

Abstract
Three series of structurally isomeric 2-benzylidene-6-(nitrobenzylidene) cyclohexanones 1–3 were prepared and evaluated against human Molt/C8 and CEM T-lymphocytes as well as murine L1210 cells. The IC50 values of the majority of compounds are less than 10 μM and in some assays, the figures for 1d and 1e are submicromolar. Correlations were discerned between cytotoxic potencies and the atomic charges on one of the olefinic carbon atoms, the torsion angles between an aryl ring, and the adjacent unsaturated group as well as logP values. Three representative compounds were examined for their effect on respiration in rat liver mitochondria.
Keywords: Unsaturated ketones, Molecular modeling, Cytotoxicity, Mitochondria
1. Introduction
The principal interest in our laboratory is the syntheses of a variety of conjugated styryl ketones as candidate antineoplastic agents. These compounds are thiol alkylators having little or no capacity to interact with amino or hydroxy groups1,2; since these latter groups, but not thiols, are found in nucleic acids, enones may be devoid of the genotoxic problems displayed by a number of anticancer drugs.3 Recently molecules have been designed to enable successive alkylation of thiols to occur since on occasion sequential reactions with cellular constituents have been claimed to be more detrimental to malignant cells than the corresponding normal tissues.4 These considerations led to the decision to prepare a number of compounds which contain the 1,5-diaryl-3-oxo-1,4-pentadienyl pharmacophore (ArCH=CR–CO–CR=CHAr)5,6 thereby allowing stepwise alkylation of cellular thiols. Recently a small number of 2,6-bis(benzylidene) cyclohexanones were prepared in which the substituents in each of the aryl rings differed in their electronic properties.7,8 In these molecules, the charges on the olefinic carbon atoms are predicted to be divergent thereby enhancing sequential reactions.
The objectives of the present study were twofold. First, an evaluation was planned of the hypotheses that cytotoxic potencies were correlated with both the charge densities and the steric environment of the olefinic carbon atoms. Second, the original series consisted of a small group of compounds which possessed widely differing potencies in the Molt 4/C8, CEM, and L1210 bioassays. 8 Hence expansion of the cluster of compounds was indicated in order to draw meaningful conclusions pertaining to those structural features which contribute to cytotoxicity.
In ring A of series 1, the strongly electron-attracting 2-nitro group was proposed which should cause the olefinic carbon atoms, designated CA and CB as indicated in Figure 1, to be electron deficient thereby enhancing thiol alkylation. Substituents with varying Hammett sigma (σ) values were considered for insertion onto ring B, including the 3,4,5-trimethoxy group due to our recent disclosure of the cytotoxicity of compounds containing the 3-(3,4,5-trimethoxyphenyl)-2-propenoyl substituent. 9 In addition, the rate of electrophilic attack with thiols will be influenced by the topography of the molecules in the environment of the CA and CB atoms. Hence the determination of the torsion angles θA and θB created between the aryl rings A and B with the adjacent olefinic carbon atoms was suggested. Such considerations led to the decision to prepare series 1. In order for these hypotheses to be examined further, the placement of the nitro group in other locations of ring A was planned leading to series 2 and 3. In addition, to assist in the understanding of those structural features in series 1–3 which contribute to cytotoxic potencies, the monobenzylidene analogs 4a–c were also proposed. The general structures of these compounds are presented in Figure 1.
Figure 1.
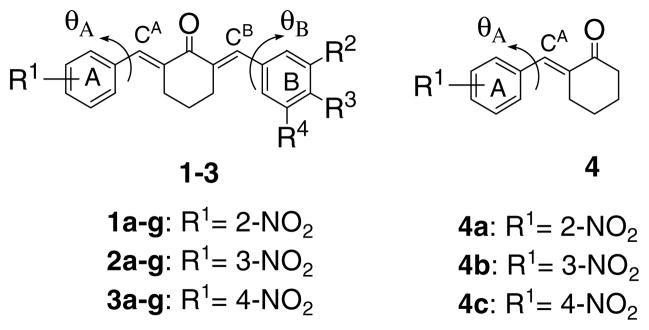
General structures of series 1–4. The R2, R3, and R4 substituents in series 1–3 are as follows, namely a: R2 = R4 =H, R3 = N(CH3)2; b: R2 = R4 =H, R3 = OCH3; c: R2 = R4 =H, R3 = CH3; d: R2 = R3 = R4 = OCH3; e: R2 = R3 = R4 = H; f: R2 = R4= H, R3 = F; g: R2 = R4 = H, R3 = Cl.
2. Results
The compounds in series 1–4 were prepared by the synthetic routes outlined in Scheme 1. The majority of the bis(benzylidene)cyclohexanones were prepared by condensation of 4a, 4b, or 4c with various aryl aldehydes under acidic conditions. However, attempts to use this procedure in the preparation of 1a, 1c, 2a, and 3a led to the formation of dark polymeric material from which no products were obtained. Under basic conditions, 2-(4-dimethylaminobenzylidene) cyclohexanone 5 and the related 4-methyl analog 6 reacted with the appropriate nitrobenzaldehyde to afford 1c and 2a. However under these conditions, reaction of 5 with both 2-nitrobenzaldehyde and 4-nitroarylaldehydes led to the formation of multiple products but under acidic conditions, both 1a and 3a were formed. Initial attempts to prepare 4a–c from cyclohexanone and the relevant nitrobenzaldehyde under acidic conditions led to isolation of the corresponding 2,6-bis(nitrobenzylidene)cyclohexanones. In the presence of sodium hydroxide solution, the 2-nitro and 4-nitro benzaldehydes condensed with cyclohexanone to produce the intermediate aldols which were dehydrated by acid to give 4a and 4c, respectively. Under basic conditions, the 3-nitroaldehyde gave only 2,6-bis(3-nitrobenzylidene)cyclohexanone. Hence 4b was prepared via the enamine route as illustrated in Scheme 1. 1H NMR spectroscopy revealed that each of the products in series 1–4 was isomerically pure. The absorptions of the olefinic protons were in the region of 7.47–7.97 ppm which is characteristic of E isomers, since compounds possessing the Z configuration absorb at higher fields.10 Furthermore, X-ray crystallography revealed that the olefinic double bonds adopted the E configuration in 3c11 and 3f12 as well as a related 2,6-bis(benzylidene)cyclohexanone.13 The assumption was made therefore that the olefinic bonds in series 1–4 adopted the E configuration. Models of the compounds in series 1–4 were built and the charge densities of the CA and CB atoms as well as the torsion angles θA and θB were determined and are presented in Table 2. In addition, the logP values of all of the compounds were obtained and are portrayed in Table 2.
