

Abstract
Novel pharmacological strategies are urgently needed to prevent or reduce radiation-induced tissue injury. Microvascular injury is a prominent feature of both early and delayed radiation injury. Radiation-induced endothelial dysfunction is believed to play a key role in the pathogenesis of post-irradiation tissue injury. Hence, strategies that could prevent or improve endothelial malfunction are expected to ameliorate the severity of radiation injury. This review focuses on the therapeutic potential of the nitric oxide synthase (NOS) cofactor 5,6,7,8-tetrahydrobiopterin (BH4) as an agent to reduce radiation toxicity. BH4 is an essential cofactor for all NOS enzymes and a critical determinant of NOS function. Inadequate availability of BH4 leads to uncoupling of the NOS enzyme. In an uncoupled state, NOS produces the highly oxidative radicals superoxide and peroxynitrite at the cost of NO. Under conditions of oxidative stress, such as after radiation exposure, BH4 availability might be reduced due to the rapid oxidation of BH4 to 7,8-dihydrobiopterin (7,8-BH2). As a result, free radical–induced BH4 insufficiency may increase the oxidative burden and hamper NO-dependent endothelial function. Given the growing evidence that BH4 depletion and subsequent endothelial NOS uncoupling play a major role in the pathogenesis of endothelial dysfunction in various diseases, there is substantial reason to believe that improving post-irradiation BH4 availability, by either supplementation with it or modulation of its metabolism, might be a novel strategy to reduce radiation-induced endothelial dysfunction and subsequent tissue injury.
Keywords: Radiation injuries, radioprotection, endothelial dysfunction, nitric oxide synthase, tetrahydrobiopterin, HMG-CoA reductase, γ-tocotrienol
INTRODUCTION
Novel pharmacological therapies are urgently needed to prevent or reduce radiation-induced injury and mortality. Radiation-induced tissue injury may occur after radiation exposure in both clinical and non-clinical settings. In patients treated with radiotherapy, normal tissue radiation toxicity remains a crucial dose-limiting factor and the most important cause of acute and chronic treatment-related side effects. In non-clinical settings, for example, in emergency situations such as a radiological/nuclear accident or attack, radiation-induced tissue injury may cause substantial mortality.
To date, pharmacological strategies to prevent or reduce radiation injury are scarce, if existent at all. Many different compounds, such as various free radical scavengers, antioxi-dants, cytokines, thiols, and steroids, have been tested as radioprotective agents [1, 2]. Only the thiol-containing compound amifostine has been proven to be an effective and applicable radioprotectant in humans. Unfortunately, the use of amifostine is hampered by a narrow therapeutic window and severe side effects, including grade 3 nausea, vomiting, and hypotension [3]. For this reason, amifostine has only been approved for clinical use in certain cancer patients undergoing radiotherapy. Because of its toxicity profile, amifostine should not be used as a radioprophylactic agent in non-clinical situations.
In order to make significant progress in the development of novel pharmacological agents that can ameliorate radiation injury, it is essential to increase our understanding of the molecular and cellular basis of radiation toxicity and to identify novel therapeutic targets.
Microvascular injury is a prominent feature of both acute and chronic radiation injury. An increasing body of evidence shows that microvascular endothelial dysfunction may play a crucial role in the development of radiation toxicity in various organ systems [4–12]. Hence, strategies that could prevent or improve post-irradiation endothelial malfunction are expected to ameliorate the severity of radiation injury.
Radiation-exposure induces different functional and morphological changes to the endothelium, including apoptosis, increased endothelial permeability, and loss of thrombo-resistance. Changes in endothelial nitric oxide synthase(eNOS) function are believed to play an important role inradiation-induced endothelial dysfunction. Radiation exposure impairs the expression of eNOS as well as endothelial NO production.
