

Abstract
Dishevelled (Dvl) PDZ domains transduce Wnt signals from the membrane-bound receptor Frizzled to the downstream. As abnormal Wnt signaling has been implicated in tumorigenesis, the Dvl PDZ domain is a potential target for small-molecule inhibitors that block Wnt signaling at the Dvl level. We expanded our in silico search to examine the chemical space near previously developed PDZ binders and identified nine additional compounds bind to the Dvl PDZ. We then performed a quantitative structure-activity relationship (QSAR) analysis of these compounds and combined these results with structural studies of the PDZ domain in complex with the compounds to design and synthesize a group of new, further optimized compounds. Two rounds of synthesis and testing yielded a total of six compounds that have greatly improved binding affinity to the Dvl PDZ domain and most potent ones competitively displace Dapper peptide from the PDZ domain. In addition to providing more potent Dvl PDZ domain inhibitors, this study demonstrates that virtual screening and structural studies can be powerful tools in guiding the chemical synthesis hit-to-lead optimization stage during the drug discovery process.
Keywords: Wnt signaling pathway, Dishevelled, PDZ domain
Introduction
Wnt signaling plays a crucial role in embryonic development and regulation of cell growth (1, 2). Inappropriate activation of Wnt signaling has been implicated in cancers and other human diseases (3). Dishevelled (Dvl) protein regulates Wnt signaling pathways by using its PDZ domain to interact with the Wnt receptor Frizzled (Fz), thereby transducing Wnt signals downstream (4). In the canonical Wnt signaling pathway, this interaction activates the β-catenin/TCF transcription pathway, which regulates the transcription of many genes, including tumor-related genes, such as Myc and Cyclin D1 (2). Dvl-Frizzled interaction mainly relies on the interaction of Dvl PDZ domain with the C-terminal intramolecular KTXXXW sequence which has a moderate binding affinity (4). Additional interaction involving Dvl DEP domain with cell membrane may facilitate the formation of Dvl-Frizzled complex (5). Transcriptional activation of Dapper, a native Dvl-PDZ inhibitor, was shown to strongly inhibit Wnt/β-catenin signaling and induce dramatic apoptosis of colon cancer cells (6), indicating the important role of Dvl in Wnt signaling and tumorigenesis. Further, up-regulation of Dvl protein was observed in Wnt-driven non-small-cell lung cancer and malignant mesothelioma, while down-regulation of Dvl through either RNA interference or Dvl mutagenesis inhibited Wnt signaling and tumor growth (7, 8). Several small molecules or peptides have been developed to target the Dvl PDZ domain and thereby regulate the Wnt/β-catenin pathway (9, 10). Such Wnt pathway inhibitors can be useful not only in dissecting signaling mechanisms but also in formulating rational approaches to the development of potential pharmaceutical agents that block specific Wnt signaling events that contribute to cancer (11).
We previously identified a PDZ domain antagonist (NSC668036) through receptor-based virtual screening of the NCI small-molecule library (9). Recently, after analyzing the complex structure of PDZ bound to NSC668036, we proposed a pharmacophore model and carried out ligand similarity screening based on the pharmacophore to identify additional PDZ antagonists (12). That study identified 15 compounds that bind to the Dvl PDZ domain with greater affinity than does NCS668036. In the current study, based on the structures of the 15 recently identified PDZ binders, we conducted an additional round of pharmacophore-based ligand similarity search and identified 9 more compounds. A 3-dimensional quantitative structure-activity relationship (3D-QSAR) analysis of the 9 new PDZ binders together with the earlier 15 compounds was consistent with our docking-based structural examination of the Dvl PDZ in complex with the compounds. Guided by the QSAR and structural studies, we designed and synthesized several novel compounds that are much more potent inhibitors of the Dvl PDZ domain.
Experimental Procedures
Virtual Screening
The UNITY module in the SYBYL software package (Tripos, Inc.) was used to screen the ChemDiv, ChemBridge and NCI databases for potential PDZ domain inhibitors.