Scheme 1.

Synthetic chemical routes in the preparation of the compounds in series 1–4. Reagents: (i) NaOH; (ii) HCl/2-NO2C6H4CHO; (iii) HCl/4-NO2C6H4CHO; (iv) NaOH/3-NO2C6H4CHO; (v) NaOH/2–NO2C6H4CHO; (vi) HCl; (vii) NaOH/4-NO2C6H4CHO; (viii) morpholine/4-CH3C6H4SO2OH; (ix) 3-NO2C6H4CHO.
Table 2.
Some physicochemical properties of compounds 1–4
| Compound | Atomic chargesa
|
Torsion anglesb
|
logP | ||
|---|---|---|---|---|---|
| qA | qB | θA | θB | ||
| 1a | −0.086 | −0.032 | 76.79 | −51.12 | 4.80 |
| 1b | −0.081 | −0.041 | 76.76 | −51.27 | 4.76 |
| 1c | −0.082 | −0.047 | −76.83 | 51.58 | 5.15 |
| 1d | −0.092 | −0.053 | 76.90 | −51.53 | 4.33 |
| 1e | −0.133 | −0.055 | 76.59 | −51.86 | 4.70 |
| 1f | −0.130 | −0.058 | −76.79 | 51.55 | 4.86 |
| 1g | −0.130 | −0.061 | −76.84 | 51.72 | 5.38 |
| 2a | −0.093 | −0.028 | −51.30 | 50.72 | 5.01 |
| 2b | −0.089 | −0.042 | −51.28 | 51.12 | 4.96 |
| 2c | −0.083 | −0.045 | −51.27 | 51.42 | 5.35 |
| 2d | −0.079 | −0.053 | −51.32 | 51.83 | 4.53 |
| 2e | −0.087 | −0.053 | −51.27 | 51.56 | 4.90 |
| 2f | −0.085 | −0.056 | −51.25 | 51.36 | 5.07 |
| 2g | −0.084 | −0.059 | −51.24 | 51.58 | 5.58 |
| 3a | −0.092 | −0.022 | −51.14 | 50.98 | 5.03 |
| 3b | −0.096 | −0.039 | −51.11 | 51.09 | 4.98 |
| 3c | −0.089 | −0.043 | −51.14 | 51.41 | 5.38 |
| 3d | −0.086 | −0.053 | 51.17 | −51.86 | 4.56 |
| 3e | −0.095 | −0.050 | −51.14 | 51.56 | 4.93 |
| 3f | −0.092 | −0.053 | −51.14 | 51.37 | 5.09 |
| 3g | −0.092 | −0.056 | −51.16 | 51.57 | 5.61 |
| 4a | −0.092 | — | −69.54 | — | 2.92 |
| 4b | −0.083 | — | −51.50 | — | 3.12 |
| 4c | −0.090 | — | −50.68 | 3.14 | |
All the compounds in series 1–4 were evaluated against human Molt 4/C8 and CEM T-lymphocytes and murine L1210 lymphoid leukemia cells. These data are summarized in Table 1. The effect of representative compounds on respiration in mitochondria isolated from rat liver cells is presented in Figure 2.
Table 1.
Cytotoxic properties of compounds 1–4
| Compound | IC50a (μM)
|
||
|---|---|---|---|
| Molt 4/C8 | CEM | L1210 | |
| 1a | 122 ± 6 | 168 ± 36 | 164± 27 |
| 1b | 8.90±0.20 | 7.45±0.08 | 42.4±1.3 |
| 1c | 7.52±0.45 | 6.09±2.12 | 7.77±0.45 |
| 1d | 0.702±0.22 | 0.402±0.033 | 1.52±0.29 |
| 1e | 1.48±0.11 | 0.925±0.056 | 4.84±0.40 |
| 1f | 1.87±0.06 | 1.51±0.04 | 8.40±0.13 |
| 1g | 3.86±1.00 | 1.75±0.14 | 9.38±0.47 |
| 2a | 10.9±0.8 | 11.7±0.8 | 156±134 |
| 2b | 7.98±0.54 | 8.22±0.12 | 29.5±9.8 |
| 2c | 9.53±1.23 | 10.1±0.6 | 41.8±3.7 |
| 2d | 44.0± 2.7 | 45.2±7.5 | 42.2±3.3 |
| 2e | 1.70±0.42 | 2.29±0.75 | 9.44±1.07 |
| 2f | 5.12±2.31 | 5.05±3.02 | 16.3±0.3 |
| 2g | 2.02±0.28 | 1.75±0.00 | 9.16±0.87 |
| 3a | >500 | >500 | >500 |
| 3bb | 300±54 | 250±6 | 240±8 |
| 3c | 8.44±0.49 | 8.53±0.31 | 8.16±0.35 |
| 3db | 6.42±1.07 | 4.61±3.89 | 6.97±1.80 |
| 3e | 8.35±0.95 | 9.32±0.20 | 9.80±0.18 |
| 3f | 17.1±4.6 | 18.6±6.9 | 26.8±2.8 |
| 3gb | 9.12±0.28 | 8.18±0.20 | 9.41±0.97 |
| 4a | 33.3±3.1 | 36.4±1.3 | 23.8±12.4 |
| 4b | 8.28±0.69 | 8.12±0.92 | 50.1±10.4 |
| 4c | 13.9±1.0 | 19.3±1.5 | 46.5±9.1 |
| Melphalanb | 3.24 ±0.79 | 2.47 ± 0.30 | 2.13±0.03 |
The IC50 values indicate the concentrations of compounds required to inhibit the growth of the cells by 50%.
The data were previously reported in Ref. 8 [copyright (2006) by Elsevier].
Figure 2.

The effect of 1d, 2d, 3d, and solvent control (dimethylsulfoxide 4 μL) on respiration in rat liver mitochondria. The figures for 1d, 2d, and 3d are different from each other and the solvent control taking standard deviations into account.
3. Discussion
The bioevaluations of 1a–g, 2a–g, 3a–g, and 4a–c toward three cell lines are presented in Table 1. The IC50 values of 1d and 1e are submicromolar in some of the bioassays and 58% of the IC50 values were less than 10 μM. In view of these promising results, various studies were initiated to seek correlations between cytotoxic potencies and different physicochemical and biochemical parameters of these molecules with the aim of obtaining guidelines for expansion of this project.
