

Abstract
All-trans-retinoic-acid (ATRA)-induced differentiation of human myeloid leukemia cells is characterized by persistent MAPK signaling. Fragmentary data suggests Src family kinase (SFK) inhibitors enhance differentiation and thus have potential therapeutic value. The present study shows that SFK inhibitors PP2 and dasatinib enhance aspects of MAPK signaling and regulate a panel of differentiation markers including CD11b and p47phox. HL-60 and NB4 myeloid leukemia cells show accelerated ATRA-induced G1/0 arrest/differentiation with inhibitor co-treatment. We also identified components of a Lyn- and c-Raf-containing MAPK signaling complex augmented by the inhibitors. PP2 and dasatinib increased ATRA-induced expression of Lyn and c-Raf (total and c-RafpS259) and their interaction. The Lyn-associated serine/threonine kinase CK2 also complexed with c-Raf and c-RafpS259, and the KSR1 scaffold protein bound c-Raf, Lyn, and ERK. c-Raf/ERK association was increased by the inhibitors, which is significant since ERK may cause c-Raf C-terminal domain (CTD) phosphorylation in a putative feedback mechanism. Consistent with this, inhibitor treatment caused more CTD phosphorylation. Lyn knockdown decreased c-Raf CTD and S259 phosphorylation. This is the first evidence suggesting SFK inhibitors enhance ATRA-induced differentiation through a possible feedback loop involving KSR1-scaffolded c-Raf and ERK complexed with Lyn and CK2.
Keywords: Src inhibitors, dasatinib, ATRA, AML differentiation
Introduction
The Src family of tyrosine kinases (SFKs) are a unique group of enzymes that have diverse functions in cell proliferation, survival, differentiation, adhesion, and migration. They play important regulatory roles in hematopoiesis, but also contribute to hematopoietic cancers. One historically prominent paradigm of SFK action is positive regulation of MAPK signaling and cell proliferation, and contribution to cell transformation [reviewed in (1)].
SFK hyperactivity is commonly associated with acute and chronic myeloid malignancies. The proliferative signals resulting from the BCR/ABL fusion tyrosine kinase in chronic myelogenous leukemia (CML) are driven by downstream SFKs including Src, Lyn, and Hck (2, 3). Lyn is the predominant active SFK expressed in AML cells (4, 5). It is often hyperactivated, is associated with iminitab resistance in CML, and may mediate the effects of the FLT3/ITD mutation found in 30% of AML cases (6–9). Blocking SFK activity has been effective in slowing leukemic cell growth (10). The inhibitor dasatinib has proven clinically successful in the treatment of CML, Philadelphia chromosome-positive acute lymphocytic leukemia (ALL) (11), and iminitab-resistant leukemias (12–14).
SFK activity and expression could also modulate ATRA differentiation induction therapy. Miranda et al. recently reported that the SFK inhibitor PP2 potentiated ATRA-induced gene expression and enhanced the differentiation marker CD11b in myeloid NB4, HL-60, and primary acute promyelocytic leukemia (APL) cells (15). Kropf et al. recently reported that dasatinib also increased ATRA-induced CD11b expression (5). In contrast, some reports show that SFKs may positively regulate ATRA-induced differentiation. Lyn and Fgr are upregulated in HL-60 and NB4 myeloid leukemia cells after ATRA treatment, and both were reported to prevent apoptosis during granulocytic differentiation (16, 17).
SFK inhibitors are capable of positive and negative regulatory effects on MAPK pathway components. PP2 enhances Ras-independent Raf-1 activation that is mediated by Raf S621 phosphorylation (18), suggesting that SFK inhibitors are able to positively regulate Raf activity. Dasatinib, however, inhibits MAPK activity in the absence of growth factors (GFs) and attenuates signaling in the presence of GFs in CML progenitors (19). MAPK augmentation may have implications for ATRA induction therapy, since retinoic acid results in sustained MAPK activity which is characteristic of HL-60 maturation (20–22).
The ability of SFKs to regulate ATRA-induced differentiation and MAPK signaling is therefore not understood. This motivates interest in how SFK inhibitors can affect the extent of ATRA-induced phenotypic conversion or modulate MAPK regulatory molecules. While ATRA is proven to be an effective intervention modality for t(15,17) positive APLs, it has not been effective in other leukemia subtypes, making means of improving its action in t(15,17) negative cells of therapeutic interest.
In this report the extent to which SFK inhibitors affect differentiation, myeloid leukemia cell phenotypic conversion, and MAPK signaling was characterized in t(15,17) negative HL-60 and t(15,17) positive NB4 cells. We specifically analyzed the effects of PP2 and dasatinib on two ATRA-regulated SFK members, Fgr and Lyn (16, 23). While Fgr activation was undetectable in HL-60 cells, we found that the inhibitors had different effects on Lyn active site phosphorylation and cellular tyrosine phosphorylation in ATRA-treated cells. Both, however, were able to enhance the ATRA-induced phenotypic conversion and cell cycle arrest in two cell lines. Both inhibitors also increased expression of Lyn and c-Raf, along with their interaction. Phosphorylation of c-Raf at S259 (c-Raf pS259) and C-terminal serine residues was increased, as well as c-RafpS259 and Lyn association. CK2 co-immunoprecipitated with c-RafpS259, possibly modulating phosphorylation. ERK, which is also capable of phosphorylating Raf, showed increased interaction with c-Raf suggesting a MAPK feedback module consistent with the observed increase in C-terminal serine phosphorylation. These activities appear to be associated with the KSR1 scaffold protein. Similar results were observed for HL-60 and NB4 cells, indicating that combination inhibitor/ATRA therapy may be effective in a variety of myeloid leukemia cell types. Our results suggest a previously unreported MAPK-linked mechanism associated with accelerated ATRA/SFK inhibitor combination therapy.