The focus of this review is the possible therapeutic potential of the NOS cofactor 5,6,7,8-tetrahydrobiopterin (BH4) to prevent and treat radiation-induced injury. The highly redox-sensitive BH4 is a critical determinant of eNOS function. To be functional, BH4 should be in its fully reduced form. Oxidation of BH4 to 7,8-dihydropbiopterin (7,8-BH2) reduces its activity as an eNOS cofactor. Decreased availability of BH4 and consequential changes in NOS enzyme function have been shown to play an important role in the pathogenesis of various pathological conditions characterized by endothelial dysfunction and increased oxidative stress, such as diabetes, hypertension, hypercholesterolemia, and cardiac ischemia [13–19]. Radiation-induced oxidative stress may decrease endothelial BH4 levels because of the rapid oxidation of BH4 to 7,8-BH2, and thereby induce eNOS dysfunction and subsequent endothelial malfunction.
EFFECTS OF RADIATION EXPOSURE ON NO-DEPENDENT ENDOTHELIAL FUNCTION
Endothelial NO is believed to be a key regulator of endothelial function, and impaired NO production by eNOS is considered to be a main cause of endothelial dysfunction [20 –23].
Many studies have been conducted to determine the effects of radiation exposure on eNOS-dependent vascular function. One of the most prominent functions of endothelial NO is the regulation of vascular tonus. NO produced by eNOS plays a crucial role in the induction of vascular relaxation. Hence, end othelium-dependent vascular relaxation responses are considered to be a reliable measure of post-irradiation eNOS function. Endothelium-dependent relaxation is determined by measuring vascular relaxation after incubation with substances that induce vasodilatation by stimulating endothelial NO production, such as acetylcholine.
Using both animal models and tissues collected from patients who had received radiotherapy, several groups of investigators have shown impaired endothelium-dependent vascular relaxation responses after radiation exposure [24–30].
The animal studies investigated the effects of various radiation doses (e.g., low and high doses) and radiation schedules (e.g., single and fractionated doses) on the occurrence of endothelial dysfunction. After high-dose irradiation with a single dose of 10 to 45 Gy, impaired relaxation of the rabbit entral ear artery was measured at different time points after radiation exposure [24, 25, 27]. The effect was dose-dependent and most pronounced after irradiation with 45 Gy. After low-dose irradiation, the effects were less clear. Using a rat total-body irradiation model and a total radiation dose of 1 Gy, Suvorava et al. [31] measured impaired aortic relaxation only after chronic exposure, i.e., irradiation with a low dose rate and an overall exposure time of 41 days. After acute exposure to 1 Gy, they found no effect on endothelium-dependent vasodilatation.
Hatoum et al. [28] investigated the effects of fractionated radiation on NO-dependent endothelial function. They assessed intestinal submucosal microvascular function in rats irradiated with nine fractions of 2.5 Gy to a total dose of 22.5 Gy. Endothelium-dependent relaxation was impaired from the third fraction onward. Their observations that microvascular reactive oxygen species (ROS) production was increased from dose 3 onward and that superoxide dismutase (SOD) mimetics partially reversed the radiation-induced decrease in endothelium-dependent relaxation and gut microxvascular NO levels suggest that radiation-induced ROS production contributes to impaired eNOS function and consequent endothelial malfunction after radiation exposure.
Sugihara et al. [26] examined radiation-induced endothelium-dependent dysfunction in human subjects treated with radiotherapy. Using tissue specimens of patients who had received either no radiotherapy or preoperative radiotherapy at doses ranging from 40 Gy to 65 Gy in 2.5-Gy fractions, they found impaired NO-dependent endothelial relaxation in human subjects treated with radiation. Remarkably, vascular relaxation was impaired without significant morphological damage of the endothelium.
The exact mechanism by which radiation exposure impairs eNOS activity and endothelial cell function has not yet been elucidated. Post-irradiation impaired eNOS function might be related to diminished eNOS expression. Several authors have observed decreases in eNOS mRNA and protein levels after radiation exposure [25, 27]. However, other factors might also be important. Radiation-induced free radical production may decrease the availability of the eNOS cofactor BH4 because of the rapid oxidation of BH4 to 7,8-BH2 under conditions of oxidative stress. Diminished BH4 availability may induce eNOS uncoupling and thereby limit NO production and increase O2− production (Fig.1). Hence, radiation-induced eNOS uncoupling due to decreased BH4 availability may also be an important cause of post-irradiation eNOS dysfunction. Preliminary data from our laboratory have confirmed that radiation exposure decreases the availability of BH4 in the early post-irradiation phase.1
Fig. (1).