Chemicals and Reagents
Compounds 19 and 20 were acquired from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (http://129.43.27.140/ncidb2/). Compounds 16 to 24 except 19 and 20 were purchased from Chemical Diversity Inc. (San Diego, CA). Fmoc-protected amino acids and HBTU were purchased from Anaspec (San Jose, CA), resins and HATU from Applied Biosystems (Foster City, CA), Fmoc-protected 4-methylphenylalanine from Advanced ChemTech (Louisville, KY), and N-(9-fluorenylmethyloxycarbonyloxy) succinimide from Novabiochem (Gibbstown, NJ). All other chemicals were purchased from Sigma-Aldrich (Milwaukee, WI).
Expression and purification of the mouse Dvl PDZ domain
The 15N-labeled mouse Dvl1 PDZ domain (residues 247-341 of mDvl1) was prepared as described previously (4, 9, 13) by the protein production facility at St. Jude Children’s Research Hospital. CYS338, a residue located outside the ligand binding site, was mutated to alanine in the expression construct to increase the solubility of the protein.
NMR studies
15N-HSQC experiments were performed by using a Varian Inova 600 MHz NMR spectrometer at 25 °C. Samples consisted of mouse Dvl1 PDZ domain (0.2 - 0.3 mM) in 100 mM potassium phosphate buffer (pH 7.5), 10% D2O, and 0.5 mM EDTA. Compounds were dissolved in the same buffer but with 5% DMSO, which did not change the spectra of the PDZ domain (data not shown). NMR spectra were processed with NMRpipe (14) and analyzed by using the Sparky program (15). The binding affinities (KD) of PDZ ligands were calculated from HSQC spectra as previously reported (16). The mean chemical-shift perturbation changes caused by ligand binding were calculated as follows: . KD was then calculated from and A = 1+R+(PR+C)KD/(PC) by applying a one-site binding model corrected for dilution, where R was the ligand:protein molar ratio, P was the protein concentration before titration, C was the ligand stock concentration, and KD was the dissociation constant. After two-parameter nonlinear least-squares fitting with the program Prism (GraphPad Software, La Jolla, CA), KD was normalized by dividing the experimentally derived KD values by the difference between the KD values obtained for NSC668036 by NMR and fluorescence methods, respectively (12).
QSAR
Two 3D-QSAR CoMFA (comparative molecular-field analysis) (17) models for the ligands of scaffold A and B were built by using Sybyl 8.1 (Tripos Inc.). Compounds identified from the first round virtual screening together with previously identified compounds whose binding affinities to the Dvl PDZ domain were known (12), total 16 total 25 compounds (9 in scaffold A and 16 scaffold in B), were used to build the 3D-QSAR CoMFA models. The NMR-derived complex structure of VWV bound to Dvl PDZ was used to derive the structures of all compounds (18). All compounds were sketched according to the Dvl PDZ–bound conformation of VWV and minimized by 100 steps of the steepest descendent method and 500 steps of the conjugate gradient method with the presence of Dvl PDZ. All energy minimizations converged before the maximum minimization steps and all the key hydrogen bonds between the C terminal carboxyl groups of ligands and the 3 N-H groups (the backbone N-H groups of Leu262, Gly263 and Ile264) of the Dvl PDZ domain were preserved. The optimized compounds were later isolated and superimposed on both scaffolds. The Gasteiger-Huckel charges were assigned to each ligand, and the steric and electrostatic energies were calculated for CoMFA modeling. The partial least squares (PLS) (19, 20) regression was performed to correlate the molecular fields and experimental binding affinity data. Leave-one-out cross-validation was used to determine the number of principal components and PLS without cross-validation was performed to build the CoMFA model. The coefficient of determination, defined as R2 = 1−SSer/SStot, where SSer is the sum of squares of residuals and SStot is the sum of total squares, was calculated and used to evaluate the correlation quality of each model.