Materials and Methods
Cell culture
HL-60 and NB4 cells were grownin RPMI 1640 with 1% antibiotic/antimycotic from Invitrogen (Carlsbad, CA) and treated with ATRA as previously described (24). PP2 and PP3 from EMD Chemicals (Gibbstown, NJ) were solubilized in dimethyl sulfoxide (DMSO) at 10 mM. Cells were treated with a final concentration of 10 μM with a 0.1% concentration of carrier DMSO. Dasatinib from Santa Cruz Biotechnology (Santa Cruz, CA) was solubilized in DMSO at 5 mM. Cells were treated with a final concentration of 300 nM. SFK activity inhibition was confirmed by Western blot. The concentrations of drugs were approximately 3–4 fold less than that found to cause overt toxicity in titrations monitoring cell growth with a hemacytometer and trypan blue exclusion.
Antibodies and reagents
Antibodies for cytometric analysis of CD11b and for CK2 Western blotting were from BD Pharmingen (San Jose, CA). Protein A/G beads used for immunoprecipitation and p-Tyr antibody were from Santa Cruz Biotechnology. GAPDH, p-RafS259, p-MEK, p-Erk1/2, ERK1/2 (rabbit), KSR1, c-Raf (rabbit), pan-SFK416, Lyn, Fgr, HRP anti-mouse, and HRP anti-rabbit were from Cell Signaling (Danvers, MA). pRafS621, c-Raf (mouse), and Lipofectamine 2000 CD were from Invitrogen. ERK1/2 (mouse) was from AbCam (Cambridge, MA). M-PER Mammalian Protein Extraction Reagent lysis buffer was from Pierce (Rockford, IL). Propidium iodide, protease and phosphatase inhibitors, and DMSO were purchased from Sigma.
Flow cytometric phenotypic analysis
Immunostaining for CD11b was performed as previously described (20) and fluorescence was detected using a Becton Dickinson LSR II flow cytometer (San Jose, CA). Cell cycle analysis was performed as previously described (20).
Western blot analysis and immunoprecipitation
For immunoprecipitation experiments, cells were lysed as previously described (24). Equal amounts of protein were pre-cleared with Protein A/G beads. The beads were pelleted and supernatant was incubated with appropriate antibodies and beads overnight. All incubations included protease and phosphatase inhibitors used for lysis with constant rotation at 4°C. Bead/antibody/protein slurries were then washed and subjected to standard SDS-PAGE analysis as previously described (24).
Creation of shLyn stable transfectants
Plasmid containing shRNA targeting Lyn (target sequence: cgagcggaagaactacatt) was from GeneCopia (Rockville, MD). 50 μg of plasmid and 50 μL of Lipofectamine 2000 CD (Invitrogen) were incubated at 25°C for 25 minutes in serum-free RPMI 1640 media. 20×106 HL-60 cells were pelleted by centrifugation and resuspended in media containing the Lipofectamine/DNA complexes. The cells were electroporated at 300mV/960 μFD capacitance and immediately transferred to prewarmed media containing 10% FBS. After 24 hours debris was removed by centrifugation and cells were cultured as described. Cells expressing shRNA targeting Lyn were isolated by FACS sorting for eGFP.
Statistics
Statistics were analyzed using Microsoft Excel statistical software.
Results
PP2 and dasatinib accelerate ATRA-induced G1/0 arrest
To determine if PP2 and dasatinib affected ATRA-induced growth inhibition, we compared population growth during different treatment conditions in HL-60 cells. A DMSO vehicle control and PP3, an inactive structural analog of PP2, were included to test for toxicity or non-specific effects. These were used separately and in lieu of PP2 and had no significant effect on cell growth alone or in combination with ATRA (supplementary data S1a). No differences were detected among treatment groups after 24 hours. After 48 hours of culture PP2 alone decreased proliferation, and cells treated with a combination of PP2 and ATRA show greater growth inhibition (Figure 1a, top). Strikingly, PP2/ATRA co-treated cells showed no proliferation after 48 hours. The cells appeared to have undergone only one doubling followed by G1/0 arrest, consistent with a G1 cell cycle block. Unlike PP2, dasatinib had no significant effect on cell population growth (Figure 1a, bottom).
Figure 1.

A: HL-60 cell counts at various time points. B: Cell cycle analysis of HL-60 and NB4 cells (*p=<0.5 significantly higher than untreated control; #p=<0.5 significantly higher than ATRA treatment only; $p=<0.5 significantly lower than ATRA treatment only.)
Next we measured cell cycle phase distribution in HL-60 and NB4 cells. After 48 and 72 hours all ATRA-treated and ATRA plus PP2 or dasatinib co-treated populations showed significantly more G1/0 compared to untreated cells or cells treated with dasatinib only (Figure 1b). PP2-treated populations showed a modest G1/0 enrichment in both cell lines at 48 hours. We also detected a corresponding decrease in S and G2/M phase at 72 hours in all ATRA plus PP2 or dasatinib co-treated samples. HL-60 cells also showed a significant decrease in G2/M within 48 hours with co-treatments, and NB4 cells with ATRA plus PP2. Strikingly, approximately 95% of HL-60 cells and 85% of NB4 cells that received co-treatment were arrested in G1/0 by 48 hours. Although PP3 or DMSO in combination with ATRA showed slight albeit statistically significant G1/0 enrichment in some HL-60 sample sets, these increases were remarkably less than PP2 plus ATRA (Figure S1b). Therefore, the effects of PP2 on cell cycle and growth are largely attributable to SFK inhibition. Together these results suggest that both inhibitors propel ATRA-induced growth arrest, but PP2 appears more effective. Trypan blue exclusion staining and cytometric analysis of sub-G1 populations indicated that cells were arrested without evidence of apoptosis.
PP2 and dasatinib modulate expression of ATRA-induced differentiation markers
To determine if PP2 or dasatinib could further enhance ATRA-induced phenotypic conversion we evaluated the expression of myeloid differentiation markers. The α-integrin receptor CD11b is upregulated after ATRA treatment and is considered a marker of granulocytic differentiation. PP2 and dasatinib increased ATRA-induced CD11b expression in HL-60 cells by 48 and 72 hours (Figure 2a). PP2 showed a more dramatic enhancement and had significant effects within 24 hours. This is consistent with previous reports (5, 15). PP3 also enhanced ATRA-induced and CD11b expression at some time points, but only modestly compared to PP2 (Figure S1c). Interestingly, within 24 hours PP2 alone caused a modest induction of CD11b expression (Figure 2a), indicating that PP2 could propel at least some differentiation. Dasatinib alone had no effect on CD11b expression. The leukocyte antigen and differentiation marker CD38 was also evaluated but neither PP2 nor dasatinib had any significant effect on expression (data not shown).