Interactions with cellular thiols are believed to occur at the olefinic carbon atoms designated CA and CB. The atomic charges on these atoms in the compounds 1–4 are presented in Table 2. The nitro group in ring A is the most electron-attracting substituent having a Taft σ* value of 0.9714 (series 1 and 4a) and Hammett σ values of 0.71 (series 2 and 4b) and 0.78 (series 3 and 4c).15 The σ constants for the ring B substituents in a–g are −0.83, −0.27, −0.17, −0.03, 0.00, 0.06, and 0.23, respectively, 16 and are arranged in sequence with a bearing the most electron-repelling group and g the most electronattracting substituent. For each compound in series 1– 3, the electron densities are lower on the CB rather than the CA atoms. Thus the polarization of the π electrons in the conjugated 1,5-diaryl-3-oxo-1,4-pentadienyl group is toward the nitro substituents, causing the CB atom to have lower electron densities than CA. Hence thiol alkylation is predicted to take place initially at CB and subsequently at CA. In order to examine whether cytotoxic potencies are correlated with the electron densities on the CA and CB atoms, linear plots were made between these values and the IC50 figures of 1a–g in each of the three bioassays. This experiment was repeated with 2a– g and finally with 3a–g. Positive correlations (p < 0.05) were noted when considering the atomic charges on the CB atoms except for the Molt 4/C8 and CEM biodata for series 2. No correlations were noted between the IC50 figures and the charges on the CA atom (p > 0.05). This evaluation was repeated except that the IC50 values were plotted against the σ constants in ring B. Negative correlations (p < 0.01) were obtained in all cases except for 2a–g in the Molt 4/C8 and CEM tests (p > 0.05). Thus potency increases (IC50 values diminish) as the electron densities on the CB atom are decreased (positive correlation) and the σ constants are elevated (negative correlation). This observation may be rationalized by considering that attack of cellular thiols at CB will be enhanced by a reduction in the atomic charges on the CB atoms. Thus in the future, compounds may be designed having substituents in ring B which have large positive sigma values.
Consideration was given to the possibility that the steric environment at the olefinic carbon atoms influences the extent of thiol alkylation and hence cytotoxic potencies. Thiolation is believed to occur initially at CB and the θB values recorded in Table 2 reveal that they are virtually constant. Thus the average θB values for series 1, 2, and 3 are 51.5°, 51.4°, and 51.4°, respectively, and there are very small variations in these torsion angles within each series. Hence the differences in cytotoxic potencies are unlikely to be influenced by the torsion angles θB. The average θA angles in series 1, 2, and 3 are 76.8°, 51.3°, and 51.1°, respectively, and minimal variation of these torsion angles was noted within each series. Since the cytotoxic potencies of the compounds in series 1 are greater than the analogs in series 2 and 3 vide infra, these torsion angles may influence the magnitude of the cytostatic effect. Hence in the future, groups with larger molecular refractivity values than nitro group should be placed in the 2 position of ring A or two ortho substituents should be employed which may lead to more potent analogs. The insertion of a second arylidene ring onto 4a–c leading to series 1–3, respectively, caused only minimal changes in the CA and θA values as the data in Table 2 indicate.
The biodata in Table 1 were examined further with a view to discerning those structural features which influence cytotoxic potencies. First, the optimal position of the nitro group in ring A was considered. A point system of 3 (highest potency), 2, and 1 (lowest potency) was used in comparing the IC50 values of compounds having the same substituents in ring B. Thus in the Molt 4/C8 assay, the IC50 figures of 1a, 2a, and 3a were compared, then 1b, 2b, and 3b and so forth. Standard deviations were taken into account and when the IC50 values were statistically indistinguishable, equal points were allocated bearing in mind that for each comparison of three compounds, a total of six points were invariably awarded. Use of this methodology indicated that the figures for series 1, 2, and 3 are 16.5, 16.5, and 9, respectively (Molt 4/C8 assay), 19.5, 13.5, and 9, respectively (CEM test) and 18, 13, and 11, respectively (L1210 screen). Hence the optimal position of the nitro group in ring A in terms of potency is the 2 position.
In order to assess whether the compounds in series 1–3, which permit sequential alkylation to occur, have increased cytotoxic potencies vis-à-vis the analogs in which this processwill not occur (series 4), the IC50 values of1a–g, 2a–g, and 3a–g were compared with those generated for 4a, 4b, and 4c, respectively. The results are summarized in Table 3. In general, structural modification of 4a, 4b, and 4c into series 1, 2, and 3, respectively, was accompanied by increases in potencies in all three bioassays except conversion of 4b into 2a–g did not lead to compounds with lower IC50 values toward CEM cells.
Table 3.
Comparison between the potencies of the bisalkylators 1a–g, 2a–g, and 3a–g with the monobenzylidene analogs 4a, 4b, and 4c, respectively
| Bioassay | Comparison of potenciesa
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1a–g | 4a | Equal | 2a–g | 4b | Equal | 3a–g | 4c | Equal | |
| Molt 4/C8 | 86 | 14 | 0 | 43 | 29 | 29 | 57 | 29 | 14 |
| CEM | 86 | 14 | 0 | 29 | 43 | 29 | 57 | 29 | 14 |
| L1210 | 71 | 29 | 0 | 57 | 0 | 43 | 71 | 29 | 0 |
| Total | 81 | 19 | 0 | 43 | 24 | 33 | 62 | 29 | 9 |
The figures represent the percentage of compounds displaying greater potency or were equipotent. The standard deviations of the IC50 figures were taken into account when making these comparisons.
The rate and extent of the ability of compounds to penetrate the cell membranes of neoplastic and transformed cells is dependent on a number of structural features including the lipophilicity of the molecules. The logP values of the compounds were calculated and are presented in Table 1. The average logP values for 1a–g, 2a–g, and 3a–g are 4.86, 5.06, and 5.08, respectively, and hence the lower lipophilicity of the compounds in series 1 may have contributed to the greater potencies than the analogs in series 2 and 3. The generally lower IC50 potencies displayed by the compounds in series 1–3 than 4a–c also reflect a negative correlation with the logP values.