Synthesis
All synthesis was done on a Symphony 12-channel peptide synthesizer (Protein Technologies, Inc; Tucson, AZ) using standard solid-phase Fmoc peptide chemistry. All compounds were synthesized from the C-terminus to the N-terminus, starting from Fmoc-protected Leu attached to resin, de-protected with 20% piperadine in NMP for 15 min at room temperature, and coupled by using Fmoc-Leu (10 eq), HBTU (9 eq), and DIEA (10 eq) in anhydrous NMP for 2 h. The second residues of J01- and J02- series compounds were Fmoc-protected 4-methylphenylalanine and phenylalanine, respectively. The last segments added to J01-007, J01-012, J01-015, J01-016, J01-019 and J01-017a were 3-fluorobenzoic acid, 3-cyanobenzoic acid, 3-methylbenzoic acid, 3-(aminomethyl)benzoic acid, 3-(phenylthio)benzoic acid and 3,4-difluoro-5-methylbenzoic acid, respectively. The last residues added to J02-001 and J02-002 were 3-fluorobenzoic acid and 4-fluorobenzoic acid, respectively. All modified benzoic acids used as the last residues were activated with HATU. Products were cleaved from the resin with 90% TFA, 5% water, and 5% TIS for 2 h at room temperature. The resulting solutions were filtered, precipitated with cold diethyl ether, centrifuged, suspended in distilled water, and lyophilized. HPLC analysis showed compounds in library J01 to be more than 90% pure. All synthesized compounds were verified by mass spectroscopy (Supplemental Table 1).
Fluorescence spectroscopy
A Fluorolog-3 spectrofluorometer (Jobin-Yvon Inc.) with a 10 × 4 mm quartz cell (Hellma Inc.) was used for competitive binding experiments. The binding affinity of Rox-DprC (ROX-SGSLKLMTTVCOOH) (4) with Dvl1 PDZ was obtained by monitoring the fluorescence polarization of 1.4 mL of 50-nM Rox-DprC in 0.1 M potassium phosphate buffer (pH=7.5) at 20 °C. The equation 1/ΔmP = a/[S]+b was used to fit the double-reciprocal plot of the fluorescence data, where ΔmP and [S] are change in fluorescence polarization and the concentration of unlabeled Dvl PDZ domain, respectively. KD was calculated as KD = a b, where a and b are the fitted values from above and it was 7.7 μM Rox-DprC, which is close to the affinity between PDZ and the Fz7 peptide (4). Considering Rox-DprC binds to the same region as Frizzled peptide does, this peptide is representative of the PDZ-Fz interaction as the PDZ-Fz interaction is mainly mediated by a binding grove formed by αA helix and βB sheet of the PDZ domain and an internal peptide of Fz. Therefore we used this peptide as a probe to test PDZ ligand binding. For competition assay, Each compound was incubated with 100 nM Rox-DprC for 10 min before titrating with PDZ protein. The KI values of 16, J01-007, J01-015 and J01-017a were determined from KDapp = KD ×(1+[I]KI, where KDapp is the apparent KD of KI PDZ binding to Rox-DprC, and [I] is the compound’s concentration. Fluorescence polarization data were analyzed by using the Prism program (GraphPad Software, Inc.).
Results and Discussion
Pharmacophore-based virtual screening
By combining structure-based virtual screening with NMR binding studies, we initially identified an organic molecule (NSC668036) in the National Cancer Institute (NCI) small-molecule library (9) that can bind to the Dvl PDZ domain. We recently used the pharmacophore model generated in that study in combination with ligand-based screening to identify 15 potent PDZ domain potential inhibitors in the ChemDiv database (12). These 15 compounds comprise two scaffolds: scaffold A (compounds 1-3) and scaffold B (the remaining compounds) (Figure 1A). In order to further understand the molecular mechanisms of PDZ-ligand binding and to further improve PDZ ligand affinity, in this study we continued virtual screening to search for compounds with similar scaffold but with more diversity in substituents. The new searches were based on the pharmacophore models derived from previously identified compounds in scaffolds A and B. The pharmacophore models indicated that three hydrogen bond donors and one hydrogen bond acceptor are important for ligand binding; in addition, at least three independent hydrophobic interactions (R1, R2, and R3, Figure 1A) are preserved in the binding model of compound 1 (Chemdiv 5435-0027) to the PDZ domain. We designed 2D and 3D similarity queries based on the above pharmacophore model to search several databases, including ChemDiv, NCI and ChemBridge, with the UNITY program in Sybyl (Tripos, Inc.). The searches returned 38 and 18 hits based on the A and B scaffolds, respectively. After docking the hits to the PDZ domain with program Glide (Schrödinger, Inc.), we visually inspected the docking complex structures and selected five scaffold A molecules and four scaffold B molecules for experimental validation. As in previous studies (12), NMR chemical shift perturbation experiments were used to examine the interactions between the Dvl PDZ domain and the nine selected molecules. All nine compounds perturbed the PDZ domain at the same sites as previously identified inhibitors, suggesting that all of them bind to the binding pocket where native PDZ ligands bind, which is consistent with our docking studies. As an example, Figures 1B and 1C show the Glide-docked structure of compound 17 in complex with the Dvl PDZ domain. Although all the newly identified recognized the PDZ domain, four of them, in particular, 18 and 20, bound to the PDZ domain with slightly better affinities than 1, the best inhibitor identified in the previous studies (Table 1).