Figure 2.

A: HL-60 cells were treated as shown and immunostained for CD11b at indicated time points. Gating was set to exclude 95% of the negative control (untreated cells). (*p=<0.5 higher than untreated cells; #p=<0.5 higher than cells treated with ATRA only.) B: Western blots for p47phox expression in HL-60 cells after 48 hours of culture. C: Western blots for p47phox expression in NB4 cells after 48 hours of culture.
Maturing myeloid cells are capable of induced respiratory burst/oxidative metabolism by an activated NADPH oxidase complex. p47phox, an NADPH oxidase component, is upregulated after ATRA. Within 48 hours of culture, cells treated with ATRA alone show enhanced expression of p47phox compared to the untreated control in NB4 and HL-60 cells (Figures 2b&c). PP2 alone modestly upregulated p47phox in HL-60 cells supporting evidence that it may induce differentiation. Co-treatment with either of the inhibitors modestly enhanced ATRA-induced p47phox expression in both cell lines. Together these results indicate that both inhibitors enhanced ATRA-induced differentiation markers.
PP2 and dasatinib enhance ATRA-induced Fgr and Lyn expression, but differentially affect Lyn activation
After ATRA treatment HL-60 cells show upregulated expression and tyrosine phosphorylation of Fgr and Lyn, while siRNA targeting Fgr or Lyn promotes apoptosis (16). Since these ATRA-regulated SFKs may participate in induced differentiation, we determined the effects of the inhibitors by comparing protein expression and active site phosphorylation with different treatments.
Fgr was not detectable in HL-60 cells until they were treated with ATRA, which resulted in clear induced expression (Figures 3a&b). Co-treatment with ATRA plus either inhibitor enhanced Fgr expression (Figure 3b). Lyn was also upregulated by ATRA, and expression was further enhanced by co-treatment with PP2 or dasatinib. Therefore, ATRA/SFK inhibitor co-treatment increased protein expression of ATRA-regulated SFKs. Using a pan-SFK antibody that detects active site phosphorylation (Y416) in all family members, we found that activation in immunoprecipitated Fgr (FgrY412) was undetectable regardless of treatment, indicating its kinase function is not involved (Figure 3a, top). This suggested that a previous report (16) of upregulated Fgr tyrosine phosphorylation may be relevant to the C-terminal autoinhibitory site.
Figure 3.

A: HL-60 cells were treated as indicated for 48 hours followed by immunoprecipitation (IP) or Western Blotting. Lyn and Fgr IP membranes were probed using a pan-SFK Y416 antibody that detects tyrosine phosphorylation at the active site for all SFK members. Membranes were stripped and reprobed for total Lyn and Fgr protein. B: HL-60 Western blots for phosphorylated SFK members (Y416) and total protein for Lyn and Fgr. C: Total cell lysate was Western blotted for tyrosine phosphorylated proteins using a p-Tyr antibody. D: NB4 cell Western blots for active and total Lyn expression.
Evaluation of immunoprecipitated Lyn showed that PP2 and dasatinib crippled basal Y416 phosphorylation (LynY397) (Figure 3a, bottom). However in ATRA-treated cells, dasatinib continued to extinguish Lyn active site phosphorylation while PP2 did not. ATRA apparently protected Lyn from PP2 in this regard. Consistent with these results pan-SFK Y416 Western blotting showed that PP2 and dasatinib both abolished basal SFK activation, but while dasatinib continued to block SFK activity after ATRA treatment PP2 failed to do so (Figure 3b). Thus, it seems Lyn is the dominant active SFK in HL-60 myeloid leukemia cells consistent with a previous report (5). This suggests that Lyn activity is dispensable for ATRA induction since cells co-treated with either inhibitor showed enhanced growth arrest and differentiation marker expression.
p-Tyr Western blotting showed that while PP2 alone decreased cellular tyrosine phosphorylation, it enhanced the p-Tyr status of at least two ATRA-modulated proteins in the range of 55–58 KD and 80 kD, upregulated the phosphorylation of a protein at approximately 150kD, and inhibited a 100kD protein (Figure 3c). In contrast, dasatinib appeared to extensively block cellular tyrosine phosphorylation alone and in combination with ATRA. This supports the argument that Lyn expression and activity plays a role in mediating ATRA-induced protein tyrosine phosphorylation.
Finally, we evaluated the effects of PP2 and dasatinib onY416 phosphorylation and Lyn expression in NB4 cells (Figure 3d). Like HL-60 cells, we found that PP2 failed to block Y416 phosphorylation in the presence of ATRA while dasatinib was effective. PP2 alone and with ATRA increased total Lyn expression; this increase was enhanced by co-treatment. Dasatinib alone did not increase Lyn expression but ATRA and co-treatment did. Therefore, treatment of NB4 and HL-60 cells with either inhibitor plus ATRA upregulated Lyn expression, coinciding with enhanced differentiation.
PP2 and dasatinib upregulate ATRA-induced c-Raf phosphorylation without affecting ERK or MEK activation
ATRA-induced differentiation is driven by a sustained MAP kinase signal such that inhibition of MEK activity blocks ERK and c-Raf activation, preventing differentiation (21, 22). Because SFKs can regulate MAPK signaling we determined the effect of PP2 and dasatinib on the Raf/MEK/ERK axis.
