

Abstract
PICK1 (protein interacting with C kinase-1) regulates the surface expression of the AMPA receptor (AMPAR) GluR2 subunit, however, the functional consequences of this interaction are not well understood. Previous work has suggested that PICK1 promotes the internalization of AMPARs. However, we found that when PICK1 is virally expressed in the CA1 region of hippocampal slices, it causes an increase in AMPAR-mediated EPSC amplitude. This effect is associated with increased AMPAR rectification and sensitivity to polyamine toxin. These effects are blocked by PKC or calcium/calmodulin-dependent protein kinase II inhibitors, indicating that the virally expressed PICK1 signals through an endogenous kinase cascade. In contrast, blockade of interactions with GluR2 at the N-ethylmaleimide-sensitive factor site did not cause a change in subunit composition, suggesting that the effects of PICK1 are not simply a nonspecific consequence of removing AMPARs from the surface. Immunocytochemical and biochemical analyses in dissociated cultured hippocampal neurons show that PICK1 causes a decrease in endogenous GluR2 surface expression but no change in GluR1 surface levels. To address the physiological role of PICK1, we virally expressed C-terminal GluR2 peptides. Blockade of endogenous PICK1 PDZ (postsynaptic density-95/Discs large/zona occludens-1) domain interactions produced opposite effects on synaptic strength and AMPAR rectification to those observed with PICK1 expression. This demonstrates that AMPAR subunit composition is physiologically regulated through a mechanism involving PICK1 PDZ domain interactions. These findings suggest that PICK1 acts to downregulate the GluR2 content of AMPARs at hippocampal CA1 synapses, thereby increasing synaptic strength at resting membrane potentials.
Keywords: glutamate, synaptic plasticity, hippocampus, interacting protein, AMPA receptor trafficking, protein interacting with C kinase
Introduction
AMPA receptor (AMPAR) subunits interact with intracellular proteins via their intracellular C-terminal domain. Much recent interest has focused on this as a mechanism regulating AMPAR function during forms of long-term synaptic plasticity (Sheng and Lee, 2001; Malinow and Malenka, 2002). The most numerous and extensively studied of these are proteins that interact with the GluR2 subunit. GluR2 is of particular interest because its presence in the edited form determines key properties of the AMPAR receptor channel: Ca2+ permeability, single-channel conductance, and rectification (Burnashev et al., 1992; Bowie and Mayer, 1995; Geiger et al., 1995; Kamboj et al., 1995; Koh et al., 1995a; Washburn et al., 1996; Swanson et al., 1997). Two sites of interaction have been identified on the GluR2 C terminus, the proximal N-ethylmaleimide-sensitive factor (NSF)-binding site and the extreme C-terminal PDZ (postsynaptic density-95/Discs large/zona occludens-1) site (Sheng and Lee, 2001; Malinow and Malenka, 2002). Three PDZ domain-containing proteins have been identified that interact at the extreme C-terminal site: GRIP (glutamate receptor-interacting protein) (Dong et al., 1997), ABP (AMPA receptor-binding protein) (Srivastava et al., 1998), and PICK1 (protein interacting with C kinase-1) (Dev et al., 1999; Xia et al., 1999).
Recent interest has focused on the PICK1-GluR2/3 interaction. PICK1 decreases GluR2 surface expression in a PKC-dependent mechanism that is associated with the phosphorylation of serine 880 on the C terminal of GluR2 (Chung et al., 2000; Perez et al., 2001). This has led to the proposal that this mechanism is involved in the expression of hippocampal NMDA receptor-dependent long-term depression (LTD) (Chung et al., 2000; Kim et al., 2001; Perez et al., 2001). Indeed, acute blockade of the PICK1-GluR2 interaction has been reported to partially block this form of LTD (Kim et al., 2001). However, another study found that blockade of this interaction did not affect this form of LTD (Daw et al., 2000; Duprat et al., 2003), and PKC activity has been shown not to be required for NMDA receptor-dependent LTD (Oliet et al., 1997; Daw et al., 2000; Kim et al., 2001). Thus, the role of PICK1 in hippocampal LTD is unclear, and the functional consequences of the GluR2-PICK1 interaction remain to be elucidated.
To investigate the role of PICK1 in regulating synaptic AMPARs, we virally expressed PICK1 in CA1 pyramidal neurons in hippocampal slices maintained for 1-2 d in culture. PICK1 caused an increase in AMPAR-mediated EPSC amplitude and an associated increase in rectification and sensitivity to polyamine toxin. Immunocytochemical and biochemical analyses in dissociated cultured neurons demonstrated that PICK1 caused a change in the subunit composition of AMPARs, resulting in surface GluR2-containing receptors being replaced by GluR2-lacking receptors. This process requires endogenous kinase activity because the effects of PICK1 expression on synaptic transmission were blocked by inhibition of PKC or CaMKII (calcium/calmodulin-dependent protein kinase II). In addition, viral expression of C-terminal GluR2 peptides that block endogenous PICK1 PDZ domain interactions caused a decrease in EPSC amplitude and a reduction in rectification, demonstrating that AMPAR subunit composition is physiologically regulated in a PICK1-dependent mechanism. These findings suggest that PICK1 acts to decreases the GluR2 content of synaptic AMPARs resulting in an increase in synaptic strength at resting membrane potentials.
Materials and Methods
Molecular biology. Point mutations in the PDZ domain of PICK1 were constructed by PCR. Flag-PICK1-digested fragments from pCIneo (either wild type or PICK1-AA mutant) or an annealed synthetic oligonucleotide for the peptides (pep2m, pep2-SVKI, pep2-EVKI, pep2-SVKE) were subcloned into pCITE-EGFP (enhanced green fluorescent protein). The pCITE-EGFP was created by subcloning CITE [internal ribosome entry site (IRES)] from pCITE4c(+) (Novagen, Madison, WI) into pEGFP-N1 (Clontech, Cambridge, UK). The construct of interest was always under the more efficient cap-dependent expression, and EGFP was under the less efficient IRES-dependent expression. The Flag-PICK1-IRES-EGFP (either wild type or PICK1-AA mutant)-digested or peptide-IRES-EGFP (pep2m, pep2-SVKI, pep2-EVKI, pep2-SVKE)-digested fragments were subcloned from pCITE-EGFP into pSinRep5 (Invitrogen, San Diego, CA). RNA for pSinRep5 constructs was prepared by in vitro translation (Invitrogen) and electroporated into baby hamster kidney cells together with the helper DH(265). The Sindbis virus was made according to the manufacturer's instructions (catalog #K750-01; Invitrogen), and integrity of constructs was verified by DNA sequencing (ABI-PerkinElmer).
Immunocytochemistry. Low-density hippocampal cultures were prepared from embryonic day 18 Sprague Dawley rats. Hippocampi were dissected in cold HBSS, then dissociated using trypsin for 10 min at 37°C. Cultures were then plated onto poly-l-lysine-coated glass coverslips and maintained at 37°C, 5% CO2 until required. Neurons were used after 14 d in vitro and were incubated with or without virus for 22 hr, after which cells were fixed using 3.7% paraformaldehyde. GluR1 and GluR2 surface expression was detected using anti-N-terminal GluR1 polyclonal rabbit (Calbiochem, La Jolla, CA) and anti-N-terminal GluR2 monoclonal mouse (Chemicon, Temecula, CA) antibodies, respectively, under nonpermeabilizing conditions. Whole-cell GluR2 expression was determined using the N-terminal antibody under permeabilizing conditions. Secondary antibodies were AlexaFluor 568 conjugated and were either goat anti-rabbit or goat anti-mouse IgG (Molecular Probes, Eugene, OR). Fluorescence was visualized under a confocal microscope (Leica, Nussloch, Germany), where the green channel was used to visualize EGFP fluorescence and the red channel was used to visualize antibody immunoreactivity. Quantification of surface puncta of GluR1 and GluR2 was performed as described previously (Pickard et al., 2000). Changes in whole-cell distribution were quantified using Adobe Photoshop and NIH ImageJ software.