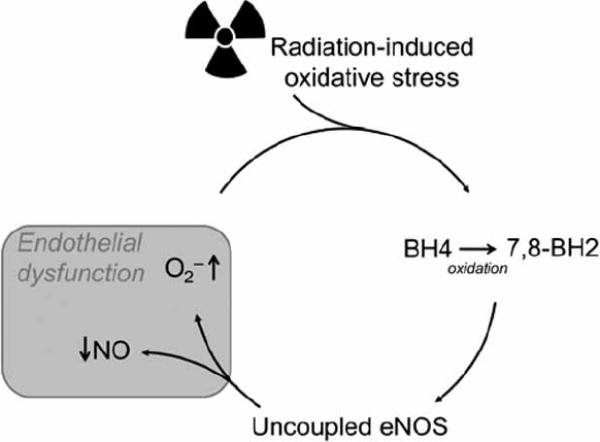
Model of radiation-induced endothelial dysfunction. Radiation-induced oxidative stress may decrease the availability of the eNOS cofactor BH4 due to rapid oxidation of BH4 to 7,8-BH2. Diminished BH4 availability may induce eNOS uncoupling and thereby limit NO production and increase O2− production. Endothelial NOS–dependent O2− may reduce BH4 levels even further.
BH4 AND NOS ENZYME FUNCTION
The NOS cofactor BH4 is required for adequate activity of various enzymes. For a long time, aromatic amino acid hydroxylases (i.e., phenylalanine, tyrosine, tryptophan hydroxylase, and glyceryl-ether monooxygenase) were believed to be the only BH4-dependent enzymes. Soon after the discovery of the NOS enzymes, however, BH4 was identified as a critical cofactor for the NOS enzymes, as well.
Since the recognition of BH4 as a NOS cofactor in 1989 [32], significant progress has been made in understanding its role in the control of NOS activity. BH4 is essential for the catalytic activity of all three NOS isoforms: the constitutively expressed neuronal (nNOS) and endothelial (eNOS) isoforms and the inducible (iNOS) isoform. The role of BH4 in NOS enzymatic function has been the focus of several recent reviews [33–35].
NOS catalyzes the conversion of l-arginine into l-citrulline and NO [36]. The reaction consumes 2 moles of oxygen and 1.5 moles of NAD PH per mole of l-citrulline formed. The NOS dimer comprises two identical monomers. Each monomer consists of a C-terminal reductase domain and an N-terminal oxygenase domain. The C-terminal contains binding sites for flavin mononucleotide and flavin adenine dinucleotide, as well as for NADPH. The oxygenase domain has binding sites for heme, BH4, and l-arginine. NO biosynthesis occurs in two discrete steps. First, l-arginine is hydroxylated to N-hydroxy-l-arginine (NHA). During this conversion step, one molecule of oxygen and two electrons are consumed. Second, NHA is further oxidized to l-citrulline and NO. This step requires one more molecule of oxygen and one more electron. The conversion of both l-arginine to NHA and NHA to l-citrulline requires the presence of adequate amounts of BH4.
BH4 was shown to regulate NOS function on multiple levels. BH4 increases enzymatic NOS activity by shifting the heme iron from a low to a high spin state [37, 38]. It substantially increases the affinity of NOS for its substrate, l-arginine [39]. Furthermore, BH4 stabilizes the NOS dimeric structure, which is essential for NOS synthetic activity [40]. It has also been proven to play a crucial role in electron transfer by donating a single electron to the heme group [41]. Lastly, BH4 may modulate NOS output by directly scavenging NOS-derived reactive nitrogen species and ROS [42, 43].