Various compounds which are thiol reagents such as N-ethylmaleimide and mersalyl interact with different mercapto groups in mitochondria.17 Furthermore, a Mannich base of a conjugated styryl ketone inhibited respiration in rat liver mitochondria and the mode of action, at least in part, was based on competition with the conjugated unsaturated ketone coenzyme Q10.18 Thus the decision was made to determine whether representative compounds interfered with respiration in mitochondria isolated from rat liver cells, and if so whether the magnitude of this effect correlated with cytotoxic potencies. Three related compounds, namely, 1d, 2d, and 3d, were chosen since they possessed markedly different potencies having average IC50 figures of 0.88, 43.8, and 6.00 μM, respectively, in the three cytotoxicity screens. A concentration of 10 μM of each compound was chosen which is in excess of the IC50 values of 1d and 3d and substantially below that of 2d. The data in Figure 2 reveal that 1d and 3d stimulated respiration. However, the magnitude of the stimulatory effect was negatively correlated with cytotoxic potencies. The least potent of these three compounds, namely 2d, had virtually no effect on respiration. Increasing the concentration of 2d to 100 μM revealed no statistically significant difference in stimulation of respiration from the solvent control (data not shown). Nevertheless if the causes for the relative cytotoxic potencies observed in this study are multifactorial, the differences in the effects on mitochondrial function may have exerted some contributions to the disparity of IC50 values.
4. Conclusions
A number of novel cytotoxic agents have been prepared, many of which display good potencies toward Molt 4/C8, CEM, and L1210 cells. The highest potencies were displayed by the compounds in series 1 and in particular 1d and 1e are lead molecules having submicromolar IC50 values in some of the assays. Factors which influence cytotoxic potencies in series 1–3 include the atomic charges on the CB atoms, the torsion angle θA, and logP values. Another factor which may have contributed to the variation in IC50 values is the differences in the effects on respiration in rat liver mitochondria. A number of guidelines for amplifying this project have been proposed.
5. Experimental
5.1. Synthesis of compounds
Melting points in Celsius degrees were determined on a Gallenkamp apparatus and are uncorrected. 1H NMR spectra were recorded using a Bruker AMX 500 FT machine while elemental analyses were obtained using an Elementer analyzer.
5.1.1. Syntheses of 1a, 2a, and 3a
2-(4-Dimethylamino-benzylidene) cyclohexanone 5 was prepared by a reported procedure19 and crystallized from ethanol at 5–6 °C to give the desired product in 45% yield, mp 130 °C [lit.19] 127–127.5 °C]. 1H NMR (CDCl3): 1.79 (p, 2H), 1.91 (p, 2H), 2.52 (t, 2H), 2.88 (t, 2H), 3.03 (s, 6H, 2× NCH3), 6.71 (d, 2H, Ar–H, J = 8.85 Hz), 7.41 (d, 2H, J = 8.83 Hz), 7.55 (s, 1H, =CH).
Dry hydrogen chloride was passed into a solution of 5 (0.005 mol) and 2-nitrobenzaldehyde (0.005 mol) in acetic acid (15 mL) and the mixture stirred at room temperature overnight. Acetic acid was removed in vacuo and the residue triturated with potassium carbonate solution (10% w/v, 20 mL) and extracted with chloroform. The organic extract was washed with water and dried. Evaporation of the solvent gave a semisolid which was purified by chromatography using a column of silica gel 60 (70–230 mesh) and an eluting solvent of 10–30% ethyl acetate in hexane to produce 1a, mp 152 °C in 41% yield. 1H NMR (CDCl3): δ 1.79 (p, 2H), 2.60 (t, 2H), 2.96 (t, 2H), 3.05 (s, 6H, 2× N(CH3)2), 6.73 (d, 2H, Ar–H, J = 8.80 Hz), 7.38 (d, 1H, Ar–H, J = 7.60 Hz), 7.49 (m, 3H, Ar–H), 7.64 (t, 1H, Ar–H), 7.84 (s, 1H, =CH), 7.95 (s, 1H, =CH), 8.13 (d, 1H, Ar–H, J = 8.20 Hz). Anal. Calcd for C22H22N2O3: C, 72.91; H, 6.12; N, 7.73. Found: C, 72.62; H, 5.98; N, 7.53%.
Aqueous sodium hydroxide solution (20% w/v, 1 mL) was added to a solution of 5 (0.005 mol) and 3-nitrobenzaldehyde (0.005 mol) in ethanol (15 mL) at 8–10 °C. The solution was stirred at room temperature for 0.5 h. The resultant precipitate was collected, washed with water (3× 15 mL), dried and crystallized from chloroform/ethanol (1:9) to give 2a, mp 169 °C in 68% yield. 1H NMR (CDCl3): δ 1.86 (p, 2H), 2.93 (t, 2H), 3.01 (t, 2H), 3.07 (s, 6H, 2× N(CH3)2), 6.75 (d, 2H, Ar–H, J = 8.84 Hz), 7.50 (d, 2H, Ar–H, J = 8.82Hz), 7.60 (t, 1H, Ar–H), 7.76 (d, 1H, Ar–H, J = 7.7 Hz), 7.79 (s, 1H, =CH), 7.84 (s, 1H, =CH), 8.20 (d, 1H, Ar–H, J = 8.18 Hz), 8.32 (s, 1H, Ar–H). Anal. Calcd for C22H22N2O3: C, 72.91; H, 6.12; N, 7.73. Found: C, 72.87; H, 6.0; N, 7.46%.
Dry hydrogen chloride was passed into a solution of 5 (0.005 mol) and 4-nitrobenzaldehyde (0.005 mol) in acetic acid (15 mL) and the mixture was stirred overnight at room temperature. The precipitate was collected, washed with diethyl ether (2× 10 mL), and potassium carbonate solution (10% w/v, 30 mL). The solid obtained was washed with water (3× 10 mL), dried and crystallized from chloroform/ethanol (1:9) to give 3a, mp 91–92 °C in 66% yield. 1H NMR (CDCl3): δ 1.86 (p, 2H), 2.91 (t, 2H), 3.0 (t, 2H), 3.07 (s, 6H, 2× N(CH3)2), 6.75 (d, 2H, Ar–H, J = 8.84 Hz), 7.50 (d, 2H, Ar–H, J = 8.84 Hz), 7.60 (d, 2H, Ar–H, J = 8.63 Hz), 7.79 (s, 1H, =CH), 7.84 (s, 1H, =CH), 8.27 (d, 2H, Ar–H, J = 8.65 Hz). Anal. Calcd for C22H22N2O3: C, 72.91; H, 6.12; N, 7.73. Found: C, 72.94; H, 6.13; N, 7.52%.