Western blotting. Cultures were lysed in solubilization buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 10 mm EDTA, 1% Triton-X100, and proteinase inhibitor mixture) for 30 min at 4°C, then centrifuged at 13,000 rpm for 15 min at 4°C to remove cell debris. Intersample total protein concentration was normalized using protein assay reagent (Bio-Rad, Hercules, CA). Samples were run on 9% SDS-PAGE gels and transferred onto polyvinylidene fluoride membrane (Immobilon P; Micropore). The primary antibodies used were anti-N-terminal GluR2 (Chemicon) and anti-C-terminal GluR1 (Upstate Biotechnology, Lake Placid, NY); chemiluminescence with subsequent densitometric measurement of band intensity was used for quantification.
Cell surface biotinylation. Nonpermeable biotin (Sulfo NHS-LC-Biotin; Pierce, Rockford, IL) was used to identify cell surface-expressed membrane proteins. Cultured hippocampal neurons were incubated with 0.5 mg/ml biotin for 30 min at 4°C, after which cells were lysed using solubilization buffer for an additional 30 min at 4°C. After centrifugation to remove cell debris, solute was removed for immunoprecipitation. Sample biotin was immunoprecipitated overnight at 4°C using streptavidin-coated agarose beads (Sigma, St. Louis, MO), then run on a standard Western blotting procedure (as described above). For determinations of the effects of PICK1 and PICK1-AA expression on GluR1 and GluR2 total and biotinylated protein levels, Western blots were compared with control sister cultures.
Acute slice culture and viral infection. Transverse hippocampal slices (300 or 350 μm thick) were prepared from Wistar rat pups (7-12 d old) under semi-sterile conditions. Slices were prepared on a microslicer at 1-4°C in a modified extracellular solution (in mm): 4 KCl, 26 NaHCO3, 1 CaCl2, 5 MgSO4, 10 glucose, and 248 sucrose, saturated with 95% O2/5% CO2. Slices were then allowed to recover for 30-60 min at 27°C before being placed in a standard sterile culture medium. The Sindbis virus was pressure ejected into a region of the CA1 pyramidal cell layer of each slice before the slices were placed in an incubator and maintained at 35°C. Slices were used for electrophysiological recordings between 20 and 48 hr after injection of virus.
Electrophysiology. Whole-cell patch-clamp recordings were made from visually identified CA1 pyramidal neurons using high-power water immersion optics and combining fluorescence and infrared microscopy. Infected neurons were identified as those expressing EGFP. Nearby non-infected neurons were used for in-slice controls. The extracellular solution during recordings was (in mm): 124 NaCl, 3 KCl, 1.25 NaHPO4, 26 NaHCO3, 2.5 CaCl2, 1.3 MgSO4, and 15 glucose, saturated with 95% O2/5% CO2. Picrotoxin (100 μm; Sigma) was also included in the extra-cellular solution, and a cut was made between CA3 and CA1 to prevent any resulting epileptiform activity. The intracellular solution was (in mm): 135 CsMeSO4, 8 NaCl, 10 HEPES, 0.5 EGTA, 4 Mg-ATP, 0.3 Na-GTP, and 5 QX-314 at pH 7.2, 285 mOsm. All recordings were performed at room temperature. EPSCs were evoked by electrical stimulation of axons in the stratum radiatum. EPSCs were collected at holding potentials of -70 and +40 mV for all cells, and in some cells, responses at additional potentials were also collected. For the pairwise comparison of control and infected neurons in the same slice, recordings were made from two neurons using the same stimulus intensity and position (Hayashi et al., 2000). For the unpaired comparison, control and infected neurons were recorded from the same slice, but stimulus intensity and position was not constant. The philanthotoxin (PhTx; Sigma) experiments were performed in the presence of d-AP-5 (100 μm; Tocris, Bristol, UK), EPSCs were evoked at a stimulation frequency of 0.1-0.2 Hz, and PhTx (1 μm) was bath applied after the collection of a baseline. Data were collected using an Axopatch 1-D amplifier (Axon Instruments, Foster City, CA), filtered at 5 kHz and digitized at 10 kHz. EPSC amplitude, DC current, input resistance, and series resistance were monitored continuously online as described previously (Daw et al., 2000).
Electrophysiology analysis. The AMPA/NMDA ratio was estimated by calculating AMPAR-mediated EPSC amplitude (estimated as the peak EPSC amplitude at a holding potential of -70 mV) divided by NMDAR-mediated EPSC amplitude (EPSC amplitude at a holding potential of +40 mV measured 50-70 msec after the AMPAR-mediated EPSC peak). The amount of rectification of the AMPAR-mediated EPSC was determined by dividing the AMPAR-mediated EPSC amplitude at -70 mV by the AMPAR-mediated EPSC amplitude at +40 mV (estimated by measuring EPSC amplitude at the same time point as the peak of the AMPAR-mediated EPSC at -70 mV). In separate control experiments (n = 19) using d-AP-5 (100 μm) and 6-nitro-7-sulfamoylbenzo-(f)quinoxaline-2,3-dione (NBQX) (2 μm), we found that this method of analysis provided reliable estimates of both the AMPA/NMDA ratio and rectification index. Furthermore, in some experiments, AMPAR-mediated EPSC rectification was measured in the presence of d-AP-5 (see Fig. 2C), and this produced very similar rectification values to those obtained in the presence of an NMDAR-mediated component. In all figures, for presentation purposes, the stimulus artifacts have been subtracted. Statistical analysis was performed using the Student's t test.
Figure 2.

The PICK1-dependent increase in AMPAR-mediated EPSC amplitude is associated with an increase in AMPAR rectification. A, Averaged EPSCs from example experiments (unpaired experiments) at a holding potential of -70 mV (bottom traces) and +40 mV (top traces) for a control noninfected neuron (left) and for a neuron infected with the PICK1 virus (right). B, Pooled data showing AMPA/NMDA ratio (B1) and rectification (AMPA EPSC-70 mV/AMPA EPSC+40 mV; B2) for neurons from uninfected slices (n = 9), noninfected neurons in slices infected with PICK1 virus (Control; n = 13), and neurons infected with the PICK1 virus (n = 13). *p < 0.05. C, Pooled data showing I-V relationships (IAMPA normalized to data at -70 mV) in the presence of 50 μm d-AP-5 for control neurons (C1; n = 7) and neurons virally expressing PICK1 (C2; n = 7). The dashed lines are linear regression fits to the data at negative holding potentials.
Results
Viral expression of PICK1 causes an increase in the amplitude and rectification of AMPAR-mediated EPSCs
We used the Sindbis virus to express recombinant proteins or peptides in hippocampal slices. An IRES construct was used to bicistronically express the protein/peptide of interest together with free EGFP. On the day of preparation, the virus was injected into the extracellular space of the CA1 pyramidal cell region of acute hippocampal slices, which were then maintained in culture for 1-2 d. This preparation was used so that acute slice connectivity and physiology were preserved. One day after infection, neurons expressing EGFP were visible under low-power magnification (Fig. 1A), and under high-power magnification, individual EGFP-expressing cells could be identified (Fig. 1B). This allowed whole-cell patch-clamp recordings to be made from neurons expressing the construct and from neighboring noninfected control neurons from the same slice (Fig. 1C). The properties of AMPAR- and NMDAR-mediated EPSCs could thus be compared for both experimental and control neurons within the same slice.
