstrated that without treatment of iTRAQ-modified peptides with hydroxylamine to remove O-acylations, substantial quantification errors will result. High coefficients of variation with iTRAQ quantification have been addressed by others10, 11 underscoring problems, some of which are likely related to O-acylation. We found that the level of O-acylation was quite significant, as shown in Figs. 1B, 2B which showed prominent O-acylated peaks, confirmed in Fig 4. Those hydroxyl side-chain residues positioned near histidyl residues were especially super-reactive to the iTRAQ reagent as were some non-histidine associated hydroxylated residues. However, clearly some hydroxylated amino acid residues were not especially reactive and further investigation will be required to distinguish sequence specificities of the reaction. It should be noted, however, that hydroxylamine can exert pleiotropic chemical effects, including Asp-Gly cleavage12, de-acylation of phospho-Asp and phospho–His13, and in general, may act as a nucleophilic reducing agent. Preliminary studies showed that it does not dephosphorylate peptide hydroxyl residues, nor do the alkaline pH reaction conditions we employed. Further studies will be required to more comprehensively evaluate the impact of hydroxylamine on protein post-translational modifications.
Figure 2. RP-HPLC of peptide 681 (ASEHATYG).
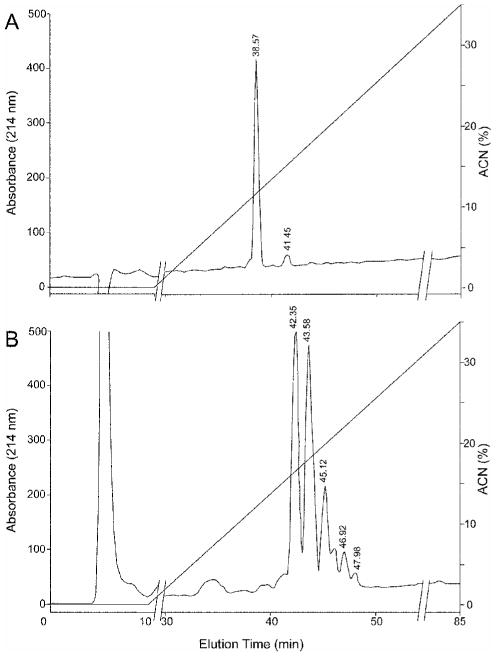
(A) Unmodified peptide. (B) Modified peptide was fractionated as for Fig. 1. The peptide fraction eluting at 45.1 min was further analyzed for iTRAQ modification(s) by MS and MS/MS (Fig. 5).
The question arises as to the prevalence of the reactive triplet sequences containing histidine within the human proteome. Using both the sequence and the pattern tools available from RoseoBase (http://roseobase.org/espsearch/index.cig) to search the human FASTA database for all possible triplets, we interrogated 38,063 protein sequences and found a surprisingly high frequency of these triplets (Table 4). This high prevalence suggested possible structural or functional significance. Just as surprising was the observation that the frequencies and occurrences of the reverse triplet sequence were nearly identical to their cognates with the exception of Thr-Xaa-His. This confirmed that it was the proximity to the His that governs the significance of these triplets. Although these residues are targets for post-translational modifications, notably phosphorylation and O-glycosylation, no great preference by kinases or glycosyltransferases for any of the triplets could be deduced from the literature14, 15. Thus, the high frequencies of these sequences in the human proteome remains unexplained, but suggest they may engage in a conserved function, perhaps in protein folding.
Table 4. Frequency of triplet amino acids in human proteinsa.
| Triplet | Triplet Hitsα | Protein Hitsb | Frequencyc | Prevalenced (%) |
|---|---|---|---|---|
| Ser-Xaa-His | 38392 | 18591 | 2.07 | 48.8 |
| Thr-Xaa-Hise | N/A | N/A | N/A | N/A |
| Tyr-Xaa-His | 13121 | 9029 | 1.45 | 23.7 |
| His-Xaa-Ser | 36988 | 18271 | 2.02 | 48.0 |
| His-Xaa-Thr | 22323 | 13813 | 1.62 | 36.3 |
| His-Xaa-Tyr | 12871 | 9032 | 1.43 | 23.7 |
38,063 protein sequences from NCBI database were interrogated.