5.1.2. Synthesis of 1c
2-(4-Methylbenzylidene)cyclohexanone 6 was prepared by a literature procedure20 and crystallized from methanol to give 6, mp 71 °C [lit.20 mp 60 °C] in 40% yield. 1H NMR (CDCl3): 1.78 (p, 2H), 1.92 (m, 2H), 2.38 (s, 3H, CH3), 2.53 (t, 2H), 2.86 (t, 2H), 7.21 (d, 2H, Ar–H, J = 7.90 Hz), 7.32 (d, 2H, Ar–H, J = 7.96 Hz), 7.50 (s, 1H, =CH).
Aqueous sodium hydroxide solution (20% w/v, 1 mL) was added to a solution of 6 (0.005 mol) and 2-nitrobenzaldehyde (0.005 mol) in ethanol (15 mL) at 8–10 °C. The solution was stirred at room temperature for 0.5 h. The reaction mixture was acidified with dilute hydrochloric acid and extracted with chloroform. Evaporation of the organic solvent gave a viscous oil which was purified by chromatography using a column of silica gel 60 (70–230 mesh) and an eluting solvent of ethyl acetate/hexane (1:9) to give 1c, mp 120 °C in 30% yield. 1H NMR(CDCl3): δ 1.78 (p, 2H), 2.40 (s, 3H, CH3), 2.62 (p, 2H), 2.94 (t, 2H), 7.24 (d, 2H, Ar–H, J = 7.94 Hz), 7.39 (t, 3H, Ar– H), 7.52 (t, 1H, Ar–H), 7.65 (t, 1H, Ar–H), 7.83 (s, 1H, =CH), 7.96 (s, 1H, =CH), 8.14 (d, 1H, Ar–H). Anal. Calcd for C21H19NO3: C, 75.66; H, 5.74; N, 4.20. Found: C, 75.42; H, 5.71; N, 4.27%.
5.1.3. Synthesis of 1b, d–g, 2b–g, and 3b–g
The enones 1b, d–g, 2b–g, and 3b–g were prepared by the following general procedure. Dry hydrogen chloride was passed into a solution of 4a, 4b, or 4c vide infra (0.005 mol) and the appropriate aryl aldehyde (0.006 mol) in ether (40 mL) and methanol (4 mL). The reaction mixture was stirred at room temperature for 24 h and the resultant solid was collected and crystallized from chloroform/methanol (1:3).
5.1.3.1. E,E-2-(4-Methoxybenzylidene)-6-(2-nitrobenzylidene) cyclohexanone (1b)
Mp 157 °C; yield 80%. 1H NMR (CDCl3): δ 1.79 (p, 2H), 2.61 (t, 2H), 2.94 (t, 2H), 3.86 (s, 3H, OCH3), 6.98 (d, 2H, Ar–H, J = 8.3 Hz), 7.38 (d, 1H, Ar–H, J = 7.65 Hz), 7.48 (d, 2H, Ar–H, J = 8.45 Hz), 7.52 (t, 1H, Ar–H), 7.65 (t, 1H, Ar–H), 7.82 (s, 1H, =CH), 7.96 (s, 1H, =CH), 8.14 (d, 1H, Ar–H, J = 8.20 Hz). Anal. Calcd for C21H19NO4: C, 72.19; H, 5.48; N, 4.01. Found: C, 71.89; H, 5.53; N, 3.90%.
5.1.3.2. E,E-2-(3,4,5-Trimethoxybenzylidene)-6-(2-nitrobenzylidene)cyclohexanone (1d)
Mp 159 °C; yield 94%. 1H NMR (CDCl3): δ 1.80 (p, 2H), 2.62 (t, 2H), 2.97 (t, 2H), 3.91 (s, 9H, 3× OCH3), 6.73 (s, 2H, Ar–H), 7.39 (d, 1H, Ar–H, J = 7.60 Hz), 7.52 (t, 1H, Ar–H), 7.66 (t, 1H, Ar–H), 7.77 (s, 1H, =CH), 7.96 (s, 1H, =CH), 8.14 (d, 1H, Ar–H, J = 8.15 Hz). Anal. Calcd for C23H23NO6: C, 67.47; H, 5.66; N, 3.42. Found: C, 67.60; H, 5.61; N, 3.58%.
5.1.3.3. E,E-2-(Benzylidene)-6-(2-nitrobenzylidene)-cyclohexanone (1e)
Mp 116 °C; yield 47%. 1H NMR (CDCl3): δ 1.78 (p, 2H), 2.63 (t, 2H), 2.95 (t, 2H), 7.40 (m, 5H, Ar–H), 6.79 (d, 2H, Ar–H, J = 7.6 Hz), 7.52 (t, 1H, Ar–H), 7.65 (t, 1H, Ar–H), 7.85 (s, 1H, =CH), 7.97 (s, 1H, =CH), 8.15 (d, 1H, Ar–H, J = 8.20 Hz). Anal. Calcd for C20H17NO3: C, 75.22, H, 5.37; N 4.39. Found: C, 74.82; H, 5.26; N, 4.04%.
5.1.3.4. E,E-2-(4-Fluorobenzylidene)-6-(2-nitrobenzylidene) cyclohexanone (1f)
Mp 136 °C; yield 52%. 1H NMR (CDCl3): δ 1.79 (p, 2H), 2.63 (t, 2H), 2.91 (t, 2H), 7.12 (t, 2H, Ar–H), 7.39 (d, 1H, Ar–H, J = 7.65 Hz), 7.47 (q, 2H, Ar–H), 7.52 (t, 1H, Ar–H), 7.65 (t, 1H, Ar–H), 7.80 (s, 1H, =CH), 7.97 (s, 1H, =CH), 8.15 (d, 1H, Ar–H, J = 8.20 Hz). Anal. Calcd for C20H16FNO3: C, 71.21; H, 4.78; N 4.15. Found: C, 70.93; H, 4.79; N 3.88%.