Figure 1.

Viral expression of PICK1 in CA1 pyramidal neurons in acute cultured hippocampal slices increases AMPAR-mediated EPSC amplitude. A, Low-power transmission (A1) and fluorescence (A2) images of a hippocampal slice cultured overnight with Sindbis virus bicistronically expressing EGFP. B, High-power images (transmission and fluorescence images superimposed) of whole-cell patch-clamp recordings from noninfected control (B1) and neighboring infected (B2) neurons in the CA1 pyramidal cell layer (same slice as in A). C, Schematic showing the experimental configuration for recording from control and infected neurons. D, Averaged EPSCs (bottom traces, -70 mV; top traces, +40 mV) from an example pairwise experiment for a control noninfected neuron (left), for a neighboring neuron infected with virus expressing PICK1 (center), and EPSCs from these two neurons superimposed (right). E, Summary analysis of all pairwise comparisons (n = 9) of the effects of viral expression of PICK1 on EPSC amplitude recorded at a holding potential of -70 mV. *p < 0.05. F, Summary analysis of all pairwise comparisons (n = 9) of effects of PICK1 overexpression on the NMDAR-mediated EPSC.
We studied the effects of virally expressing full-length PICK1 on synaptic currents by comparing transmission evoked in pairs of infected and neighboring noninfected cells using the same stimulus position and intensity (Hayashi et al., 2000). Using this pairwise comparison, we found that PICK1 caused an increased AMPAR-mediated EPSC amplitude (Fig. 1D,E) but had no effect on the NMDAR-mediated component of transmission in the same cells (Fig. 1D,F). The EPSCs recorded at -70 mV in neurons virally expressing PICK1 could be completely blocked by NBQX (2 μm; EPSC amplitude in NBQX, 4 ± 3% of baseline; n = 5), confirming that they were mediated by AMPARs.
In a second experimental paradigm, we performed within-cell comparisons of the effects of PICK1 by comparing the ratio of the AMPAR-mediated EPSC to the NMDAR-mediated EPSC at the same input onto each cell (AMPA/NMDA ratio). We also estimated the amount of rectification of the AMPAR-mediated EPSC (expressed as the ratio of AMPA EPSC-70 mV/AMPA EPSC+40 mV). These parameters were compared between infected neurons and neighboring noninfected neurons (Fig. 2A,B). The noninfected neurons were good in-slice controls because their synaptic properties were unaffected by the viral infection procedure compared with neurons in cultured slices that had not undergone the infection procedure (“uninfected”) (Fig. 2B).
In these experiments, viral expression of PICK1 caused an increase in the AMPA/NMDA ratio (289 ± 81% control; p < 0.05; n = 13) (Fig. 2 A,B) and an increase in rectification (188 ± 60% control; p < 0.05; n = 13) (Fig. 2 A,B).
We also studied the rectification properties of the isolated AMPAR-mediated EPSC in experiments in which the full I-V relationship was determined in the presence of bath-applied d-AP-5 (50 μm) (Fig. 2C). This showed that in control cells AMPARs exhibited a small amount of rectification, whereas in PICK1-expressing neurons the AMPARs rectified strongly at positive potentials. There was no difference in the reversal potential between these two data sets (control, 3.1 ± 2.1 mV, n = 7; PICK1, 0.1 ± 3 mV, n = 7; p = 0.6). The change in rectification is consistent with PICK1, causing a reduction in the proportion of surface-expressed AMPARs containing GluR2 (Bowie and Mayer, 1995; Kamboj et al., 1995; Koh et al., 1995a). AMPARs lacking GluR2 have a higher single-channel conductance than GluR2-containing receptors (Swanson et al., 1997); thus, the PICK1-dependent increase in the size of the AMPAR-mediated EPSC at -70 mV is likely attributable, at least in part, to this increase in single-channel conductance.
In control experiments, we investigated the effects of expressing a mutant form of PICK1 in which a double amino acid substitution was introduced into the PDZ domain (K27, D28→ A, A; “PICK1-AA”) (Fig. 3A). This mutant has been shown in yeast two-hybrid and biochemical assays not to interact with GluR2/3 (Xia et al., 1999; Perez et al., 2001) or PKCα (Staudinger et al., 1997). PICK1-AA had no effect either on the AMPA/NMDA ratio (105 ± 37%; p = 0.3; n = 12) (Fig. 3B,C) or on rectification (95 ± 32%; p = 0.9; n = 12) (Fig. 3B,C). This demonstrates that, as expected, the effects of PICK1 on AMPARs require a PDZ interaction. Importantly, these data also show that Sindbis viral infection itself does not have nonspecific effects on AMPAR-mediated synaptic transmission in neurons, over the time course of our experiments.
Figure 3.

A functional PDZ domain on PICK1 is required for the regulation of AMPARs by PICK1. A, Schematic showing the domain structure of wild-type PICK1 and the position of the amino acid substitutions in the mutant form, PICK1-AA, that lacks a functional PDZ domain. B, Averaged EPSCs from an example control neuron (left) and a neuron expressing PICK-AA (right) at a holding potential of -70 mV (bottom traces) and +40 mV (top traces). C, Pooled data (n = 12) showing the effects of PICK1-AA on the AMPA/NMDA ratio (C1) and rectification (C2).
PICK1 causes an increase in the sensitivity of AMPAR-mediated EPSCs to PhTx
The change in rectification caused by PICK1 suggests that the GluR2 content of AMPARs is altered. To further test this, we used the polyamine toxin PhTx-433 (Koh et al., 1995b; Washburn and Dingledine, 1996; Tóth and McBain, 1998), which blocks Ca2+-permeable AMPARs lacking edited GluR2. AMPAR-mediated EPSCs evoked in control cells in cultured slices showed little sensitivity to PhTx (1 μm) when it was bath applied during recordings (EPSC amplitude 15 min after application of PhTx, 87 ± 7% of baseline; n = 11) (Fig. 4A,C). This is in agreement with the small amount of inward rectification we observed for AMPARs in control neurons and consistent with previous work (Tóth and McBain, 1998; Tóth et al., 2000). However, in neurons virally expressing PICK1, EPSCs showed increased sensitivity to PhTx (EPSC amplitude percentage of baseline, 59 ± 9%; n = 5; p < 0.05 vs control) (Fig. 4B,C), providing additional evidence that PICK1 causes a reduction in the proportion of surface-expressed synaptic AMPARs containing GluR2.
Figure 4.