Number of times the given triplet was found in entire database.
Average frequency of triplet per protein hit.
Prevalence of proteins containing the triplets in entire database.
Reactivity with Thr-Xaa-His triplets was not observed although His-Xaa-Thr was likely related to the branched nature of the threonyl side-chain.
It should be noted that the HPLC did not fully resolve the multiply iTRAQ-modified peptides. For example, when analyzed by MS, the single HPLC peak at 46.5 min (Fig. 1B) yielded four precursor ions, representing one to four iTRAQ modifications. We interpret this to be due to chromatographic analyte carry-over as the peptides move through the column. It is unlikely that isobaric peptides were removed during the first MS of the MALDI analysis since no iTRAQ 114-117 reporter ions were observed. Furthermore, repeat analysis of the 46.5 min peak containing four iTRAQ modifications gave essentially the same results after nano-LC/MS/MS on an Applied Biosystems'; 4000 QTrap, which utilizes a weaker ionization protocol than the MALDI-TOF/TOF. This indicated that the cleavage of an iTRAQ residue during or subsequent to HPLC processing was unlikely.
In conclusion, the principle underlying stable isotope labeled peptide quantification requires that identical peptides from different sources co-elute from the fractionation step and appear in the same MS scan for quantification to occur. Accuracy is dependent upon the assumption that all the co-eluting peptide copies from one source are equally labeled, so that the ratio of the cognate peptides from both or multiple sources will accurately reflect the relative abundance of the peptides. Moreover, since stable isotopic labeling does not increase MS sensitivity, dilution of the peptides into multiple species decreases their detectability. Partial, variable modification of the same peptide from one sample to the next may preclude co-elution, resulting in a loss of signal from that species, or may cause non-cognate, but isobaric peptide species to co-elute. In either case, the accuracy and reproducibility will be diminished in proportion to the degree of altered elution/modification. Since the reporter group carries the quantification label and MS/MS demonstrates that a single HPLC fraction can contain from 1-4 iTRAQ tags on the same peptide, there is no assurance that the same peptide from a different comparative sample will be modified identically. The outcome is that the collection of isobaric peptides will yield differing ratios, thereby impacting accuracy and reproducibility. Furthermore, the distribution of a given peptide across several variably eluting HPLC peaks decreases the amount of peptide analyzed by the MS/MS system, decreasing overall detection sensitivity as well. Thus, the occurrence of O-acylation side reactions may compromise accuracy by greatly reducing the molar excess ratio of reagent to amino groups. This reduction could be significant in many peptide mixtures containing a number of reactive side-chain hydroxyl groups. Also, reagent loss due to hydrolysis is another additive factor, decreasing the molar excess of reactive iTRAQ. Finally, it should be underscored that the removal of problematic O-acyl modifications can readily be achieved by reaction with hydroxylamine. It is important to note also that although we have concentrated our efforts on the iTRAQ reagent, our results apply equally to other commercially available stable isotope labeling reagents that utilize NHS-ester chemistry.