Figure 1.

Complex-structure-based pharmacophore model. (A) 2D structures of scaffolds A and B. (B) Complex structure of Dvl PDZ domain bound to scaffold-A compound 17. (C) Protein surface plotted in color to represent PDZ domain pockets P1 (orange), P2 (gray) and P3 (blue) that interact with R1, R2 and R3 fragments of 17 in the complex.
Table 1.
Structures and binding affinities of the PDZ binders identified by virtual screening
| Compound | Scaffold | R1 | R2 | R3 | KD’ (μM)a | |
|---|---|---|---|---|---|---|
| No. | ID | |||||
| 16 | 5435-0028 | A | -ph-4-CH3 | -ph | -i-Bu | 6.1 |
| 17 | 5435-0029 | A | -ph-4-CH3 | -ph | -Bn | 6.9 |
| 18 | 3865-0112 | A | -ph | -ph | -Bn | 4.7 |
| 19 | NSC350589 | A | -ph | -CH2NHCOCH2NH2 | -i-Bu | 19.3 |
| 20 | NSC334018 | A | -ph | -O-Bn | -i-Bu | 2.8 |
| 21 | 3865-0014 | B | -ph | -ph | -i-Bu | 14.1 |
| 22 | 3865-0016 | B | -ph | -ph | -Bn | 20.7 |
| 23 | 5613-0123 | B | -ph | -ph-4-CH3 | -CH3 | 187.5 |
| 24 | 5613-0128 | B | -ph | -ph-4-CH3 | -i-Bu | 18.7 |
Normalized KD values obtained by dividing the NMR-derived KD values by the difference between the KD values of NSC668036 measured by NMR and fluorescence methods (12).
Structure-activity relationships and structural analysis
The above virtual screening designed based on previous SAR studies allowed us to develop nine more PDZ binders with increased diversity of the molecules in both scaffold groups. Together with the PDZ binders identified previously, there were 8 molecules in scaffold A group and 16 molecules in scaffold B group. In order to aid the design of more potent PDZ inhibitors, we carried out 3D-QSAR analyses of the molecules in both groups. Comparative molecular-field analysis (CoMFA) (17) models were built for scaffolds A and B by using Sybyl 8.1 (Tripos, Inc.). The molecular fields and experimental binding data were highly correlated in both models; R2 values were 0.934 and 0.836 for scaffolds A and B, respectively (Table 2), indicating that both CoMFA models are highly accurate and predictive (17).
Table 2.
PLS statistics of CoMFA 3D-QSAR models for compounds in Scaffold A and B
| PLS statistics (LOO) | Scaffold A | Scaffold B |
|---|---|---|
| q2(cross validated correlation coefficient) | 0.513 | 0.243 |
| Number of components | 5 | 4 |
| r2 (correlation coefficient) | 0.998 | 0.874 |
| SEE (standard error of estimation) | 0.037 | 0.134 |
| F [ratio of r2 explained to unexplained = r2 /(1-r2)] | 259.754 | 12.469 |
| Pr2=0 | 0.000 | 0.001 |
| Contribution | ||
| steric | 0.642 | 0.899 |
| electrostatic | 0.358 | 0.101 |
The CoMFA contour maps for scaffolds A and B are illustrated in Figure 2, with 16 used as the reference for scaffold A (Figure 2A) and 21 as the reference for scaffold B (Figure 2B). The contour map of scaffold B, unlike that of scaffold A, shows large sterically unfavorable areas (yellow) in all three positions, with a sterically favorable area (green) only at the R1 position. This marked difference between the two contour maps is probably caused by the rigidity and steric effects arising from the sp2 carbons at the R1 position of scaffold B, which make this scaffold less promising for further optimization. We therefore focused on the scaffold A molecules. For the first model, compound 16 was selected as a reference because it was the compound which we wanted to use as a starting point to synthesize more PDZ inhibitors. Although compound 20 has a lower binding affinity, it was not chosen because its -O-Bn group might be more susceptible to water hydrolysis and synthesis conditions. In addition, compound 18 was not chosen because comparing 16 and 17 suggesting that -Bn is not favored at R3 while the relative high binding affinity of 16 was possibly due to the ortho-methyl group on the benzyl at R1.