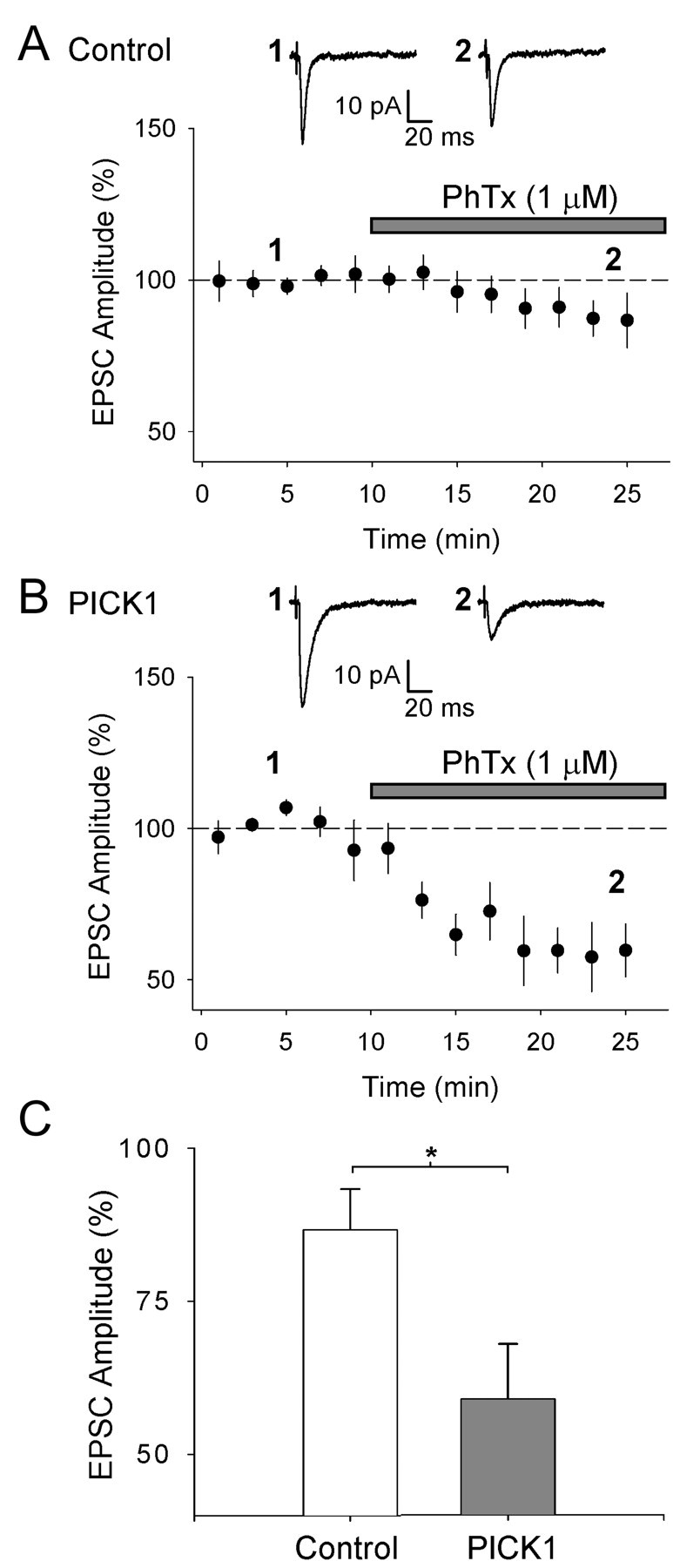
PICK1 causes an increase in the sensitivity of AMPAR-mediated EPSCs to PhTx. A, Pooled data (n = 11) for EPSC amplitude (normalized to mean amplitude before PhTx application) versus time for experiments in which PhTX (1 μm) was bath applied to cultured hippocampal slices during recordings from control (uninfected) neurons. B, Pooled data for the effects of PhTx on neurons infected with the PICK1 virus (n = 5). For A and B, the inset shows averaged EPSCs from an example experiment taken at the time points indicated. C, Summary analysis for the effects of PhTx on EPSC amplitude (averaged at 25 min as percentage of baseline) under both conditions. *p < 0.05.
PICK1 reduces GluR2, but not GluR1, surface expression
To directly investigate the effects of PICK1 on AMPAR surface expression, we studied AMPAR subunit distributions using immunocytochemistry in cultured hippocampal neurons. Using subunit specific N-terminal antibodies, we found that PICK1 caused a dramatic reduction in the number of endogenous GluR2 surface puncta (Fig. 5A,D), consistent with previous results for the effects of PICK1 on recombinant GluR2 (Perez et al., 2001). However, in contrast to GluR2, PICK1 had no effect on the number of GluR1 surface puncta (Fig. 5B,D). To investigate the locus of action of PICK1, we also studied the whole-cell distribution of GluR2 using the N-terminal antibody under permeabilizing conditions. PICK1 had no effect on the intracellular distribution or total levels of GluR2 (Fig. 5C,E), indicating that PICK1 did not prevent expression of GluR2 or its transport to the vicinity of synapses.
Figure 5.

PICK1 causes a reduction in surface-expressed endogenous GluR2 puncta but not GluR1 puncta in cultured hippocampal neurons. A, Examples of EGFP fluorescence (left) and surface GluR2 staining (N-terminal GluR2 antibody, nonpermeabilizing conditions; right) for a control neuron (top) and a neuron infected with the PICK1 virus (bottom). ForA-C, right panels are close-up images of antibody staining of the region of the dendrite indicated by the box (for all images, scale bars are 20 μm for low-power and 2 μm for close-up). B, Examples of EGFP fluorescence (left) and surface GluR1 staining (N-terminal GluR1 antibody, nonpermeabilizing conditions; right) for a control neuron (top) and a neuron infected with the PICK1 virus (bottom). C, Examples of EGFP fluorescence (left) and whole-cell GluR2 staining (N-terminal GluR2 antibody, permeabilizing conditions; right) for a control neuron (top) and a neuron infected with the PICK1 virus (bottom). D, Quantitative analysis of the effects of PICK1 viral expression on GluR2 and GluR1 surface levels (n = 10 neurons from three separate experiments for all experimental groups). ****p < 0.001. E, Quantitative analysis of the effects of PICK1 viral expression on GluR2 whole-cell levels in absolute intensity [arbitrary units (AU)] values for soma and dendrites (left) and ratio of dendritic/somatic fluorescence (right; n = 10 neurons from three separate experiments for control; n = 9 neurons from three separate experiments for PICK1).
We further investigated the effects of PICK1 on AMPAR subunit expression in cultured hippocampal neurons using biochemical assays. PICK1 caused no change in the total levels of either GluR1 or GluR2 (Fig. 6A). However, consistent with the immunocytochemistry data, cell surface biotinylation assays demonstrated that PICK1 caused a large decrease in GluR2 surface levels (Fig. 6B), whereas GluR1 surface levels were unchanged (Fig. 6C). Moreover, PICK1-AA, which lacks a functional PDZ domain, had no effect on surface levels of either GluR1 or GluR2 (Fig. 6B,C). This is also consistent with a lack of effect of this mutant on GluR2 surface levels as shown previously using immunocytochemistry (Perez et al., 2001). These immunocytochemical and biochemical analyses demonstrate that PICK1 greatly reduces GluR2 surface levels but does not cause a reduction in GluR1 surface expression. Consistent with the results from the electrophysiology, these data indicate that PICK1 decreases the GluR2 content of surface-expressed AMPARs.
Figure 6.

PICK1 causes a reduction in surface GluR2 but not in surface GluR1. A, Western blot analyses of total GluR2 (left) and total GluR1 (right) levels in cultured hippocampal neurons under control conditions and in sister cultures infected with the PICK1 virus (n = 3 experiments for both; for this and subsequent quantifications, values are expressed as a percentage of levels in sister control cultures). B, Western blot analysis of cell surface biotinylation of GluR2 in control cultures (n = 7) and in sister cultures infected with either the PICK1 virus (n = 7) or the PICK-AA virus (n = 3). **p < 0.01. C, Western blot analysis of cell surface biotinylation of GluR1 in control cultures (n = 6) and in sister cultures infected with either the PICK1 virus (n = 6) or the PICK-AA virus (n = 3).