Like ATRA, PP2 alone upregulated c-Raf expression in HL-60 cells, and co-treatment showed further enhancement (Figure 4a). PP2 did not affect ATRA-induced phosphorylation of ERK or MEK or total ERK or MEK expression. We then evaluated the phosphorylation status of c-Raf regulatory residues. ATRA induces c-Raf S621 phosphorylation by 48 hours and coincides with nuclear migration (25), and ectopic expression of the Raf CR3 domain containing S621 enhances ATRA-induced differentiation (26). PP2 alone and with ATRA increased c-Raf phosphorylation at S621 within 48 hours and also upregulated c-RafpS259 phosphorylation (Figure 4b). PP2 by itself and with ATRA strongly enhanced S259 phosphorylation. To our knowledge this is the first report of c-RafpS259 modification after ATRA or SFK inhibitor treatment.
Figure 4.

A: HL-60 cells were treated as indicated for 48 hours. Western blotting shows phospho- and total ERK and MEK, and total c-Raf. B: HL-60 Western blots for phosphorylated c-RafpS259 and S621 after 48 hours of culture. C: Western blots for various MAPK signaling molecules and their phosphorylation status in HL-60 cells after 48 hours of treatment as indicated. D: Phosphorylation status of c-Raf in NB4 cells after 48 hours of treatment as indicated.
We then determined if dasatinib had similar effects. Dasatinib alone showed little enhancement of c-Raf expression or phosphorylation of c-Raf at S621 and S259 (Figure 4c). However, co-treatment increased expression and phosphorylation at S621, with the most significant increase in c-RafpS259. This suggests that PP2 and dasatinib have similar effects on the Raf/MEK/ERK axis after ATRA treatment. Like PP2 dasatinib did not cause detectable differences in MEK or ERK expression or phosphorylation after ATRA (data not shown). Consistent with this, experiments with NB4 cells showed an increase in c-RafpS259 and c-Raf expression after ATRA or inhibitor treatment (Figure 4d). c-RafpS259 also increased with ATRA or PP2 (but not dasatinib alone), and combination treatment with either inhibitor plus ATRA further enhanced phosphorylation. These results focused attention on c-Raf as a downstream target of PP2 and dasatinib. This motivated interest in identifying c-Raf and c-RafpS259 partners, particularly if there were associations between c-Raf and Lyn or other MAPK signaling regulators.
PP2 and dasatinib regulate interactions between Lyn/c-Raf and ERK/c-Raf, and c-Raf phosphorylation
Since the above results suggest a linkage between Lyn and c-Raf, there was interest in exploring associations between c-RafpS259 and Lyn, and also with CK2 and KSR1, two Raf regulating kinases (27–30). We first evaluated if the inhibitors affected MAPK signaling molecule associations in HL-60 cells. Lyn and c-Raf immunoprecipitation experiments showed that these two molecules complex with each other after ATRA treatment, and that this interaction is increased by the addition of either PP2 or dasatinib (Figure 5a&b). Immunoprecipitated Fgr did not show significant interaction with c-Raf (data not shown).
Figure 5.

A: After 48 hours, Lyn was immunoprecipitated from HL-60 samples treated as shown. Western blotting shows interaction with partner molecules and Lyn confirms the IP. B: After 48 hours, c-Raf was immunoprecipitated from HL-60 samples treated as shown. Western blotting shows interaction with partner molecules and c-Raf confirms the IP. Interaction with Lyn and Lyn-complexed proteins detected in (A) was confirmed. C: After 48 hours, c-RafpS259 was immunoprecipitated from HL-60 samples treated as shown. Protein interaction with Lyn, CK2, and KSR1 was confirmed. Western blotting for CK2 and KSR1 show total protein expression. D: ERK was immunoprecipitated from HL-60 samples after 48 hours, and membranes were probed to confirm interaction with c-Raf and KSR1, suggesting a KSR1 scaffolded ERK-mediated feedback loop. E: HL-60 cell Western blot for c-Raf CTD phosphorylation of either S289, S296, or S301. F: Immunoprecipitation experiments with NB4 cells confirm increased interaction with Lyn/c-Raf, Lyn/c-RafpS259, c-Raf/ERK, and potential KSR1 scaffolding. Western blotting confirms increases in CTD phosphorylation.
The serine/threonine (S/T) kinase, casein kinase II (CK2), is known to complex with and be phosphorylated by Lyn and Fgr (31, 32). CK2 also interacts with KSR1, a scaffold protein that modulates MAPK signaling [(30) and reviewed in (33)]. CK2/KSR1 binding facilitates Raf phosphorylation, which is dependent on SFK-mediated Raf Y341 phosphorylation (28). Therefore, Lyn may be linked to MAPK signaling and c-Raf binding through interactions with CK2 and KSR1. We immunoprecipitated Lyn and c-Raf and found that CK2 and KSR1 complex with c-Raf and Lyn (Figure 5a&b). We also detected ERK binding to Lyn and c-Raf, which is also reported to interact with KSR1 (30, 34). These results raised the possibility that Lyn/c-Raf binding may facilitate a CK2/c-Raf complex that takes place on a KSR1 scaffold, which may include ERK. MEK also precipitated with c-Raf and Lyn in ATRA-treated cells but the amount of interaction was unaffected by the inhibitors (data not shown).
Since CK2 is an S/T kinase and we observed a significant increase in c-Raf phosphorylation at S259, we precipitated c-RafpS259 and probed for Lyn, CK2, and KSR1 (Figure 5c). Co-treatment with either inhibitor plus ATRA increased interaction between c-RafpS259 and Lyn. We were also able to detect CK2 and KSR1 interaction with pRaf259, consistent with these proteins existing in a KSR1-scaffolded complex. Total protein expression of CK2 and KSR1 did not change after inhibitor co-treatment (Figure 5c).
KSR1/ERK interactions can control MAPK signaling specificity and duration, as well as cell proliferation (30, 35, 36). We found that the inhibitors caused a notable enhancement in ERK interaction with c-Raf in ATRA-treated cells (Figure 5d). We also observed KSR1/ERK interaction. Although reciprocal experiments with immunoprecipitated c-Raf did not show such a pronounced increase in ERK interaction, this could be attributed to differences in protein abundance and different proportions of c-Raf or ERK protein that are heterodimerized with each other.