Figure 2.

CoMFA contour maps of (A) scaffold A and (B) scaffold B binders. Greater affinity in electrostatic contours is related with a more positive charge near blue and a more negative charge near red. Greater affinity in steric contours is correlated with more bulky groups near green and less bulky groups near yellow.
The CoMFA contour map for scaffold A was highly consistent with the Glide-docked complex structure of the Dvl PDZ domain bound to 16. The steric map showed a small number of few unfavorable areas (yellow) in R1, indicating that the binding affinity may not benefit from increasing the molecular sizes of these two positions, consistent with the above SAR analysis. There is a large sterically favorable area at the R2 position, which points toward the hydrophobic site composed by the sidechains of Ile266, Ile278 and Val318 (Figure 3). In addition, the R2 position also possesses both positively (blue) and negatively (red) charged favorable areas, which correspond to the electrostatic interactions with the backbone carboxyl group of Ile266 and the distant charge effects from the side-chain of Arg322, respectively (Figure 3).
Figure 3.
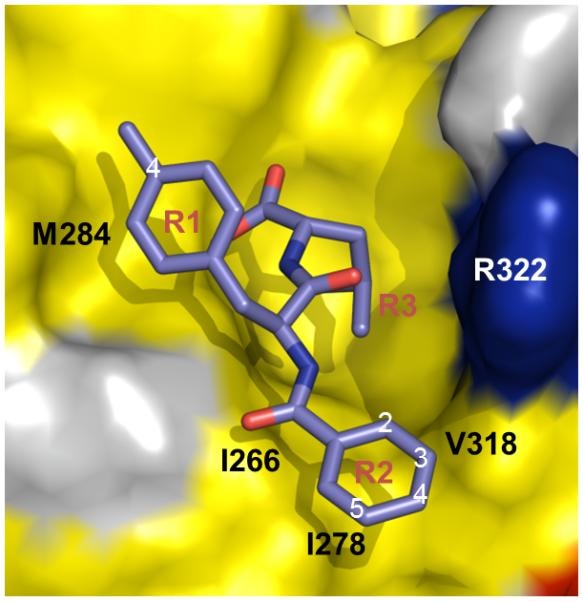
Schematic of model for new compound design based on docking structure of 16 bound to Dvl PDZ domain and QSAR. Positions on R1 and R2 for modification are labeled.
Design and synthesis of additional compounds
To optimize the hits obtained from the virtual screens and verified by NMR experiments, we decided to use QSAR and structural analyses to guide the synthesis of additional scaffold. A compounds that could not be found in the existing small-molecule libraries. To reduce the burden of the synthesis, we first virtually generated all of the potential compounds that could be synthesized in a combinatorial fashion.
As the QSAR model based on 16 (Figure 3) indicated that R2 is the hot spot for modification, we first generated a group of compounds by using CombiLibMaker in Sybyl (Tripos, Inc.) with the following alterations on 16: i) the electronegative atoms/groups F, Cl, Br, I, -CH2F, -CH2Cl, -CH2Br, -CH2I, -CH2NO2 or -CH2OH were attached at position 2; ii) the electronegative atoms/groups F, Cl, Br, I, -CN, -OH or -NO2 were placed at position 3 to maximize the likelihood of favorable electrostatic interactions with Arg322; iii) a methyl group was added at position 4 and iv) methyl, ethyl, -S-ph or -O-ph groups were added at position 5 for favorable hydrophobic interactions with the residues in pocket P2. We termed this set of compounds the J01 group. As the QSAR analysis also indicated that the methyl group at the 4 position on the benzyl ring of R1 might not be ideal for modification, we also generated a second set of molecules, the J02 group, using the same method with the exception that the R1 was a phenyl ring instead of a -ph-4-CH3.