Blockade of interactions at the NSF site on GluR2 does not affect rectification
We also studied the effects of virally expressing the peptide pep2m (KRMKVAKNAQ), which blocks interactions at the NSF site on GluR2 (Nishimune et al., 1998), to investigate whether interactions at this site also affects AMPAR rectification. Pep2m caused a reduction in the AMPA/NMDA ratio (67 ± 6%; p < 0.05, n = 16) (Fig. 7A,B) but had no effect on AMPAR rectification (101 ± 15%; p = 1.0; n = 16) (Fig. 7A,B). We also tested whether pep2m alters the sensitivity of AMPAR-mediated EPSCs to PhTx. Consistent with the lack of effect on rectification, pep2m expression did not increase the sensitivity of AMPAR-mediated EPSCs to PhTx (EPSC amplitude, 81 ± 5%; p = 0.5 vs control; n = 5) (Fig. 7C,D). Thus, in agreement with previous studies (Nishimune et al., 1998; Song et al., 1998), blockade of interactions at the NSF site causes a reduction in the AMPAR-mediated EPSC. The lack of effect of pep2m on rectification and PhTx sensitivity shows that, in contrast to the effects of PICK1 and consistent with previous immunocytochemical data (Lüscher et al., 1999; Noel et al., 1999), the receptors removed from the synapse are not replaced by GluR2-lacking receptors. These data, therefore, indicate that the effects of PICK1 involve a specific mechanism and are not simply an indirect consequence of removing surface-expressed AMPARs.
Figure 7.

Blockade of interactions at the NSF site on GluR2 reduces AMPAR-mediated EPSC amplitude but does not affect rectification. A, Averaged EPSCs from example experiments at a holding potential of -70 mV (bottom traces) and +40 mV (top traces) for a control uninfected neuron (left) and for a neuron expressing pep2m (right). B, Pooled data (n = 16) for the effects of pep2m expression on AMPA/NMDA ratio (B1) and rectification (B2). *p < 0.05. C, Pooled data of EPSC amplitude versus time for experiments in which PhTx (1 μm) was bath applied during recordings from neurons expressing pep2m (n = 5). D, Summary data showing the effects of PhTx on EPSC amplitude (averaged at 25 min as percentage of baseline) in neurons expressing pep2m compared with interleaved controls (same control data set as shown in Fig. 4).
PICK1 regulation of AMPARs requires PKC and CaMKII, but not protein kinase A, activity
Although it has been shown that PICK1 can bind to PKCα (Staudinger et al., 1997), little else is known about the signal transduction pathway(s) involving PICK1. We set out to investigate this by culturing slices continuously in the presence of blockers of various candidate kinases, while infecting slices with the PICK1 virus. Viral expression of PICK1 in the continuous presence of the PKC inhibitor bisindolylmaleimide I (BIS; 1 μm) (Toullec et al., 1991; Daw et al., 2000) strongly reduced the effects of PICK1 both on the AMPA/NMDA ratio (157 ± 17%; p < 0.05 vs control; n = 10; p < 0.05 vs PICK1 in the absence of BIS) (Fig. 8A) and on rectification (131 ± 16%; p = 0.1 vs control; n = 10; p < 0.01 vs PICK1 in the absence of BIS) (Fig. 8A), although in interleaved control experiments PICK1 had the usual effect on these parameters.
Figure 8.

The regulation of AMPARs by PICK1 requires endogenous PKC and CaMKII activity. A1, Averaged EPSCs from example experiments at a holding potential of -70 mV (bottom traces) and +40 mV (top traces) for a control uninfected neuron (left) and for a neuron virally expressing PICK1 (right) in slices cultured in the continuous presence of the PKC inhibitor BIS (1 μm). Summary data for the AMPA/NMDA ratio (A2) and rectification (A3) for these experiments (right pair of bars: n = 10 for Control+BIS; n = 10 for PICK1+BIS). Also shown are data from interleaved experiments for control neurons and neurons virally expressing PICK1 cultured under control conditions (left pair of bars: n = 22 for Control; n = 22 for PICK1). B, Similar experiments showing the results of CaMKII inhibition by KN62 (10 μm) on the effects of PICK1 expression (n = 12 for Control+KN62; n = 11 for PICK1+KN62; interleaved control experiments: n = 17 for Control; n = 7 for PICK1). *p < 0.05; ***p < 0.005.
Recent studies have provided evidence for a role of CaMKII and protein kinase A (PKA) in NMDAR-dependent AMPAR insertion (Hayashi et al., 2000; Shi et al., 2001; Esteban et al., 2003). We, therefore, studied the role of these kinases in the PICK1-induced changes in synaptic function. The CaMKII inhibitor KN62 (10 μm) (Fig. 8B) blocked the PICK1-induced changes in both the AMPA/NMDA ratio (121 ± 22% control; p = 0.6; n = 11) and rectification (112 ± 19% control; p = 0.6; n = 11), although in interleaved controls a robust effect of PICK1 was observed. Inhibition of PKA either with KT5720 (1 μm) or with Rp-cAMPS (100 μm), however, did not prevent the PICK1-induced changes in AMPAR EPSCs [for KT5720 (data not shown): AMPA/NMDA ratio, 175 ± 28% of control; p < 0.05; n = 7; rectification, 210 ± 33% of control; p < 0.05; n = 7; for Rp-cAMPS (data not shown): AMPA/NMDA ratio, 188 ± 29% of control; p < 0.05; n = 7; rectification, 227 ± 64% of control; p < 0.05; n = 7). These data show that the PICK1 mechanism for altering the subunit composition of AM-PARs requires a transduction cascade involving endogenous PKC and CaMKII.
Endogenous regulation of AMPAR rectification and EPSC amplitude
The data presented so far demonstrate that virally expressed PICK1 regulates AMPAR subunit composition and EPSC amplitude. To determine whether such a regulation occurs endogenously, we virally expressed peptides that disrupt PDZ protein interactions with GluR2. We used pep2-SVKI (YNVYGIESVKI), which is the extreme C terminal of GluR2, and two single-point mutants, pep2-EVKI (YNVYGIEEVKI) and pep2-SVKE (YNVYGIESVKE). Pep2-SVKI contains the type II C-terminal PDZ-binding motif of GluR2 and prevents PDZ protein interactions (PICK1, GRIP, and ABP) with GluR2. pep2-EVKI binds PICK1 but not GRIP or ABP, thus selectively blocking PICK1 PDZ domain interactions, and pep2-SVKE is a control peptide that does not block PDZ protein interactions (Li et al., 1999; Daw et al., 2000). Expression of either pep2-SVKI or pep2-EVKI caused a reduction in the AMPA/NMDA ratio and a decrease in rectification (Fig. 9A,B), effects opposite to that observed with PICK1 expression. However, expression of the control peptide pep2-SVKE (data not shown) had no effect on the AMPA/NMDA ratio (control, 2.10 ± 0.30; SVKE, 2.38 ± 0.20; p = 0.6; n = 8) or rectification (control, 3.42 ± 0.56; SVKE, 3.25 ± 0.47; p = 0.6; n = 8). This antagonistic effect of the active peptides demonstrates that there is an endogenous regulation of AMPAR subunit composition. The finding that pep2-EVKI causes changes in the AMPA/NMDA ratio and rectification suggests that it is endogenous PICK1 that is regulating AMPARs in this way.
Figure 9.