5.1.3.5. E,E-2-(4-Chlorobenzylidene)-6-(2-nitrobenzylidene) cyclohexanone (1g)
Mp 149 °C; yield 41%. 1H NMR (CDCl3): δ 1.79 (p, 2H), 2.63 (t, 2H), 2.84 (t, 2H), 7.40 (m, 5H, Ar–H), 7.53 (t, 1H, Ar–H), 7.66 (t, 1H, Ar–H), 7.77 (s, 1H, =CH), 7.97 (s, 1H, =CH), 8.15 (d, 1H, Ar–H, J = 8.2 Hz). Anal. Calcd for C20H16ClNO3: C, 67.90; H, 4.56; N, 3.96. Found: C, 67.70; H 4.61; N 3.78%.
5.1.3.6. E,E-2-(4-Methoxybenzylidene)-6-(3-nitrobenzylidene) cyclohexanone (2b)
Mp 114 °C; yield 68%. 1H NMR (CDCl3): δ 1.85 (p, 2H), 2.94 (t, 2H), 2.98 (t, 2H), 6.96 (d, 2H, Ar–H, J = 8.50 Hz), 7.49 (d, 2H, Ar–H, J = 8.45 Hz), 7.60 (t, 1H, Ar–H), 7.75 (d, 1H, Ar–H, J = 7.7 Hz), 7.79 (s, 1H, =CH), 7.81 (s, 1H, =CH), 8.19 (d, 1H, Ar–H, J = 8.10 Hz), 8.32 (s, 1H, Ar–H). Anal. Calcd for C21H19NO4: C, 72.19; H, 5.48; N 4.01. Found: C, 71.91; H, 5.46; N, 3.90%.
5.1.3.7. E,E-2-(4-Methylbenzylidene)-6-(3-nitrobenzylidene) cyclohexanone (2c)
Mp 156 °C; yield 60%. 1H NMR (CDCl3): δ 1.84 (p, 2H), 2.94 (t, 2H), 2.98 (t, 2H), 7.25 (t, 2H, Ar–H), 7.41 (d, 2H, Ar–H, J = 7.85 Hz), 7.60 (t, 1H, Ar–H), 7.76 (d, 2H, Ar–H, J = 7.65 Hz), 7.81 (s, 1H, =CH), 7.82 (s, 1H, =CH), 8.20 (d, 1H, Ar–H, J = 8.20 Hz), 8.32 (s, 1H, Ar–H). Anal. Calcd for C21H19NO3: C, 75.66; H, 5.74; N, 4.20. Found: C, 75.58; H, 5.88; N, 3.92%.
5.1.3.8. E,E-2-(3,4,5-Trimethoxybenzylidene)-6-(3-nitrobenzylidene)cyclohexanone (2d)
Mp 172 °C; yield 82%. 1H NMR (CDCl3): δ 1.86 (p, 2H), 2.95 (t, 2H), 3.00 (t, 2H), 3.91 (s, 9H, 3× OCH3), 6.74 (s, 1H, Ar–H), 7.61 (t, 1H), 7.76 (d, 2H, Ar–H, =CH, J = 7.70 Hz), 7.80 (s, 1H, =CH), 8.21 (d, 1H, Ar–H, J = 8.15 Hz), 8.29 (s, 1H, Ar–H). Anal. Calcd for C23H23NO6: C, 67.47; H, 5.66; N, 3.42. Found: C, 67.29; H, 5.57; N, 3.30%.
5.1.3.9. E,E-2-(Benzylidene)-6-(3-nitrobenzylidene)-cyclohexanone (2e)
Mp 120 °C; yield 22%. 1H NMR (CDCl3): δ 1.85 (p, 2H), 2.97 (m, 4H), 7.38 (t, 1H, Ar–H), 7.44 (m, 2H, Ar–H), 7.49 (d, 2H, Ar–H, J = 7.65 Hz), 7.61 (t, 1H, Ar–H), 7.76 (d, 1H, Ar–H, J = 7.65 Hz), 7.80 (s, 1H, =CH), 7.84 (s, 1H, =CH), 8.21 (d, 1H, Ar–H, J = 8.25 Hz), 8.33 (s, 1H, Ar–H). Anal. Calcd for C20H17NO3: C, 75.22, H 5.37; N, 4.39. Found: C, 75.28; H, 5.44; N, 4.28%.
5.1.3.10. E,E-2-(4-Fluorobenzylidene)-6-(3-nitrobenzylidene) cyclohexanone (2f)
Mp 126 °C; yield 63%. 1H NMR (CDCl3): δ 1.85 (p, 2H), 2.81 (t, 2H), 7.13 (t, 2H, Ar–H), 7.48 (dd, 2H, Ar–H), 7.61 (t, 1H, Ar–H), 7.76 (d, 1H, Ar–H, J = 7.65 Hz), 7.80 (s, 2H, =CH), 8.21 (d, 1H, Ar–H, J = 8.15 Hz), 8.32 (s, 1H, Ar–H). Anal. Calcd for C20H16FNO3: C, 71.21; H, 4.78; N, 4.15. Found: C, 70.97; H, 4.70; N, 4.08%.
5.1.3.11. E,E-2-(4-Chlorobenzylidene)-6-(3-nitrobenzylidene) cyclohexanone (2g)
Mp 138 °C; yield 68%. 1H NMR (CDCl3): δ 1.99 (p, 2H), 2.96 (q, 4H), 7.42 (m, 4H), 7.62 (t, 1H), 7.77 (d, 2H, Ar–H & =CH, J = 8.80 Hz), 7.81 (s, 1H, =CH), 7.84 (s, 1H, =CH), 8.16 (d, 1H, Ar–H, J = 8.16 Hz), 8.34 (s, 1H, Ar–H). Anal. Calcd for C22H16ClNO3: C, 67.90; H, 4.56; N, 3.96. Found: C, 67.33; H, 4.58; N, 3.84%.
5.1.4. Compounds 3b–g
The synthesis of 3b, d, and g has been reported previously.8
5.1.4.1. E,E-2-(4-Methylbenzylidene)-6-(4-nitrobenzylidene) cyclohexanone (3c)
Mp 136 °C; yield 54%. 1H NMR (CDCl3): δ 1.84 (p, 2H), 2.41 (s, 3H, CH3), 2.92 (t, 2H), 2.98 (t, 2H), 7.25 (d, 2H, Ar–H, J = 7.8Hz), 7.41 (d, 2H, Ar–H, J = 7.80 Hz), 7.60 (d, 2H, Ar–H, J = 8.35 Hz), 7.79 (s, 1H, =CH), 7.82 (s, 1H, =CH), 8.27 (d, 2H, Ar–H, J = 8.40 Hz). Anal. Calcd for C21H19NO3: C, 75.66; H, 5.74; N, 4.20. Found: C, 75.45; H, 5.75; N, 4.13%.