Since compounds in libraries J01 and J02 were derivatives of 16 with small substitutions, it was likely that their binding characteristics would be similar to those of 16. Indeed, when they were superimposed on the docked 16, they showed no major impediment to binding with the PDZ domain. After energy minimization of these compounds at the binding site, the binding potency of these molecules with the Dvl PDZ domain was predicted by using the Cscore Subset module in SYBYL (Tripos, Inc.). Scored docking poses were then extracted and ranked as previously described (9, 12). On the basis of this ranking, seven compounds (Supplemental Table S1) were selected and synthesized by using a relative standard solid-phase synthesis protocol (Supplemental Scheme S1). Due to solubility issues with two compounds, five of the synthesized compounds were tested by NMR chemical shift perturbation experiments. All of the five tested compounds perturbed the same binding sites as previously identified inhibitors, suggesting that they all bind to the same binding pocket. Binding affinities (Table 3) were determined by monitoring chemical shift perturbations as previously reported (9, 12). Four compounds (J01-007, -012 and -015, J02-002) bound to the Dvl PDZ domain with greater affinity than 16 (Table 3), indicating that the proposed substitutions interact favorably with the P2 pocket of the PDZ domain.
Table 3.
Structures and binding affinities of synthesized compounds
| Compound | R1 | R2 | R3 | KD’ (μM)a |
|---|---|---|---|---|
| J01-007 | -ph-4-CH3 | -ph-3-F | -i-Bu | 2.27 |
| J01-012 | -ph-4-CH3 | -ph-3-CN | -i-Bu | 1.83 |
| J01-015 | -ph-4-CH3 | -ph-5-CH3 | -i-Bu | 2.96 |
| J02-001 | -ph | -ph-3-F | -i-Bu | 7.67 |
| J02-002 | -ph | -ph-4-F | -i-Bu | 3.49 |
| J01-017a | -ph-4-CH3 | -ph-3,4-2F-5-CH3 | -i-Bu | ____b |
KD values obtained directly by NMR titration (12).
KD could not be obtained by NMR titration due to slow exchange.
To take full advantage of this group (R2) within the compound scaffold, we carried out a second round of synthesis and generated J01-017a (Tables 3 and 4), which combined three components shown to enhance binding in compounds J01-007, J01-015 and J02-002 (comparing J01-007 and J01-015 to 16, as well as J02-002 to 21). Docking studies of J01-017a suggested that it would interact with both the hydrophobic groove and the positively charged Arg322 residue around pocket P2 (Figure 1). Indeed, NMR titration generated 1H-15N-HSQC spectra different from those of previously identified compounds. In particular, peak Arg319 which locates right at the binding site on helix αA shifted continuously when the PDZ domain was titrated with increasing concentrations of compound J01-007 (Figure 4A). However in sharp contrast, when titrated with J01-017a (Figure 4B), the peak did not shift but instead the intensity decreased (see decreased magenta contours) when the ligand to protein ratio went up to one; when the ratio reached three, not only the intensity further decreased but also the peak appeared at a new position; and the intensity further increased with increasing concentrations of ligand until saturated. This suggests J01-017a is in slow exchange with PDZ domain on the NMR time scale (Figure 4), which are the classical behavior of compounds with sub-micromolar binding affinities (21). In addition to Arg319, many other residues on the binding site also show slow exchange, e.g, Arg322, Val318 on the αA helix and Ile266 on the βB sheet as well. We choose to show Arg319 simply for clarity because it is well isolated from other signals.
Table 4.
Fluorescence polarization competitive binding assay
| Compound | R1 | R2 | R3 | KI(μM) |
|---|---|---|---|---|
| J01-017a | -ph-4-CH3 | -ph-3,4-2F-5-CH3 | -i-Bu | 1.5 ± 0.52 |
| J01-015 | -ph-4-CH3 | -ph-5-CH3 | -i-Bu | 3.5 ± 0.21 |
| J01-007 | -ph-4-CH3 | -ph-3-F | -i-Bu | 4.2 ± 0.46 |
| 16 | -ph-4-CH3 | -ph | -i-Bu | 7.1 ± 2.8 |
Figure 4.