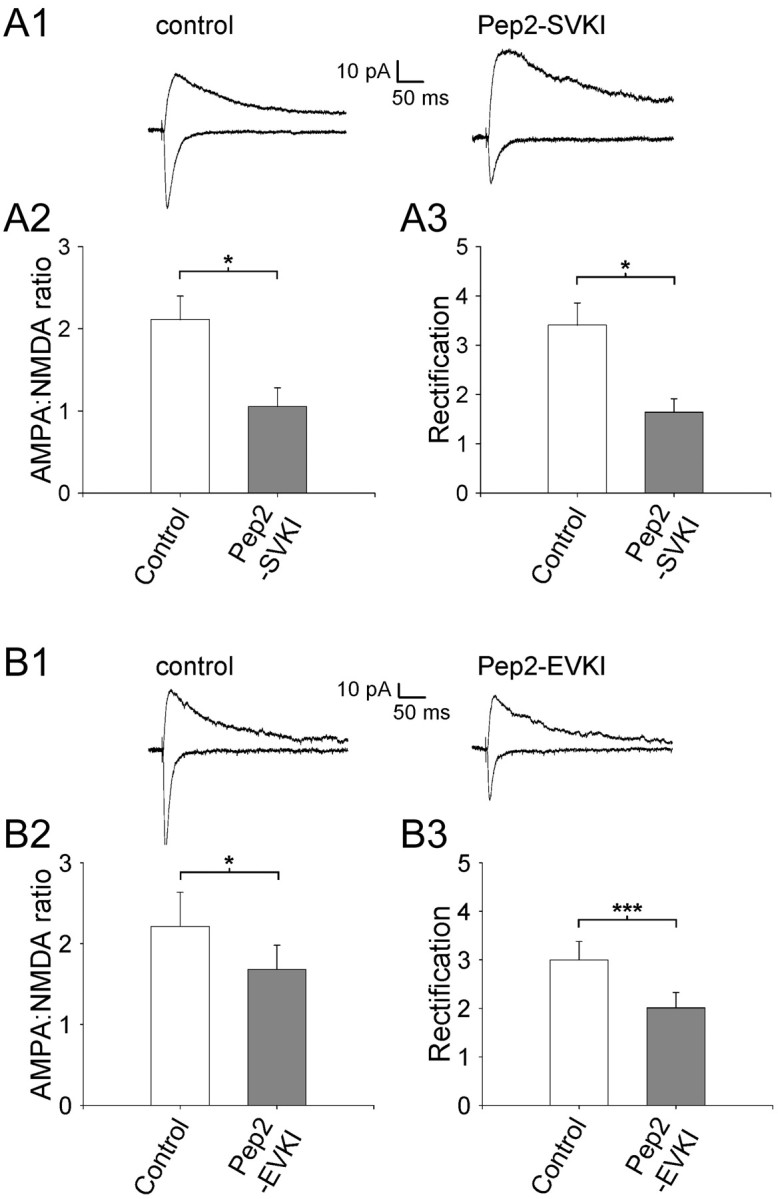
Endogenous regulation of AMPAR subunit composition by a mechanism involving PDZ domain interactions. A1, Averaged EPSCs from example experiments at a holding potential of -70 mV (bottom traces) and +40 mV (top traces) for a control uninfected neuron (left) and for a neuron expressing pep2-SVKI (right). Pooled data (n = 7) for the effects of pep2-SVKI expression on the AMPA/NMDA ratio (A2) and rectification (A3). B1, Averaged EPSCs from example experiments at a holding potential of 70 mV (bottom traces) and +40 mV (top traces) for a control uninfected neuron (left) and for a neuron expressing pep2-EVKI (right). Pooleddata (n = 8) for the effects of pep2-EVKI expression on the AMPA/NMDA ratio (B2) and rectification (B3). *p < 0.05; ***p < 0.005.
Discussion
We investigated the role of PICK1 in regulating endogenous AMPARs at CA1 synapses in hippocampal slices. Viral expression of PICK1 caused an increase in AMPAR-mediated EPSC amplitude, which was associated with increased rectification and sensitivity to polyamine toxin. This suggests that PICK1 expression causes a loss of GluR2-containing AMPARs that are replaced by GluR2-lacking AMPARs. This was confirmed by biochemical and immunocytochemical analyses of dissociated cultured hippocampal neurons that showed that PICK1 expression caused a profound loss of surface-expressed GluR2 but no change in surface GluR1. In contrast to PICK1, blockade of interactions at the NSF site on GluR2 with pep2m decreased EPSC amplitude but had no effect on rectification. This is consistent with previous studies showing that pep2m causes a general decrease in all surface-expressed AMPAR subunits (Lüscher et al., 1999; Noel et al., 1999).
In addition, we show that blockade of PKC and CaMKII, but not PKA, prevented viral-expressed PICK1 from exerting its effects. Moreover, disruption of endogenous PDZ interactions with pep2-SVKI (the extreme C terminus of GluR2), or a point mutant (pep2-EVKI) that selectively blocks PICK1 interactions, caused the opposite effects on synaptic AMPAR function to PICK1 expression. These findings demonstrate that the activation of an endogenous signal transduction cascade involving PKC and CaMKII is required for the effects of PICK1 to occur and that AMPAR GluR2 content is physiologically regulated at CA1 synapses. This suggests that endogenous PICK1 regulates synaptic strength and the GluR2 content of AMPARs in a CaMKII- and PKC-dependent mechanism.
Our data using the C-terminal GluR2 peptides indicates that the GluR2 content of AMPARs at CA1 synapses is under ongoing physiological regulation by PICK1. The active peptides (pep2-SVKI, pep2-EVKI) caused a reduction in the AMPA/NMDA ratio and a decrease in rectification. Under our experimental conditions, we found that there was some inward rectification of AMPARs under control conditions and that these receptors showed some sensitivity to PhTx, as has been shown previously (Tóth and McBain, 1998; Tóth et al., 2000). This indicates that some fraction of the AMPARs under control conditions lacks edited GluR2 or has a low copy number of this subunit (Washburn et al., 1997) and, thus, there is scope for increasing the GluR2 content of AMPARs at CA1 synapses. Moreover, AMPARs containing a high copy number of edited GluR2 show outward rectification (Verdoorn et al., 1991; Liu and Cull-Candy, 2000, 2002). Therefore, the observed decrease in rectification with pep2-SVKI and pep2-EVKI is consistent with an increase in GluR2 content and suggests that PICK1 acts to limit the abundance of this subunit at synapses.
An additional possibility that has been proposed for the actions of pep2-SVKI and pep2-EVKI is that their effects can be attributed to a blockade of PKC activity because they are potential PKC substrates (Li et al., 1999; Daw et al., 2000). However, that they act through this mechanism is unlikely because pep2-SVKE, which also contains the consensus sequence (S/T-X/K/R) for PKC phosphorylation but lacks the PDZ-binding motif, was without effect.
It is likely that part or all of the potentiation of AMPAR-mediated transmission by PICK1 is attributable to the decrease in the GluR2 content of receptors. Such a reduction in GluR2 content will cause an increase in AMPAR-mediated EPSC amplitude because of the higher single-channel conductance exhibited by such receptors compared with those containing GluR2 (Swanson et al., 1997). In this regard, strongly rectifying, Ca2+-permeable AMPARs in hippocampal and neocortical interneurons, which express low levels of GluR2 (Geiger et al., 1995; Lambolez et al., 1996), have a twofold to threefold higher single-channel conductance than pyramidal neurons that have predominantly GluR2-containing AMPARs (Hestrin, 1993; Koh et al., 1995a; Spruston et al., 1995). This difference is very similar to the increase in EPSC amplitude we observed with expression of PICK1. It is also possible, however, that an increase in AMPAR number at synaptic sites also contributes to the potentiation we observed. Such an increase would not be detected in our immunocytochemistry experiments because they were designed to detect changes in surface puncta number. Nor would they be detected in the cell surface biotinylation experiments because this assay cannot distinguish between extrasynaptic and synaptic receptors. In addition, overexpression of a number of postsynaptic density proteins (e.g., PSD95, GluR2) has been shown to cause an increase in the size of spines (El-Husseini et al., 2000; Passafaro et al., 2003). Therefore, it is also possible that PICK1 may cause an increase in spine size, allowing the accommodation of more AMPARs at the synapse.