5.1.4.2. E,E-2-(Benzylidene)-6-(4-nitrobenzylidene)cyclohexanone (3e)
Mp 141 °C; yield 40%. 1H NMR (CDCl3): δ 1.84 (p, 2H), 2.93 (t, 2H), 2.98 (t, 2H), 7.38 (t, 1H, Ar–H), 7.44 (t, 1H, Ar–H), 7.49 (d, 2H, Ar–H, J = 7.75 Hz), 7.60 (d, 2H, Ar–H, J = 8.30 Hz), 7.80 (s, 1H, =CH), 7.84 (s, 1H, =CH), 8.27 (d, 2H, Ar–H, J = 8.40 Hz). Anal. Calcd for C20H17NO3: C, 75.22; H, 5.37; N, 4.39. Found: C, 75.02; H, 5.29; N, 4.19%.
5.1.4.3. E,E-2-(4-Fluorobenzylidene)-6-(4-nitrobenzylidene) cyclohexanone (3f)
Mp 167 °C; yield 33%. 1H NMR (CDCl3): δ 1.85 (p, 2H), 2.94 (t, 4H), 7.13 (t, 2H), 7.48 (t, 2H), 7.60 (d, 2H), 7.79 (s, 2H, =CH), 8.27 (d, 2H, Ar–H, J = 8.5 Hz) Anal. Calcd for C20H16FNO3: C, 71.21; H, 4.78; N, 4.18. Found: C, 70.88; H, 4.70; N, 4.10%.
5.1.5. Synthesis of 4a
A solution of sodium hydroxide (0.6 g, 0.015 mol) in water (5 mL) was added dropwise to a mixture of cyclohexanone (3.0 g, 0.02 mol) and 2-nitrobenzaldehyde (5.85 g, 0.056 mol) at room temperature for 0.25 h and the stirring was continued for 4 h. The solid was collected, dried, and recrystallized from chloroform/methanol to yield 2-(α-hydroxy-2-nitrobenzyl) cyclohexanone (7), mp 126 °C in 28% yield. 1H NMR (CDCl3): δ 1.71 (m, 4H), 2.13 (m, 1H), 2.45 (m, 2H), 2.82 (m, 1H), 4.18 (d, 1H), 5.46 (t, 1H), 7.44 (t, 1H), 7.65 (t, 1H), 7.78 (d, 1H, J = 7.9 Hz), 7.86 (d, 1H, J = 8.15 Hz).
Hydrochloric acid (2 mL) was added to a solution of 7 (10.5 g, 0.045 mol) in ethanol (30 mL) and the reaction mixture was heated at 40–45 °C for 4 h. The solvent was removed in vacuo at 40–45 °C and water (100 mL) was added to the residue. The solid was collected and dried to give 4a, mp 92 °C in a yield of 64%. 1H NMR (CDCl3): δ 1.75 (p, 2H), 1.95 (p, 2H), 2.54 (t, 2H), 2.58 (t, 2H), 2.58 (t, 2H), 7.33 (d, 1H, Ar–H, J = 7.63 Hz), 7.51 (t, 1H, Ar–H), 7.60 (s, 1H, =CH), 7.64 (t, 2H, Ar–H), 8.12 (d, 1H, Ar–H, J = 8.21Hz). Anal. Calcd for C13H13NO3: C, 67.52; H, 5.67; N, 6.06. Found: C, 67.40; H, 5.49; N, 6.29%.
5.1.6. Synthesis of 4b
A solution of cyclohexanone (4.9 g, 0.05 mol), morpholine (4.75 g, 0.055 mol), 4-toluenesulfonic acid (0.02 g) in toluene (50 mL) was heated under reflux using a Dean-Stark apparatus until the stoichiometric amount of water separated (~8 h). 3-Nitrobenzaldehyde (6.8 g, 0.045 mol) was added to the reaction mixture and heating under reflux was continued for 12 h. Water (25 mL) was added to the reaction mixture which was heated at 50–55 °C for ~1 h. The organic phase was separated, washed with hydrochloric acid (5%, 20 mL) and water (3× 50 mL), and dried. Toluene was removed in vacuo at 50–55 °C to give a viscous oil which was purified by chromatography using a column of silica gel 60 (70–230 mesh) and an eluting solvent of ethyl acetate/hexane (1:9) to give 4b, mp 51–52 °C in 45% yield. 1H NMR(CDCl3): 1.82 (p, 2H), 1.98 (p, 2H), 2.59 (t, 2H), 2.86 (t, 2H), 7.52 (s, 1H, =CH), 7.58 (t, 1H, Ar–H), 7.69 (d, 1H, Ar–H, J = 7.6 Hz), 8.19 (d, 1H, Ar–H, J = 8.15 Hz), 8.25 (s, 1H, Ar–H). Anal. Calcd for C13H13NO3: C, 67.52; H, 5.67; N, 6.06. Found: C, 67.30; H, 5.48; N, 6.41%.
5.1.7. Synthesis of 4c
This compound was prepared by a literature procedure21 to give 4c, mp 119 °C [lit.21] mp 118–120 °C] in 72% yield with respect to 4-nitrobenzaldehyde. 1H NMR(CDCl3): 1.82 (p, 2H), 1.98 (p, 2H), 2.59 (t, 2H), 2.83 (m, 2H), 7.47 (s, 1H), 7.53 (d, 2H), 8.25 (d, 2H).
5.2. Molecular modeling
Models of the compounds in series 1–4 were built using a BioMedCache program.22 The lowest energy conformers were generated using the CONFLEX program and optimized by mechanics using augmented MM2 parameters.