Inadequate availability of BH4 can lead to a form of NOS dysfunction referred to as “NOS uncoupling.” In an uncoupled reaction, NADPH oxidation and oxygen reduction are uncoupled from l-arginine hydroxylation and NO formation, resulting in the production of superoxide anions (Fig.2).
Fig. (2).
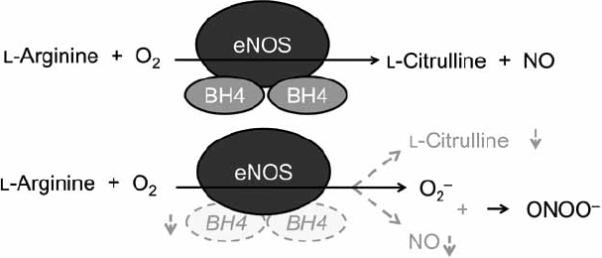
Schematic representation of NOS function under coupled and uncoupled conditions. Under coupled conditions, NOS enzymes mainly produce l-citrulline and NO. Suboptimal concentrations of BH4 may cause NOS uncoupling. Under such conditions, O2− is produced at the cost of NO.
BH4 BIOSYNTHESIS
The unconjugated pterin analog BH4 is synthesized by two different pathways: a de novo pathway and a salvage pathway [44]. The first pathway produces BH4 from the precursor guanidine triphosphate (GTP); the second pathway regenerates BH4 from its oxidized forms (Fig. 3).
Fig. (3).
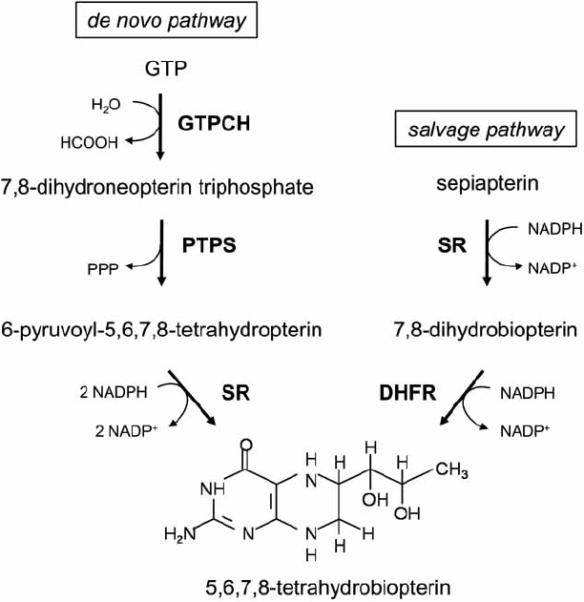
BH4 synthesis. BH4 is synthesized from GTP by a de novo pathway. The conversion of GTP to 7,8-dihydroneopterin triphosphate by GTPCH is the rate-limiting step of this pathway. The salvage pathway generates BH4 from its oxidized forms. The salvage pathway is also necessary to convert exogenous sepiapterin into BH4. GTPCH: GTP cyclohydrolase I; PTPS: 6-pyruvoyl-tetrahydropterin synthase; SR: sepiapterin reductase; DHFR: dihydrofolate reductase.
De Novo Pathway of BH4 Synthesis
In the first and rate-limiting step of de novo synthesis, GTP cyclohydrolase I (GTPCH) catalyzes the formation of dihydroneopterin triphosphate from GTP in a zinc-, magnesium-, and NADPH-dependent way. Subsequently, dihydroneopterin triphosphate is converted to 6-pyruvoyl-tetrahydrobiopterin by the action of 6-pyruvoyl-tetrahydrobiopterin synthase. In the last step of the de novo pathway, sepiapterin reductase catalyzes the conversion of 6-pyruvoyltetrahydrobiopterin into BH4. The rate of BH4 synthesis by the de novo pathway depends of the activity of its rate-limiting enzyme GTPCH. GTPCH activity can be regulated at multiple levels. Transcriptional and post-translational changes, as well as protein-protein interactions, are known to affect the activity of GTPCH.