ERK participates in Raf feedback phosphorylation of serine residues in the C-terminal domain (CTD), which include S289, S296, and S301 (37, 38). Therefore we investigated CTD phosphorylation status. We found that PP2 or dasatinib alone increased p-CTD, which was enhanced by ATRA and further increased by co-treatment (Figure 5e).
Immunoprecipitation experiments with NB4 cells were consistent with these results (Figure 5f). c-Raf and c-RafpS259 showed increased interaction with Lyn after ATRA/inhibitor co-treatment within the context of a potential KSR1 scaffold. Co-treatment also increased interaction between ERK and c-Raf. Finally, p-CTD was increased by ATRA combined with PP2 and to a lesser extent dasatinib, although these increases were not as striking compared to HL-60 cells. Together, these results suggest that the inhibitor-induced increase in c-Raf and Lyn expression facilitates an increase in their interaction, which is accompanied by CK2 and KSR1 binding. This may be consistent with CK2 kinase activity toward c-Raf that results in c-RafpS259 phosphorylation, and the ability of KSR1 to act as a scaffold. PP2 and dasatinib enhanced ERK association with c-Raf which is consistent with ERK feedback phosphorylation within the CTD, also in the context of the KSR1 scaffold.
Lyn knockdown decreases c-RafpS259 and CTD phosphorylation
To evaluate whether downregulating Lyn expression would interfere with ATRA-induced c-Raf phosphorylation, we created a stably transfected cell line expressing shRNA targeted against Lyn (shLyn). Lyn expression in untreated shLyn cells was similar to wild-type HL-60 cells, but ATRA could no longer upregulate Lyn (Figure 6a). After ATRA treatment, transfectants were still capable of c-Raf upregulation but showed decreased phosphorylation of c-RafS259 and CTD serine residues (Figure 6b). Therefore, blocking ATRA-induced Lyn upregulation interfered with c-RafpS259 and p-CTD that is characteristic of HL-60 myeloid differentiation. These results suggest that the ATRA-induced increases in Lyn expression modulates MAPK signaling through c-Raf.
Figure 6.
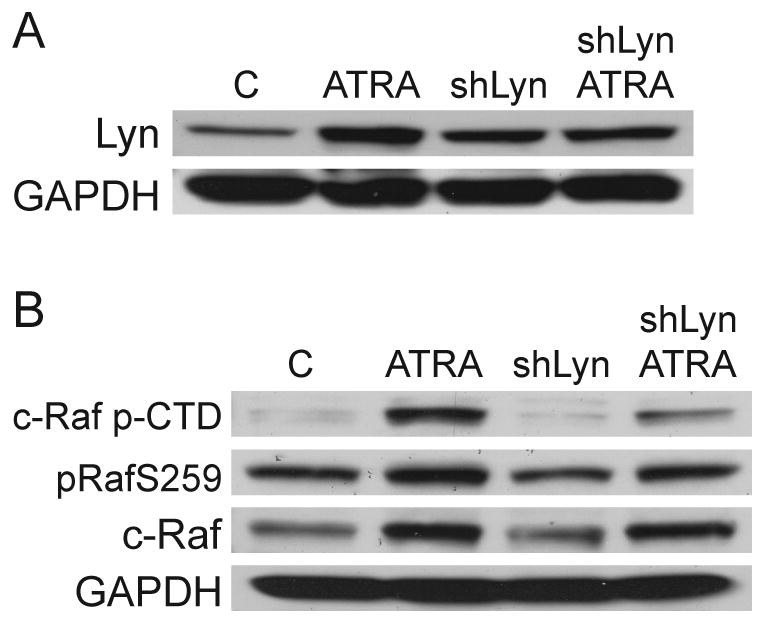
A: HL-60 cells and shLyn stable transfectants were incubated with ATRA for 48 hours followed by Western blotting for total Lyn expression. B: Western blotting for c-Raf CTD serine phosphorylation, c-RafpS259, and total c-Raf after 48 hours of culture with or without ATRA.
We also evaluated if Lyn knockdown affected two additional differentiation markers: G1/0 arrest or CD11b expression. There were no significant differences between parental HL-60 cells and shLyn transfectants, which is likely a result of achieving only partial Lyn knockdown. Partial Lyn suppression does not seem to affect features of myeloid differentiation that may not be directly or solely related to Lyn in the knockdowns. In sum, shRNA targeting Lyn affected c-Raf phosphorylation, which is a putative signaling molecule that regulates ATRA-induced myeloid differentiation. The effects we did observe in the transfectants that were specific to c-Raf are likely most evident because Lyn has a specific role in increased phosphorylation of S259 and CTD residues.
Discussion
ATRA has been successfully used to treat APL for many years. However, patients can develop resistance to treatment, and those presenting t(15,17) negative AML have not been responsive to ATRA therapy alone. Therefore alternative or combination therapies could improve prognosis and survival.
We found that co-treating t(15,17) negative HL-60 or t(15,17) positive NB4 cells with ATRA plus either dasatinib or PP2 (SFK inhibitors) caused significant G1/0 DNA enrichment within 48 hours compared to ATRA alone. The inhibitor-induced cell cycle arrest led to an investigation of differentiation marker effects. Both dasatinib and PP2 enhanced the ATRA-induced upregulation of the α-integrin receptor CD11b and NADPH oxidase protein p47phox. PP2 by itself also appeared able to induce some cell differentiation. These results show that SFK inhibitors increase cell cycle arrest and differentiation markers in ATRA-treated cells that are either t(15,17) positive or negative, suggesting that combination therapy may improve ATRA effectiveness in different types of leukemia.
We then investigated the effects of the inhibitors on Lyn and Fgr, which are upregulated and tyrosine phosphorylated after ATRA treatment (16, 23) and therefore may regulate differentiation. ATRA increased Lyn and Fgr expression in HL-60 cells, and co-treatment with either inhibitor plus ATRA further enhanced expression. NB4 cells also showed Lyn upregulation. Fgr active site phosphorylation was not detectable in any samples, suggesting that the previously reported increase in phosphorylation after ATRA (16) may be specific to the inhibitory C terminal region. Since Fgr activity seemed irrelevant to induced differentiation, we turned our attention toward Lyn.