5.3. Determination of logP values
The logP values for enones 1–4 were generated with the JME molecular editor.23
5.4. Cytotoxicity assays
A literature procedure was employed to examine the cytotoxicity of 1a–g, 2a–g, 3a–g, and 4a–c toward human Molt 4/C8 and CEM T-lymphocytes as well as murine L1210 cells.24 In brief, different concentrations of compounds were incubated with the cells in RPMI 1640 medium at 37 °C for 72 h (Molt 4/C8 and CEM T-lymphocytes) or 48 h (L1210 cells). The correct IC50 values for 4c are presented in Table 1 which replaces the figures quoted previously.25
5.5. Evaluation of 1d, 2d, and 3d on respiration in rat liver mitochondria
Rats were anesthetized with isoflurane and decapitated. A previously reported procedure was employed to isolate mitochondria from the liver.26 The consumption of oxygen by mitochondria was determined by polarography using a literature methodology.27
5.6. Statistical analyses
The linear, semilogarithmic and logarithmic plots were constructed using a statistical software package.28
References and notes
- 1.Mutus B, Wagner JD, Talpas CJ, Dimmock JR, Phillips OA, Reid RS. Anal Biochem. 1989;177:237–243. doi: 10.1016/0003-2697(89)90045-6. [DOI] [PubMed] [Google Scholar]
- 2.Baluja G, Municio AM, Vega S. Chem Ind. 1964:2053–2054. [Google Scholar]
- 3.Benvenuto JA, Connor TH, Monteith DK, Laidlaw JA, Adams SC, Matney TS, Theiss JC. J Pharm Sci. 1993;82:988–991. [PubMed] [Google Scholar]
- 4.Dimmock JR, Sidhu KK, Chen M, Reid RS, Allen TM, Kao GY, Truitt GA. Eur J Med Chem. 1993;28:313–322. [Google Scholar]
- 5.Dimmock JR, Padmanilayam MP, Zello GA, Nienaber KH, Allen TM, Santos CL, De Clercq E, Balzarini J, Manavathu EK, Stables JP. Eur J Med Chem. 2003;38:169–177. doi: 10.1016/s0223-5234(02)01444-7. [DOI] [PubMed] [Google Scholar]
- 6.Dimmock JR, Padmanilayam MP, Zello GA, Quail JW, Oloo EO, Prisciak JS, Kraatz HB, Cherkasov A, Lee JS, Allen TM, Santos CL, Manavathu EK, De Clercq E, Balzarini J, Stables JP. Eur J Med Chem. 2002;37:813–824. doi: 10.1016/s0223-5234(02)01402-2. [DOI] [PubMed] [Google Scholar]
- 7.Dimmock JR, Das U, Gul HI, Kawase M, Sakagami H, Baráth Z, Ocsovsky I, Molnár J. Bioorg Med Chem Lett. 2005;15:1633–1636. doi: 10.1016/j.bmcl.2005.01.054. [DOI] [PubMed] [Google Scholar]
- 8.Das U, Gul HI, Alcorn J, Shrivastav A, George T, Sharma RK, Nienaber KH, De Clercq E, Balzarini J, Kawase M, Kan N, Tanaka T, Tani S, Werbovetz KA, Yakovich AJ, Manavathu EK, Stables JP, Dimmock JR. Eur J Med Chem. 2006;41:577–585. doi: 10.1016/j.ejmech.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 9.Das U, Kawase M, Sakagami H, Ideo A, Shamada J, Molnár J, Baráth Z, Bata Z, Dimmock JR. Bioorg Med Chem. 2007;15:3373–3380. doi: 10.1016/j.bmc.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 10.Hassner A, Mead TC. Tetrahedron. 1964;20:2201–2210. [Google Scholar]
- 11.Quail JW, Doroudi A, Pati HN, Das U, Dimmock JR. Acta Cryst. 2005;E61:1795–1797. [Google Scholar]
- 12.Quail JW, Doroudi A, Pati HN, Das U, Dimmock JR. Acta Cryst. 2005;E61:1774–1776. [Google Scholar]
- 13.Quail JW, Das U, Dimmock JR. Acta Cryst. 2005;E61:1150–1152. [Google Scholar]
- 14.Taft RW., Jr . In: Steric Effects in Medicinal Chemistry. Newman MS, editor. John Wiley and Sons, Inc; New York: 1956. p. 591. [Google Scholar]
- 15.Hansch C, Leo AJ. Substituent Constants for Correlation Analysis in Chemistry and Biology. John Wiley and Sons; New York: 1979. p. 49. [Google Scholar]
- 16.Hansch C, Leo AJ. Substituent Constants for Correlation Analysis in Chemistry and Biology. John Wiley and Sons; New York: 1979. pp. 49–50. [Google Scholar]
- 17.Bryla J. In: Inhibition of Mitochondrial Function. Erecińska M, Wilson DF, editors. Pergamon Press; Oxford: 1981. pp. 243–246. [Google Scholar]
- 18.Hamon NW, Bassendowski DL, Wright DE, Dimmock JR, Noble LM. J Pharm Sci. 1978;67:1539–1542. doi: 10.1002/jps.2600671112. [DOI] [PubMed] [Google Scholar]
- 19.Shriner RL, Teeters WO. J Am Chem Soc. 1938;60:936–939. doi: 10.1021/ja01210a010. [DOI] [PubMed] [Google Scholar]
- 20.Colonge J, Sibeud J. J Bull Soc Chim France. 1952:786–789. [Google Scholar]
- 21.Vieweg H, Wagner G. Pharmazie. 1979;34:785–788. [PubMed] [Google Scholar]
- 22.BioMedCache 6.1 Windows, BioMedCache. Fujitsu America; Inc; 2003. [Google Scholar]
- 23.JME Molecular. 2006 Apr; Version; http://www.molinspiration.com.
- 24.Baraldi PB, Nunez M, Del C, Tabrizi MA, De Clercq E, Balzarini J, Bermejo J, Estévez F, Romagnoli R. J Med Chem. 2004;47:2877–2886. doi: 10.1021/jm031104y. [DOI] [PubMed] [Google Scholar]
- 25.Dimmock JR, Chamankhah M, Das U, Zello GA, Quail JW, Yang J, Nienaber KH, Sharma RK, Selvakumar P, Balzarini J, De Clercq E, Stables JP. J Enzyme Inhib Med Chem. 2004;19:1–10. doi: 10.1080/14756360310001624975. [DOI] [PubMed] [Google Scholar]
- 26.Kowaltowski AJ, Castilho RF, Grijalba MT, Bechara EJ, Vercesi AE. J Biol Chem. 1996;271:2929–2934. doi: 10.1074/jbc.271.6.2929. [DOI] [PubMed] [Google Scholar]
- 27.Estabrook RW. Methods Enzymol. 1967;X:41–47. [Google Scholar]
- 28.Statistical Package for Social Sciences. SPSS for Windows, Release 14.0.0. SPSS Inc; Chicago: 2005. [Google Scholar]