NMR binding studies. NMR titration spectra for J01-007 (A) and J01-017a (B). Spectra of free PDZ domain are shown in black; spectra of PDZ domain with increasing concentration of compounds (ligand: protein ratio = 1, 3, 5, 7, and 11) are shown in magenta, blue, green, yellow and red, respectively.
To determine the binding affinity between the synthesized compounds and the PDZ domain, we carried out competitive PDZ binding assays of structurally related compounds, using fluorescence polarization as previously described (22) (Figure 5). J01-017a had the highest binding affinity, it competed with the binding of Dapper peptide, a Dvl PDZ domain binding peptide (22), with inhibition constant 1.5 ± 0.2 μM, as compared to 7.1 ± 0.5 μM for 16. In the same binding assay study, the precursors of J01-017a (J01-007, J01-015) also showed slightly greater affinity than 16, coinciding with the NMR titration analysis that J01-017a is the first PDZ inhibitor entered the slow exchange regime. Therefore, J01-017a offers much better binding affinity than its parent compounds.
Figure 5.

Competitive binding of structurally related compounds to the PDZ domain. Binding affinity was measured by fluorescence polarization.
Conclusions
We have extended our previous studies to identify additional, more potent PDZ domain inhibitors. By exhaustive virtual screening of libraries for derivatives of PDZ binders and close inspection of their docking structures, we identified compounds that not only provide scaffolds for subsequent synthesis but can also be used to guide the synthesis of additional, more optimal compounds. We constructed two series of new compounds with similar scaffolds based on the QSAR derived from virtually identified PDZ binders and the complex structure of these compounds with PDZ. After docking and scoring the compounds in the first series, we synthesized seven compounds and confirmed five of them to be more potent inhibitors than the template, 16. We then further optimized the leads and synthesized compound J01-017a, whose R2 group has several hydrophobic contacts with the P2 pocket residues on the surface of the Dvl PDZ domain that are more favorable than those of 16. In addition, the fluoride on position 3 of J01-017a is able to form a polar interaction with the side chain of Arg322 (Figure 6). As the result, the binding affinity of J01-07a to the Dvl PDZ domain is about 5 times lower that of 16.
Figure 6.

The Dvl PDZ domain in complex with 16 (A) and J01-017a (B). Carbon atoms in 16 and J01-017a are shown in magenta. Surface of PDZ protein is shown in brown. PDZ domain residues composing the P2 pocket are labeled and the side-chains are shown.
To our knowledge, J01-017a is the most potent inhibitor of the Dvl PDZ domain that has been produced. Testing of its biological effects is currently under way. Because its binding affinity is much lower than the native ligands of the Dvl PDZ domain (4), we believe that the compound could be a very useful tool for various biological studies. We are also testing the selectivity of this compound against other PDZ domains. Although there are many copies of PDZ domain in human, it is possible to achieve selectivity. One of our previously identified compound NSC668036 is selective to the mDvl PDZ domain against two other PDZ domains, a class I PDZ domain and a class II PDZ domain (8). Our studies also demonstrate that virtual screening has an additional important use beyond identifying potential lead compounds from existing compound libraries for drug discovery. When combined with other experimental methods to validate compound potency, it can be used for structure-activity relationship studies that lead to the design and synthesis of feasible compounds with potentially higher binding potency.
Supplementary Material
Acknowledgment
We thank Dr. Weixing Zhang for assistance with NMR experiments, Dr. Charles Ross and Mr. Scott Malone for computer support, and Sharon Naron for editing the manuscript. This work was supported by grants CA21765 and GM061739 from the National Institutes of Health, by the American Lebanese Syrian Associated Charities (ALSAC), and by an American Heart Association Predoctoral Fellowship (J. Shan).
Footnotes
Supporting Information Available. The identities and purities of the synthesized compounds, and the synthetic scheme are available in the online version of this article.