The GTPCH gene contains a cAMP response element that plays a predominant role in the regulation of GTPCH transcription. In vitro studies using hepatocytes, inflammatory cells, and endothelial cells showed increased GTPCH expression and BH4 synthesis after stimulation with different cytokines, insulin, hydrogen peroxide, and lipopolysaccharide (LPS) [44]. LPS treatment has also been shown to increase GTPCH mRNA levels in rat tissue [45]. More research is needed to determine which factors are most important in the regulation of GTPCH transcription in vivo under physiological conditions.
Post-translational regulation of GTPCH activity appears to occur by protein phosphorylation. Phosphorylation of GTPCH by protein kinase CK2 (casein kinase II) increases GTPCH activity without increasing GTPCH protein levels [46, 47]. Widder et al. [46] showed that laminar shear stress increased endothelial BH4 levels by stimulating phosphorylation of GTPCH serine 81.
Protein–protein interaction is another important regulatory mechanism controlling the activity of GTPCH. Interaction between GTPCH and the GTPCH regulatory feedback protein (GFRP) can either stimulate or inhibit GTPCH activity [48–50]. The binding of GFRP to GTPCH enables end-product feedback inhibition by BH4. Conversely, phenylalanine can stimulate GTPCH enzymatic activity via GFRP. The latter process may be most important in hepatocytes.
Salvage Pathway of BH4 Synthesis
The salvage pathway generates BH4 from its oxidized forms via the enzymes sepiapterin reductase and dihydrofolate reductase. Dihydrofolate reductase, which is also involved in folate metabolism, regenerates BH4 from inactive 7,8-BH2. The importance of the salvage pathway in regulating endothelial BH4 availability both in vitro and in vivo has been demonstrated by Cai's group [51, 52]. They showed that decreased expression of dihydrofolate reductase resulted in decreased endothelial BH4 levels. In contrast, stimulating dihydrofolate reductase improved BH4 availability and NO production, thus ameliorating the production of superoxide.
The salvage pathway is essential for the conversion of the BH4 precursor sepiapterin to BH4. Although it is not considered a physiological metabolite in mammals, sepiapterin can be used as an exogenous source to increase BH4 levels in mammals. Sepiapterin is reduced by sepiapterin reductase to 7,8-BH2, which is then converted to BH4 by dihydrofolate reductase.
POSSIBLE STRATEGIES TO INCREASE POST-IRRADIATION BH4 AVAILABILITY
In view of the possible importance of BH4-dependent eNOS uncoupling in the development of post-irradiation endothelial dysfunction, restoring BH4 supplies or preventing BH4 deficiency could be a novel and interesting approach to ameliorate radiation injury. BH4 levels can be raised either directly by administering BH4 or its precursor sepiapterin or indirectly by influencing the key regulators of BH4 metabolism.
To date, several agents are known to affect the regulatory proteins of BH4 metabolism or to improve BH4 stability. Remarkably, some of these agents have already been reported to have radioprotective properties [53, 54].
BH4/Sepiapterin
The most obvious way to increase BH4 availability is supplementation of BH4 itself. BH4 supplementation has been shown to ameliorate eNOS uncoupling and restore endothelial function in animal models of hypertension, diabetes, hypercholesterolemia, and organ transplantation [16, 55–57]. Studies have shown that administration of BH4 has beneficial effects in patients with hypercholesterolemia, diabetes, and hypertension, as well as after ischemia–reperfusion [17, 18, 58–61].
When used in human subjects, BH4 is generally administered orally. The small intestines are the principal site of absorption after oral administration of BH4. Peak plasma levels are reached 1–4 hours after oral administration, and the elimination half-life of BH4 is in the range of 3–5 hours [62].
It is believed that most of exogenous BH4 does not reach intracellular compartments as BH4 itself [63,64]. Exogenous BH4 is first oxidized to 7,8-BH2, and it is this oxidized form that is taken up by tissues. After reaching the intracellular compartment, 7,8-BH2 is then reduced back to BH4. The pharmacological efficacy of exogenous BH4 may depend on the activity of the salvage pathway.