Acknowledgments
*This work was supported in part by the National Heart, Lung, and Blood Institute's Proteomics Initiative NO1-HV-00245 (AK), National Institute for Environmental Health Sciences Center grant P30-ES006676 (C. Elferink), and the National Institute of Allergy and Infectious Diseases grant P01-AI062885 (A. Brasier). The authors wish to acknowledge Steven Serabin for peptide synthesis, Drs. Anthony Haag for his MS consultation, and Kizhake Soman for his assistance in quantifying the frequency of triplet sequences in the human genome. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Abbreviations
The abbreviations used are:
- NHS
N-hydroxysuccinimide
- iTRAQ™
isobaric tag for relative and absolute quantitation
- Fmoc
fluorenylmethyloxycarbonyl
- RP
reversed-phase
- HPLC
high performance liquid chromatography
- TFA
trifluoroacetic acid
- MS
mass spectrometry
- MALDI TOF/TOF
matrix-assisted laser desorption/ionization time-of-flight/time-of-flight
- RT
retention time
- ACN
acetonitrile
- Q1, 2, and 3
quadrupole 1, 2, or 3, respectively
- XIC
extracted ion chromatogram
- ESI
electrospray ionization
- ITMS
ion trap MS
- FTMS
Fourier transform MS
References
- 1.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin D. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Gan CS, Chong PK, Pham TK, Wright PC. Technical, Experimental, and Biological Variations in Isobaric Tags for Relative and Absolute Quantitation (iTRAQ) J Proteome Res. 2007;6(2):821–827. doi: 10.1021/pr060474i. [DOI] [PubMed] [Google Scholar]
- 3.Quaglia M, Pritchard C, Hall Z, O'Connor G. Amine-reactive isobaric tagging reagents: requirements for absolute quantification of proteins and peptides. Anal Biochem. 2008;379(2):164–9. doi: 10.1016/j.ab.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Choe LH, Aggarwal K, Franck Z, Lee KH. A comparison of the consistency of proteome quantitation using two-dimensional electrophoresis and shotgun isobaric tagging in Escherichia coli cells. Electrophoresis. 2005;26(12):2437–2449. doi: 10.1002/elps.200410336. [DOI] [PubMed] [Google Scholar]
- 5.Wiese S, Reidegeld KA, Meyer HE, Warscheid B. Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research. Proteomics. 2007;7(3):340–350. doi: 10.1002/pmic.200600422. [DOI] [PubMed] [Google Scholar]
- 6.Wu WW, Wang G, Baek SJ, Shen RF. Comparative Study of Three Proteomic Quantitative Methods, DIGE, cICAT, and iTRAQ, Using 2D Gel- or LC-MALDI TOF/TOF. J Proteome Res. 2006;5:651–658. doi: 10.1021/pr050405o. [DOI] [PubMed] [Google Scholar]
- 7.Miller BT, Kurosky A. Elevated intrinsic reactivity of seryl hydroxyl groups within the linear peptide triads His-Xaa-Ser or Ser-Xaa-His. Biochem Biophys Res Commun. 1993;196(1):461–7. doi: 10.1006/bbrc.1993.2272. [DOI] [PubMed] [Google Scholar]
- 8.Miller BT, Rogers ME, Smith JS, Kurosky A. Identification and characterization of O-biotinylated hydroxy amino acid residues in peptides. Anal Biochem. 1994;219(2):240–8. doi: 10.1006/abio.1994.1263. [DOI] [PubMed] [Google Scholar]
- 9.Okayasu T, Ikeda M, Akimoto K, Sorimachi K. The amino acid composition of mammalian and bacterial cells. Amino Acids. 1997;13(3):379–391. [Google Scholar]
- 10.Aggarwal K, Choe LH, Lee KH. Quantitative analysis of protein expression using amine-specific isobaric tags in Escherichia coli cells expressing rhsA elements. Proteomics. 2005;5(9):2297–308. doi: 10.1002/pmic.200401231. [DOI] [PubMed] [Google Scholar]
- 11.Hill EG, Schwacke JH, Comte-Walters S, Slate EH, Oberg AL, Eckel-Passow JE, Therneau TM, Schey KL. A statistical model for iTRAQ data analysis. J Proteome Res. 2008;7(8):3091–101. doi: 10.1021/pr070520u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bornstein P, Balian G. Cleavage at Asn-Gly bonds with hydroxylamine. Methods in Enzymol. 1977;47:132–45. doi: 10.1016/0076-6879(77)47016-2. [DOI] [PubMed] [Google Scholar]
- 13.Ventrella V, Elvir JR, Borgatti AR, Trigari G, Proverbio T, Pagliarani A, Trombetti F, Pirini M, Marin R, Proverbio F. Phosphorylated intermediate of the ouabain-insensitive, Na(+)-stimulated ATPase in rat kidney cortex and rainbow trout gills. Biochimie. 2010;92(2):128–35. doi: 10.1016/j.biochi.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 14.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294(5):1351–62. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 15.Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8(7):530–41. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]