As with all studies using an overexpression paradigm, in the present experiments a possibility that has to be considered is that the overexpressed recombinant PICK1 exerts its effects in some nonspecific way because of aberrant trafficking or compartmentalization, or by interfering with the trafficking of other endogenous proteins. We think that it is unlikely that the effects we described are attributable to such effects because (1) the inactive mutant, PICK1-AA, has no effect on synaptic transmission, arguing against a nonspecific disruption in trafficking; (2) the effects of PICK1 expression are blocked by PKC or CaMKII inhibitors, demonstrating that PICK1 is not simply acting through its role as a GluR2-binding protein but rather requires an endogenous kinase cascade to exert its effects; and (3) blockade of endogenous type II PDZ interactions, and more specifically PICK1 PDZ interactions, in the absence of overexpressed PICK1 causes changes in synaptic strength and AMPAR rectification. Thus, we have strong evidence that the PICK1-dependent regulation of the GluR2-content of synaptic AMPARs is a physiological mechanism.
Mechanisms of regulation of AMPAR subunit composition by PICK1
Our data indicate that PICK1 regulates the subunit composition of synaptic receptors by removing GluR2-containing AMPARs from the surface, which are then replaced by GluR2-lacking receptors in a mechanism involving the activation of PKC and CaMKII. We found that PICK1 did not alter surface levels of GluR1, suggesting that the new receptors are either composed of GluR3 or GluR4 subunits and/or are recruited from an extrasynaptic population.
Analysis of the PICK1-AA mutant shows that a PDZ interaction is required for the regulation of AMPARs by PICK1. In addition to GluR2/3 (Dev et al., 1999; Xia et al., 1999) and PKCα (Staudinger et al., 1997), PICK1 interacts with a number of other proteins via its PDZ domain [e.g., ephrin receptors (Torres et al., 1999), the kainate receptor subunits GluR5 and GluR6 (Hirbec et al., 2003), and mGluR7 (Boudin et al., 2000; Dev et al., 2000; El Far et al., 2000)]. We cannot exclude the possibility that some of the effects of PICK1 involve one or some of these other interactors. However, because in the present study PICK1 selectively affected AMPAR function and blockade of PICK1 PDZ interactions with peptides had the opposite effect to PICK1 expression, it seems likely that the effects we observed were primarily attributable to the direct interaction of PICK1 with GluR2/3.
Although previous studies have provided evidence that PICK1 acts at synapses to regulate AMPARs (Chung et al., 2000; Kim et al., 2001; Perez et al., 2001), a recent study suggested that it is also required for the efficient export of GluR2 from the endoplasmic reticulum (Greger et al., 2002). Our immunocytochemical and biochemical data show that PICK1 does not detectably alter the intracellular distribution of GluR2, or affect total GluR2 and GluR1 protein levels. These data, therefore, argue against any PICK1-dependent gross change in the intracellular levels or assembly of these subunits in our experiments.
Role of GluR2 regulation by PICK1
The presence of edited GluR2 confers key properties to the AMPAR by determining its single-channel conductance, rectification properties, and permeability to Ca2+ (Burnashev et al., 1992; Bowie and Mayer, 1995; Geiger et al., 1995; Kamboj et al., 1995; Koh et al., 1995a; Washburn and Dingledine, 1996; Swanson et al., 1997; Washburn et al., 1997). The critical importance of the GluR2 subunit has been demonstrated using transgenic mice that either lack GluR2 (Jia et al., 1996) or the appropriate editing of this subunit (Brusa et al., 1995). There is also evidence that one important mechanism for excitotoxicity involves a decrease in the GluR2 content of synaptic AMPARs, leading to increased Ca2+ entry into neurons (Tanaka et al., 2000). Moreover, the regulation of the GluR2-dependent properties of synaptic AMPARs has been demonstrated to occur acutely during a form of synaptic plasticity in the cerebellum (Liu and Cull-Candy, 2000, 2002) and chronically during development at hippocampal and neocortical synapses (Pickard et al., 2000; Kumar et al., 2002). In none of these studies, however, has the molecular mechanism for these changes in AMPAR GluR2 content been determined. Our data now provide evidence for a novel molecular mechanism that could allow such a dynamic regulation of the GluR2 content of AMPARs.
Footnotes
This work was supported by the Medical Research Council (J.M.H., G.L.C.) and the Wellcome Trust (A.T., K.K.D., F.D., S.Z., J.M.H., G.L.C., J.T.R.I.). We are grateful to Bill Anderson for supplying the data acquisition software (www.ltp-program.com). We also thank Jon Hanley, Werner Hoch, and Elek Molnar for critical reading of this manuscript.
Correspondence should be addressed to Dr. John Isaac, Medical Research Council Centre for Synaptic Plasticity, Department of Anatomy, University of Bristol, Bristol BS8 1TD, UK. E-mail: j.t.r.isaac@bris.ac.uk.
K. K. Dev's present address: Novartis Pharma AG, Nervous Systems Research, WSJ-386.7.43, CH-4002 Basel, Switzerland.
G. Meyer's present address: Max-Planck-Institute for Experimental Medicine, Hermann-Rein-Strasse 3, D-37075 Göttingen, Germany.