Dasatinib inhibited Lyn phosphorylation alone and after ATRA treatment in HL-60 and NB4 cells, but while PP2 alone was inhibitory ATRA treatment rescued Lyn activity in co-treated cells. It is noteworthy that Lyn is the dominant active SFK in HL-60 and NB4 myeloid leukemia cells (5), yet the failure of PP2 to inhibit Lyn after ATRA still coincided with accelerated G1/0 arrest and phenotypic conversion. The mechanism by which ATRA protects Lyn from inhibition remains unknown. Together, these results suggest that SFK kinase activity is not necessary for differentiation. One could speculate that SKFs such as Lyn provide scaffolding functions similar to ERK, which can act as a scaffold independent of its kinase activity (39).
ATRA induction is characterized by MAPK activation, and inhibitors of MEK and c-Raf block differentiation (21, 22). Neither PP2 nor dasatinib affected ATRA-induced ERK or MEK phosphorylation. However both inhibitors further enhanced ATRA-upregulated c-Raf expression in both cell lines. The increase in c-Raf is significant, since expression of activated c-Raf drives ATRA-induced differentiation (26). Dasatinib and PP2 also enhanced ATRA-induced c-Raf phosphorylation at S621, which is associated with differentiation (25). Most strikingly, co-treatment with ATRA alone and ATRA plus either inhibitor increased c-RafpS259 phosphorylation in NB4 and HL-60 cells, implicating a previously unreported role for c-RafpS259 in ATRA-induced differentiation.
Dual phosphorylation of c-Raf S621 and S259 is characteristic of quiescent cells and associated with 14-3-3 binding which can inactivate Raf (38, 40, 41). Mutated c-RafpS259A has higher basal activity than wild-type (38), which supports the argument that Raf S259 phosphorylation may attenuate mitogenic signaling. Although we did not observe any significant interruptions in ERK and MEK activation, MAPK signaling depends on the finely-tuned orchestration of interactions with scaffolds, regulator molecules, and positive and negative feedback loops that include direct regulators such as MEK and ERK. This motivated interest in whether there was interaction between c-RafpS259, SFKs, and other MAPK signaling proteins.
KSR1 is a scaffolding molecule that modulates interactions between Raf, MEK, and ERK, and fine-tunes the specificity of MAPK signaling (30). It also interacts with casein kinase II (CK2), an S/T kinase known to complex with and be phosphorylated by Lyn and Fgr (32). CK2 phosphorylates KSR1 and is part of the scaffolding complex that regulates MAPK signaling. Specifically, c-Raf Y341 is phosphorylated by SFKs; a modification that is reported to be a prerequisite for CK2 c-Raf S338 phosphorylation and is dependent on KSR1 scaffolding (28). Our results showed that Lyn was able to complex with CK2 and KSR1, providing a link to MAPK signaling proteins. Consistent with this, ATRA plus inhibitor treatment increased interaction between c-Raf and Lyn in HL-60 and NB4 cells. c-Raf also co-immunoprecipitated CK2 and KSR1 in HL-60 cells, suggesting a Lyn-containing CK2/MAPK complex scaffolded by KSR1. A potential KSR1 scaffolding function was also detected in NB4 cells. c-RafpS259 also showed increased interaction with Lyn, CK2, and KSR1. One could speculate that c-Raf phosphorylation at S259 is a result of CK2 kinase activity facilitated by Lyn interaction. Alternatively, c-Raf/Lyn binding could localize CK2 to one of its targets, many of which include key cell cycle regulators [reviewed in (42)].
Consistent with a MAPK feedback mechanism, we found that after PP2 or dasatinib/ATRA co-treatment ERK showed more interaction with c-Raf, and also bound KSR1. This coincided with upregulated serine phosphorylation at the C terminus of c-Raf. ERK can directly mediate feedback phosphorylation of c-Raf on S287, S296, and S301, which controls Raf activation and modulates signaling (37, 38). Thus co-treatment could be attenuating proliferative MAPK signaling through a KSR1 scaffolding complex containing a Lyn/CK2/c-Raf/ERK module. Knockdown of Lyn by shRNA decreased ATRA-induced c-RafpS259 and CTD phosphorylation, suggesting that Lyn regulates c-Raf post-translational modifications that are important for differentiation. Therefore it is possible that alterations in c-Raf phosphorylation status and interactions facilitated by ATRA and Lyn are changing the character of the MAPK signaling cascade to regulate induced growth arrest and differentiation. Lyn-regulated MAPK orchestration though c-Raf modifications, specifically phosphorylation of S259 and the CTD, may reflect differential involvement of feedback loops with ERK that significantly slow proliferation and expedite cell cycle arrest and differentiation. For example, MAPK stimulation by different growth factors creates positive or negative feedback loops that result in differentiation or proliferation respectively by fine-tuning magnitude and longevity (43, 44).
In conclusion, SFK inhibitors may have the potential to modulate MAPK signaling to enhance the therapeutic efficacy of ATRA in the treatment of a variety of myeloid leukemias. Co-treatment with ATRA plus PP2 or dasatinib may accelerate the phenotypic conversion of AML and APL cells to differentiating myeloid cells by targeting specific molecular markers that are characteristic of ATRA-induced differentiation. The identification of potential molecular targets and mechanisms that may improve the clinical benefit of ATRA encourages further evaluation of ATRA/SFK inhibitor combination therapy.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH) CA033505 (Yen), CA152870 (Yen), 1U54 CA143876 (Shuler), and New York State Stem Cell Science (NYSTEM) (Yen).
Footnotes
The authors acknowledge there are no competing financial interests in relation to the work described. Supplementary data is available at http://www.nature.com/leu/index.html.