References
- 1.Logan CY, Nusse R. The Wnt Signaling Pathway in Development and Disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 2.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–98. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 4.Wong HC, Bourdelas A, Krauss A, Lee HJ, Shao YM, Wu D, et al. Direct binding of the PDZ domain of Dishevelled to a conserved internal sequence in the C-terminal region of Frizzled. Mol Cell. 2003;12:1251–60. doi: 10.1016/s1097-2765(03)00427-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simons M, Gault WJ, Gotthardt D, Rohatgi R, Klein TJ, Shao Y, et al. Electrochemical cues regulate assembly of the Frizzled/Dishevelled complex at the plasma membrane during planar epithelial polarization. Nature cell biology. 2009;11:286–94. doi: 10.1038/ncb1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang X, Tan J, Li J, Kivimae S, Yang X, Zhuang L, et al. DACT3 is an epigenetic regulator of Wnt/beta-catenin signaling in colorectal cancer and is a therapeutic target of histone modifications. Cancer Cell. 2008;13:529–41. doi: 10.1016/j.ccr.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uematsu K, He B, You L, Xu Z, McCormick F, Jablons DM. Activation of the Wnt pathway in non small cell lung cancer: evidence of dishevelled overexpression. Oncogene. 2003;22:7218–21. doi: 10.1038/sj.onc.1206817. [DOI] [PubMed] [Google Scholar]
- 8.Uematsu K, Kanazawa S, You L, He B, Xu Z, Li K, et al. Wnt pathway activation in mesothelioma: evidence of Dishevelled overexpression and transcriptional activity of beta-catenin. Cancer Res. 2003;63:4547–51. [PubMed] [Google Scholar]
- 9.Shan J, Shi DL, Wang J, Zheng J. Identification of a specific inhibitor of the dishevelled PDZ domain. Biochemistry. 2005;44:15495–503. doi: 10.1021/bi0512602. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Appleton BA, Wiesmann C, Lau T, Costa M, Hannoush RN, et al. Inhibition of Wnt signaling by Dishevelled PDZ peptides. Nat Chem Biol. 2009;5:217–9. doi: 10.1038/nchembio.152. [DOI] [PubMed] [Google Scholar]
- 11.Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov. 2006;5:997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 12.Shan J, Zheng JJ. Optimizing Dvl PDZ domain inhibitor by exploring chemical space. J Comput Aided Mol Des. 2009;23:37–47. doi: 10.1007/s10822-008-9236-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.London TB, Lee HJ, Shao Y, Zheng J. Interaction between the internal motif KTXXXI of Idax and mDvl PDZ domain. Biochem Biophys Res Commun. 2004;322:326–32. doi: 10.1016/j.bbrc.2004.07.113. [DOI] [PubMed] [Google Scholar]
- 14.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 15.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
- 16.Worrall JA, Reinle W, Bernhardt R, Ubbink M. Transient protein interactions studied by NMR spectroscopy: the case of cytochrome C and adrenodoxin. Biochemistry. 2003;42:7068–76. doi: 10.1021/bi0342968. [DOI] [PubMed] [Google Scholar]
- 17.Gramer RDP,DE, Bunce JD. Comparative molecular-field analysis (CoMFA). Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc. 1988;110:5959–67. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 18.Lee HJ, Wang NX, Shao Y, Zheng JJ. Identification of tripeptides recognized by the PDZ domain of Dishevelled. Bioorg Med Chem. 2009;17:1701–8. doi: 10.1016/j.bmc.2008.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cramer RDB,JD, Patterson DE, Frank IE. Cross-validation, bootstrapping, and partial least squares compared with multiple regression in conventional QSAR studies. Quant Struct-Act Relat. 1988;7:18–25. [Google Scholar]
- 20.Dunn WJ, III, Wold S, Edlund U, Hellberg S, Gasteiger J. Multivariate structure-activity relationships between data from a battery of biological tests and an ensemble of structure descriptors: the PLS method. Quant Struct-Act Relat. 1984;3:131–7. [Google Scholar]
- 21.Wemmer DE, Williams PG. Use of nuclear magnetic resonance in probing ligand-macromolecule interactions. Methods Enzymol. 1994;239:739–67. doi: 10.1016/s0076-6879(94)39028-2. [DOI] [PubMed] [Google Scholar]
- 22.Lee HJ, Wang NX, Shi DL, Zheng JJ. Sulindac inhibits canonical Wnt signaling by blocking the PDZ domain of the protein Dishevelled. Angew Chem Int Ed Engl. 2009;48:6448–52. doi: 10.1002/anie.200902981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.