F. Duprat's present address: Institut de Pharmacologie Moléculaire et Cellulaire, 06560 Valbonne, France.
S. Zaman's present address: Guy's Hospital, 47 Weston Street, London SE1 3RR, UK.
Copyright © 2004 Society for Neuroscience 0270-6474/04/245381-10$15.00/0
A.T. and L.C. contributed equally to this work
References
- Boudin H, Doan A, Xia J, Shigemoto R, Huganir RL, Worley P, Craig AM (2000) Presynaptic clustering of mGluR7a requires the PICK1 PDZ domain binding site. Neuron 28: 485-497. [DOI] [PubMed] [Google Scholar]
- Bowie D, Mayer ML (1995) Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron 15: 453-462. [DOI] [PubMed] [Google Scholar]
- Brusa R, Zimmermann F, Koh DS, Feldmeyer D, Gass P, Seeburg PH, Sprengel R (1995) Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science 270: 1677-1680. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B (1992) Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron 8: 189-198. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL (2000) Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with the PDZ domain-containing proteins. J Neurosci 20: 7258-7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, Collingridge GL, Isaac JTR (2000) PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron 28: 873-886. [DOI] [PubMed] [Google Scholar]
- Dev KK, Nishimune A, Henley JM, Nakanishi S (1999) The protein kinase Cα binding protein PICK1 interacts with short but not long form alternative splice variants of AMPA receptor subunits. Neuropharmacology 38: 635-644. [DOI] [PubMed] [Google Scholar]
- Dev KK, Nakajima Y, Kitano J, Braithwaite SP, Henley JM, Nakanishi S (2000) PICK1 interacts with and regulates PKC phosphorylation of mGLUR7. J Neurosci 20: 7252-7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, O'Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL (1997) GRIP: a synaptic PDZ-containing protein that interacts with AMPA receptors. Nature 386: 279-284. [DOI] [PubMed] [Google Scholar]
- Duprat F, Daw MI, Lim W, Collingridge GL, Isaac JTR (2003) GluR2 protein-protein interactions and the regulation of AMPA receptors during synaptic plasticity. Proc R Soc Lond B 358: 715-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Far O, Airas J, Wischmeyer E, Nehring RB, Karschin A, Betz H (2000) Interaction of the C-terminal tail region of the metabotropic glutamate receptor 7 with the protein kinase C substrate PICK1. Eur J Neurosci 12: 4215-4221. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000) PSD-95 involvement in maturation of excitatory synapses. Science 290: 1364-1368. [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R (2003) PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6: 136-143. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Melcher TD, Koh S, Sakmann B, Seeburg PH, Jonas P, Monyer H (1995) Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 15: 193-204. [DOI] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Ziff EB (2002) RNA editing at Arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron 34: 759-772. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi S-H, Esteban JA, Piccini A, Poncer J-C, Malinow R (2000) Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287: 2262-2267. [DOI] [PubMed] [Google Scholar]
- Hestrin S (1993) Different glutamate receptor channels mediate fast excitatory synaptic currents in inhibitory and excitatory cortical neurons. Neuron 11: 1083-1091. [DOI] [PubMed] [Google Scholar]
- Hirbec H, Francis JC, Lauri SE, Braithwaite SB, Coussen F, Mulle C, Dev KK, Couthino V, Meyer G, Isaac JTR, Collingridge GL, Henley JM (2003) Rapid and differential regulation of AMPA and kainate receptors at hippocampal mossy fibre synapses by PICK1 and GRIP. Neuron 37: 625-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J (1996) Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron 17: 945-956. [DOI] [PubMed] [Google Scholar]
- Kamboj SK, Swanson GT, Cull-Candy SG (1995) Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. J Physiol (Lond) 486: 297-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, Lee HK, Huganir RL (2001) Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci USA 98: 11725-11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh DS, Geiger JR, Jonas P, Sakmann B (1995a)) Ca(2+)-permeable AMPA and NMDA receptor channels in basket cells of rat hippocampal dentate gyrus. J Physiol (Lond) 485: 383-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh DS, Burnashev N, Jonas P (1995b)) Block of native Ca(2+)-permeable AMPA receptors in rat brain by intracellular polyamines generates double rectification. J Physiol (Lond) 486: 305-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Bacci A, Kharazia V, Huguenard JR (2002) A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci 22: 3005-3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambolez B, Ropert N, Perrais D, Rossier J, Hestrin S (1996) Correlation between kinetics and RNA splicing of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors in neocortical neurons. Proc Natl Acad Sci USA 93: 1797-1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Kerchner GA, Sala C, Wei F, Huettner JE, Sheng M, Zhuo M (1999) AMPA receptor-PDZ interactions in facilitation of spinal sensory synapses. Nat Neurosci 2: 972-977. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Cull-Candy SG (2002) Activity-dependent change in AMPA receptor properties in cerebellar stellate cells. J Neurosci 22: 3881-3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SQ, Cull-Candy SG (2000) Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature 405: 454-458. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA (1999) Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron 24: 649-658. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103-126. [DOI] [PubMed] [Google Scholar]
- Nishimune A, Isaac JTR, Molnar E, Noel J, Nash SR, Tagaya M, Collingridge GL, Nakanishi S, Henley JM (1998) NSF binding to GluR2 regulates synaptic transmission. Neuron 21: 87-97. [DOI] [PubMed] [Google Scholar]
- Noel J, Ralph GS, Pickard L, Willimans J, Molnar E, Uney JB, Collingridge GL, Henley JM (1999) Surface expression of AMPA receptors in hippocampal neurons is regulated by an NSF-dependent mechanism. Neuron 23: 365-376. [DOI] [PubMed] [Google Scholar]
- Oliet SHR, Malenka RC, Nicoll RA (1997) Two distinct forms of long-term depression coexist in CA1 pyramidal cells. Neuron 18: 969-982. [DOI] [PubMed] [Google Scholar]
- Passafaro M, Nakagawa T, Sala C, Sheng M (2003) Induction of dendritic spines by an extracellular domain of AMPA receptor subunit GluR2. Nature 424: 677-681. [DOI] [PubMed] [Google Scholar]
- Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB (2001) PICK1 targets activated protein kinase C alpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci 21: 5417-5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard L, Noel J, Henley JM, Collingridge GL, Molnar E (2000) Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA receptor subunit composition in living hippocampal neurons. J Neurosci 20: 7922-7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Lee SH (2001) AMPA receptor trafficking and the control of synaptic transmission. Cell 105: 825-828. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R (2001) Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105: 331-343. [DOI] [PubMed] [Google Scholar]
- Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL (1998) Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron 21: 393-400. [DOI] [PubMed] [Google Scholar]
- Spruston N, Jonas P, Sakmann B (1995) Dendritic glutamate receptor channels in rat hippocampal CA3 and CA1 pyramidal neurons. J Physiol (Lond) 482: 325-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Osten P, Vilim FS, Khatri L, Inman G, States B, Daly C, DeSouza S, Abagyan R, Valtschanoff JG, Weinberg RJ, Ziff EB (1998) Novel anchorage of GluR2/3 to the postsynaptic density by AMPA receptor-binding protein ABP. Neuron 21: 581-591. [DOI] [PubMed] [Google Scholar]
- Staudinger J, Lu J, Olson EN (1997) Specific interaction of the PDZ domain protein PICK1 with the COOH terminus of protein kinase C-alpha. J Biol Chem 272: 32019-32024. [DOI] [PubMed] [Google Scholar]
- Swanson GT, Kamboj SK, Cull-Candy SG (1997) Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci 17: 58-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Grooms SY, Bennett MV, Zukin RS (2000) The AMPAR subunit GluR2: still front and center-stage. Brain Res 886: 190-207. [DOI] [PubMed] [Google Scholar]
- Torres R, Firestein BL, Dong H, Staudinger J, Olson EN, Huganir RL, Bredt DS, Gale NW, Yancopoulos GD (1998) PDZ proteins bind, cluster, and synaptically colocalize with Eph receptors and their ephrin ligands. Neuron 21: 1453-1463. [DOI] [PubMed] [Google Scholar]
- Tóth K, McBain CJ (1998) Afferent-specific innervation of two distinct AMPA receptor subtypes on single hippocampal interneurons. Nat Neurosci 1: 572-578. [DOI] [PubMed] [Google Scholar]
- Tóth K, Suares G, Lawrence JJ, Philips-Tansey E, McBain CJ (2000) Differential mechanisms of transmission at three types of mossy fiber synapse. J Neurosci 20: 8279-8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamel L, Charon D, Kirilovsky J (1991) The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem 266: 15771-15781. [PubMed] [Google Scholar]
- Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B (1991) Structural determinants of ion flow through recombinant glutamate receptor channels. Science 252: 1715-1718. [DOI] [PubMed] [Google Scholar]
- Washburn MS, Dingledine R (1996) Block of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by polyamines and polyamine toxins. J Pharmacol Exp Ther 278: 669-678. [PubMed] [Google Scholar]
- Washburn MS, Numberger M, Zhang S, Dingledine R (1997) Differential dependence on GluR2 expression of three characteristic features of AMPA receptors. J Neurosci 17: 9393-9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Zhang X, Staudinger J, Huganir RL (1999) Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron 22: 179-187. [DOI] [PubMed] [Google Scholar]