A different way to increase BH4 levels by the salvage pathway is administration of the BH4 precursor sepiapterin. Sepiapterin treatment has been shown to reduce endothelial dysfunction ex vivo in human and porcine atherosclerotic coronary arteries [65]. Moreover, it has improved endothelial dysfunction in vivo in animal models of diabetes and cardiac ischemia [14, 66].
It should be noted that several authors have raised concerns about the use of high doses of sepiapterin. At high concentrations, sepiapterin may compete with BH4 for eNOS binding, thereby inducing eNOS uncoupling [67, 68].
To date, no studies have been published investigating the effect BH4 or sepiapterin administration on radiation injury. Preliminary data from our laboratory show that treatment with BH4 may decrease post-irradiation vascular oxidative stress. Further research is needed to determine whether treatment with BH4 or sepiapterin can ameliorate radiation injury.
Statins
3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, the so-called statins, can increase endothelial BH4 levels by stimulation of de novo BH4 synthesis [69, 70]. Using endothelial cell cultures, Hattori et al. [69, 70] showed that statin treatment increased the transcription of GTPCH, the rate-limiting enzyme in de novo BH4 synthesis. Moreover, they found that statin treatment augmented intracellular BH4 levels and NO formation, indicating that statin-induced upregulation of GTPCH prevented the occurrence of eNOS uncoupling by increasing BH4 availability in vitro. Later on, Wenzel et al. [69, 70] demonstrated that the regulation of BH4 levels may also be an important aspect of the mechanism by which statins reduce endothelial dysfunction in vivo. Statin-treated diabetic rats showed increased expression of GTPCH together with increased vascular BH4 levels and decreased vascular oxidative stress.
Statins have been known to confer radioprotection and reduce radiation injury for several years now. Early preclinical studies showed that statins ameliorated the development of lung injury and intestinal radiation toxicity after localized irradiation of the thorax and gut, respectively [53, 54, 71, 72]. Lovastatin was shown to reduce postirradiation pulmonary macrophage and lymphocyte populations, as well as collagen deposition. Both pravastatin and simvastatin have been shown to ameliorate the development of delayed intestinal radiation injury. Delayed radiation enteropathy is characterized by progressive intestinal wall fibrosis and vascular sclerosis. Haydont et al. [54, 72] showed that pravastatin given before and just after irradiation ameliorated post-irradiation fibrosis and delayed radiation-induced enteropathy in rats. Wang et al. [53] reported further evidence for beneficial effects of statins on radiation-induced intestinal injury. They showed that simvastatin prevented the development of late radiation enteropathy after localized, fractionated intestinal radiation in rats. They hypothesized that statins might exert an effect on radiation injury by improving endothelial dysfunction and vascular thromboresistance through upregulation of endothelial thrombomodulin. All statins strongly upregulate thrombomodulin gene expression and protein levels [73]. Radiation exposure induces a decrease in thrombomodulin expression [74], and deficient levels of thrombomodulin are known to cause excessive activation of cellular thrombin receptors by thrombin and insufficient activation of protein C, a plasma protein with anticoagulant, anti-inflammatory, and cytoprotective properties.
Overall, the modulating effects of statins on radiation injury appear to be mediated by their anti-inflammatory, antifibrotic, and vasculoprotective properties. A more recent study by Holler et al. [75] confirmed that statin treatment reduced radiation injury by ameliorating NO-dependent vascular dysfunction. Pravastatin was shown to reduce skin lesions and vascular functional activation after dorsal radiation in mice. Most interestingly, these effects were shown to be eNOS-dependent. Pravastatin did not have an effect in eNOS-deficient mice.
Further research is needed to investigate whether the radioprotective properties of statins indeed depend on improving BH4 availability and preventing eNOS uncoupling.
Ascorbic Acid
Ascorbic acid is known to increase endothelial BH4 levels and NO synthesis. In contrast to the original understanding of its action, ascorbic acid appears, not to stabilize BH4 by preventing its oxidation, but to facilitate the reduction of the intermediate BH3• radical back to BH4 [76, 77]. Mice studies have shown that long-term treatment with ascorbic acid improves vascular endothelial function by protecting BH4 and restorating eNOS enzymatic activity [78].