References
- 1.Kim MP, Park SI, Kopetz S, Gallick GE. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res. 2009 Jan;335(1):249–259. doi: 10.1007/s00441-008-0682-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Danhauser-Riedl S, Warmuth M, Druker BJ, Emmerich B, Hallek M. Activation of Src kinases p53/56lyn and p59hck by p210bcr/abl in myeloid cells. Cancer Res. 1996 Aug 1;56(15):3589–3596. [PubMed] [Google Scholar]
- 3.Klejman A, Schreiner SJ, Nieborowska-Skorska M, Slupianek A, Wilson M, Smithgall TE, et al. The Src family kinase Hck couples BCR/ABL to STAT5 activation in myeloid leukemia cells. EMBO J. 2002 Nov 1;21(21):5766–5774. doi: 10.1093/emboj/cdf562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dos Santos C, Demur C, Bardet V, Prade-Houdellier N, Payrastre B, Recher C. A critical role for Lyn in acute myeloid leukemia. Blood. 2008 Feb 15;111(4):2269–2279. doi: 10.1182/blood-2007-04-082099. [DOI] [PubMed] [Google Scholar]
- 5.Kropf PL, Wang L, Zang Y, Redner RL, Johnson DE. Dasatinib promotes ATRA-induced differentiation of AML cells. Leukemia. 2010 Mar;24(3):663–665. doi: 10.1038/leu.2009.267. [DOI] [PubMed] [Google Scholar]
- 6.Small D. FLT3 mutations: biology and treatment. Hematology Am Soc Hematol Educ Program. 2006:178–184. doi: 10.1182/asheducation-2006.1.178. [DOI] [PubMed] [Google Scholar]
- 7.Okamoto M, Hayakawa F, Miyata Y, Watamoto K, Emi N, Abe A, et al. Lyn is an important component of the signal transduction pathway specific to FLT3/ITD and can be a therapeutic target in the treatment of AML with FLT3/ITD. Leukemia. 2007 Mar;21(3):403–410. doi: 10.1038/sj.leu.2404547. [DOI] [PubMed] [Google Scholar]
- 8.Robinson LJ, Xue J, Corey SJ. Src family tyrosine kinases are activated by Flt3 and are involved in the proliferative effects of leukemia-associated Flt3 mutations. Exp Hematol. 2005 Apr;33(4):469–479. doi: 10.1016/j.exphem.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Wu J, Meng F, Lu H, Kong L, Bornmann W, Peng Z, et al. Lyn regulates BCR-ABL and Gab2 tyrosine phosphorylation and c-Cbl protein stability in imatinib-resistant chronic myelogenous leukemia cells. Blood. 2008 Apr 1;111(7):3821–3829. doi: 10.1182/blood-2007-08-109330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guerrouahen BS, Futami M, Vaklavas C, Kanerva J, Whichard ZL, Nwawka K, et al. Dasatinib inhibits the growth of molecularly heterogeneous myeloid leukemias. Clin Cancer Res. Feb 15;16(4):1149–1158. doi: 10.1158/1078-0432.CCR-09-2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinberg M. Dasatinib: a tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia and philadelphia chromosome-positive acute lymphoblastic leukemia. Clin Ther. 2007 Nov;29(11):2289–2308. doi: 10.1016/j.clinthera.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Papageorgiou SG, Pappa V, Economopoulou C, Tsirigotis P, Konsioti F, Ionnidou ED, et al. Dasatinib induces long-term remission in imatinib-resistant Philadelphia chromosome-positive acute megakaryoblastic leukemia but fails to prevent development of central nervous system progression. Leuk Res. 2010 Sep;34(9):e254–256. doi: 10.1016/j.leukres.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 13.Stein B, Smith BD. Treatment options for patients with chronic myeloid leukemia who are resistant to or unable to tolerate imatinib. Clin Ther. 2010 May;32(5):804–820. doi: 10.1016/j.clinthera.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klamova H, Faber E, Zackova D, Markova M, Voglova J, Cmunt E, et al. Dasatinib in imatinib-resistant or -intolerant CML patients: data from the clinical practice of 6 hematological centers in the Czech Republic. Neoplasma. 2010;57(4):355–359. [PubMed] [Google Scholar]
- 15.Miranda MB, Redner RL, Johnson DE. Inhibition of Src family kinases enhances retinoic acid induced gene expression and myeloid differentiation. Mol Cancer Ther. 2007 Dec;6(12 Pt 1):3081–3090. doi: 10.1158/1535-7163.MCT-07-0514. [DOI] [PubMed] [Google Scholar]
- 16.Katagiri K, Yokoyama KK, Yamamoto T, Omura S, Irie S, Katagiri T. Lyn and Fgr protein-tyrosine kinases prevent apoptosis during retinoic acid-induced granulocytic differentiation of HL-60 cells. J Biol Chem. 1996 May 10;271(19):11557–11562. doi: 10.1074/jbc.271.19.11557. [DOI] [PubMed] [Google Scholar]
- 17.Notario V, Gutkind JS, Imaizumi M, Katamine S, Robbins KC. Expression of the fgr protooncogene product as a function of myelomonocytic cell maturation. J Cell Biol. 1989 Dec;109(6 Pt 1):3129–3136. doi: 10.1083/jcb.109.6.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee M, Kim JY, Anderson WB. Src tyrosine kinase inhibitor PP2 markedly enhances Ras-independent activation of Raf-1 protein kinase by phorbol myristate acetate and H2O2. J Biol Chem. 2004 Nov 19;279(47):48692–48701. doi: 10.1074/jbc.M403132200. [DOI] [PubMed] [Google Scholar]
- 19.Konig H, Copland M, Chu S, Jove R, Holyoake TL, Bhatia R. Effects of dasatinib on SRC kinase activity and downstream intracellular signaling in primitive chronic myelogenous leukemia hematopoietic cells. Cancer Res. 2008 Dec 1;68(23):9624–9633. doi: 10.1158/0008-5472.CAN-08-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen M, Yen A. c-Cbl interacts with CD38 and promotes retinoic acid-induced differentiation and G0 arrest of human myeloblastic leukemia cells. Cancer Res. 2008 Nov 1;68(21):8761–8769. doi: 10.1158/0008-5472.CAN-08-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yen A, Roberson MS, Varvayanis S, Lee AT. Retinoic acid induced mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase-dependent MAP kinase activation needed to elicit HL-60 cell differentiation and growth arrest. Cancer Res. 1998 Jul 15;58(14):3163–3172. [PubMed] [Google Scholar]