Several studies have been performed to evaluate the effect of ascorbic acid, given alone or in combination with other antioxidants, on various forms of radiation injury [79–81]. Overall, the results of these studies conflict, and the usefulness of ascorbic acid as a radioprotector or mitigator remains unclear.
Folates
Folic acid and its active metabolite 5-methyltetrahydrofolate (5-MTHF) have been shown to ameliorate endothelial dysfunction in patients suffering from diabetes mellitus, hypercholesterolemia, and hyperhomocysteinemia [82–84]. These effects appear not to derive from folic acid–dependent remethylation of homocysteine to methionine, but rather on improvement of eNOS function. Folic acid and 5-MTHF improve eNOS function in a BH4-dependent way [85]. They have been shown to reverse eNOS uncoupling by (1) direct interaction with eNOS, (2) enhancement of BH4–eNOS binding, (3) chemical stabilization of BH4, and (4) increased regeneration of BH4 from inactive 7,8-BH2. The latter effect appears to derive from folate-dependent upregulation of dihydrofolate reductase [51, 86].
Further research is needed to determine the effects of folate supplementation on radiation-induced injury.
Angiotensin II Type I Receptor Blockers
Angiotensin II type I receptor (AT1R) blockers (ARBs) have been known for more than 10 years to confer radioprotection [87]. Animal experiments have shown that the AT1R antagonist L-158809 can ameliorate radiation-induced injury to the kidney and lungs. L-158809 prevents and/or reduces radiation-induced nephropathy, when started both before or after radiation exposure [88, 89]. In the lung, L-158809 ameliorates radiation-induced pneumonitis and lung fibrosis [90, 91]. More recently, ARBs have been shown to ameliorate whole-brain irradiation–induced cognitive impairment [92].
Little is known about the mechanisms by which ARBs protect against radiation injury, but recent evidence suggests a role for BH4 in this activity [93]. ARBs have been shown to protect against diabetic nephropathy by increasing GTPCH protein levels and the availability of BH4, thereby preventing eNOS uncoupling.
Further research is needed to determine whether ARBs indeed confer radioprotection by increasing BH4 availability.
Gamma-Tocotrienol
The vitamin E analog γ-tocotrienol is a potent protector against radiation injury. In studies in mice, a single dose of 400mg/kg γ-tocotrienol greatly reduced radiation-induced injury and subsequent mortality [94, 95]. Gamma-tocotrienol also reduced vascular and intestinal radiation injury and improved hematopoietic recovery after exposure of mice to total-body irradiation. Gamma-tocotrienol exerts its radio-prophylactic effect not only by virtue of its antioxidant properties but also by its inhibition of HMG-CoA reductase. In contrast to statins, which directly inhibit the activity of HMG-CoA reductase, γ-tocotrienol reduced HMG-CoA reductase activity by enhancing proteasomal degradation of the enzyme [96, 97].
Gamma-tocotrienol prevents radiation-induced vascular peroxynitrite production in a HMG-CoA reductase–dependent manner. As mentioned before, inhibition of HMG-CoA reductase has been shown to increase BH4 availability by stimulating de novo BH4 synthesis. Hence, it seems plausible that γ-tocotrienol reduces post-irradiation vascular oxidative stress by stimulating BH4 production and thereby preventing eNOS uncoupling.
More research is needed to elucidate whether and how γ-tocotrienol affects BH4 metabolism and to determine the exact importance of BH4 in radioprotection by γ-tocotrienol.
CONCLUSIONS
Novel strategies are urgently needed to ameliorate both early and delayed radiation injury.
Decreased availability of the redox-sensitive NOS cofactor BH4 is known to play an important role in the pathogenesis of different conditions characterized by increased oxidative stress. Adequate availability of BH4 is crucial to preventing NOS-dependent O2− production or socalled NOS uncoupling.