- 22.Wang J, Yen A. A MAPK-positive feedback mechanism for BLR1 signaling propels retinoic acid-triggered differentiation and cell cycle arrest. The Journal of biological chemistry. 2008 Feb 15;283(7):4375–4386. doi: 10.1074/jbc.M708471200. [DOI] [PubMed] [Google Scholar]
- 23.Katagiri K, Katagiri T, Koyama Y, Morikawa M, Yamamoto T, Yoshida T. Expression of src family genes during monocytic differentiation of HL-60 cells. J Immunol. 1991 Jan 15;146(2):701–707. [PubMed] [Google Scholar]
- 24.Congleton J, Jiang H, Malavasi F, Lin H, Yen A. ATRA-induced HL-60 myeloid leukemia cell differentiation depends on the CD38 cytosolic tail needed for membrane localization, but CD38 enzymatic activity is unnecessary. Experimental cell research. 2011 Apr 15;317(7):910–919. doi: 10.1016/j.yexcr.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith J, Bunaciu RP, Reiterer G, Coder D, George T, Asaly M, et al. Retinoic acid induces nuclear accumulation of Raf1 during differentiation of HL-60 cells. Exp Cell Res. 2009 Aug 1;315(13):2241–2248. doi: 10.1016/j.yexcr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yen A, Williams M, Platko JD, Der C, Hisaka M. Expression of activated RAF accelerates cell differentiation and RB protein down-regulation but not hypophosphorylation. Eur J Cell Biol. 1994 Oct;65(1):103–113. [PubMed] [Google Scholar]
- 27.Zafrullah M, Yin X, Haimovitz-Friedman A, Fuks Z, Kolesnick R. Kinase suppressor of Ras transphosphorylates c-Raf-1. Biochem Biophys Res Commun. 2009 Dec 18;390(3):434–440. doi: 10.1016/j.bbrc.2009.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ritt DA, Zhou M, Conrads TP, Veenstra TD, Copeland TD, Morrison DK. CK2 Is a component of the KSR1 scaffold complex that contributes to Raf kinase activation. Curr Biol. 2007 Jan 23;17(2):179–184. doi: 10.1016/j.cub.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 29.Hagemann C, Kalmes A, Wixler V, Wixler L, Schuster T, Rapp UR. The regulatory subunit of protein kinase CK2 is a specific A-Raf activator. FEBS Lett. 1997 Feb 17;403(2):200–202. doi: 10.1016/s0014-5793(97)00011-2. [DOI] [PubMed] [Google Scholar]
- 30.McKay MM, Ritt DA, Morrison DK. Signaling dynamics of the KSR1 scaffold complex. Proc Natl Acad Sci U S A. 2009 Jul 7;106(27):11022–11027. doi: 10.1073/pnas.0901590106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donella-Deana A, Cesaro L, Sarno S, Brunati AM, Ruzzene M, Pinna LA. Autocatalytic tyrosine-phosphorylation of protein kinase CK2 alpha and alpha’ subunits: implication of Tyr182. Biochem J. 2001 Jul 15;357(Pt 2):563–567. doi: 10.1042/0264-6021:3570563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donella-Deana A, Cesaro L, Sarno S, Ruzzene M, Brunati AM, Marin O, et al. Tyrosine phosphorylation of protein kinase CK2 by Src-related tyrosine kinases correlates with increased catalytic activity. Biochem J. 2003 Jun 15;372(Pt 3):841–849. doi: 10.1042/BJ20021905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005 Nov;6(11):827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 34.Filbert EL, Nguyen A, Markiewicz MA, Fowlkes BJ, Huang YH, Shaw AS. Kinase suppressor of Ras 1 is required for full ERK activation in thymocytes but not for thymocyte selection. Eur J Immunol. 2010 Nov;40(11):3226–3234. doi: 10.1002/eji.201040349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Razidlo GL, Kortum RL, Haferbier JL, Lewis RE. Phosphorylation regulates KSR1 stability, ERK activation, and cell proliferation. J Biol Chem. 2004 Nov 12;279(46):47808–47814. doi: 10.1074/jbc.M406395200. [DOI] [PubMed] [Google Scholar]
- 36.Kortum RL, Lewis RE. The molecular scaffold KSR1 regulates the proliferative and oncogenic potential of cells. Mol Cell Biol. 2004 May;24(10):4407–4416. doi: 10.1128/MCB.24.10.4407-4416.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brummer T, Naegele H, Reth M, Misawa Y. Identification of novel ERK-mediated feedback phosphorylation sites at the C-terminus of B-Raf. Oncogene. 2003 Dec 4;22(55):8823–8834. doi: 10.1038/sj.onc.1207185. [DOI] [PubMed] [Google Scholar]
- 38.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005 Jan 21;17(2):215–224. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 39.Hong SK, Yoon S, Moelling C, Arthan D, Park JI. Noncatalytic function of ERK1/2 can promote Raf/MEK/ERK-mediated growth arrest signaling. J Biol Chem. 2009 Nov 27;284(48):33006–33018. doi: 10.1074/jbc.M109.012591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morrison DK, Heidecker G, Rapp UR, Copeland TD. Identification of the major phosphorylation sites of the Raf-1 kinase. J Biol Chem. 1993 Aug 15;268(23):17309–17316. [PubMed] [Google Scholar]
- 41.Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996 Mar 22;84(6):889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 42.St-Denis NA, Litchfield DW. Protein kinase CK2 in health and disease: From birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci. 2009 Jun;66(11–12):1817–1829. doi: 10.1007/s00018-009-9150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santos SD, Verveer PJ, Bastiaens PI. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nature cell biology. 2007 Mar;9(3):324–330. doi: 10.1038/ncb1543. [DOI] [PubMed] [Google Scholar]
- 44.Brightman FA, Fell DA. Differential feedback regulation of the MAPK cascade underlies the quantitative differences in EGF and NGF signalling in PC12 cells. FEBS letters. 2000 Oct 6;482(3):169–174. doi: 10.1016/s0014-5793(00)02037-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.