Radiation-induced oxidative stress may decrease endothelial BH4 levels and thereby prolong or even amplify post-irradiation oxidative stress. Post-irradiation decreased BH4 availability may play an important role in the pathogenesis of radiation-induced endothelial dysfunction and subsequent tissue injury.
Further research is needed to determine the exact role of decreased BH4 availability and NOS uncoupling in the pathogenesis of radiation injury. Nevertheless, it is reasonable to believe that enhancing the availability of BH4 either by supplementing it or by modulating its metabolism could be a novel approach to reduce radiation-induced oxidative stress and subsequent tissue injury. Further research is also needed to determine the therapeutic potential for reducing radiation injury with BH4, its precursor sepiapterin, and various drugs known to modulate BH4 metabolism, such as statins, folates, ascorbic acid, angiotensin II type I receptor antagonists, and γ-tocotrienol, as well as to determine whether these agents exert their effect by increasing BH4 availability.
ACKNOWLEDGEMENT
We thank the Office of Grants and Scientific Publications at the University of Arkansas for Medical Sciences for editorial assistance in the preparation of the manuscript.
FINANCIAL SUPPORT National Institutes of Health/National Cancer Institute (grant CA83719 to MH-J) and Defense Threat Reduction Agency (grant HDTRA1-07-C-0028 to MH-J and H.10027-07-AR-R to KSK).
ABBREVIATIONS
- ARB
Angiotensin II type I receptor blocker
- AT1R
Angiotensin II type I receptor
- 7,8-BH2
7,8-Dihydropbiopterin
- BH4
5,6,7,8-Tetrahydrobiopterin
- eNOS
Endothelial nitric oxide synthase
- GFRP
GTPCH regulatory feedback protein
- GTP
Guanidine triphosphate
- GTPCH
Guanidine triphosphate cyclohydrolase I
- HMG-
3-Hydroxy-3-methylglutaryl-coenzyme A CoA
- iNOS
Inducible nitric oxide synthase
- LPS
Lipopolysaccharide
- 5-MTHF
5-Methyltetrahydrofolate
- nNOS
Neuronal nitric oxide synthase
- NHA
N-hydroxy-L-arginine
- NO
Nitric oxide
- ROS
Reactive oxygen species
- SOD
Superoxide dismutase
Footnotes
Berbée M, Fu Q, Werner ER, Kumar KS, Hauer-Jensen M. The effect of total body irradiation on the availability of the NOS cofactor tetrahydrobiopterin. Presented at the 55th annual meeting of the Radiation Research Society, Savannah, GA; October 4–7, 2009.
REFERENCES
- [1].Weiss JF, Landauer MR. Radioprotection by antioxidants. Ann NY Acad Sci. 2000;899:44–60. [PubMed] [Google Scholar]
- [2].Hosseinimehr SJ. Trends in the development of radioprotective agents. Drug Discov Today. 2007;12:794–805. doi: 10.1016/j.drudis.2007.07.017. [DOI] [PubMed] [Google Scholar]
- [3].Koukourakis MI, Kyrias G, Kakolyris S, et al. Subcutaneous administration of amifostine during fractionated radiotherapy: a randomized phase II study. J Clin Oncol. 2000;18:2226–33. doi: 10.1200/JCO.2000.18.11.2226. [DOI] [PubMed] [Google Scholar]
- [4].Wang J, Boerma M, Fu Q, Hauer-Jensen M. Significance of endothelial dysfunction in the pathogenesis of early and delayed radiation enteropathy. World J Gastroenterol. 2007;13:3047–55. doi: 10.3748/wjg.v13.i22.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Paris F, Fuks Z, Kang A, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001;293:293–7. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- [6].Maj JG, Paris F, Haimovitz-Friedman A, Venkatraman E, Kolesnick R, Fuks Z. Microvascular function regulates intestinal crypt response to radiation. Cancer Res. 2003;63:4338–41. [PubMed] [Google Scholar]
- [7].Baker DG, Krochak RJ. The response of the microvascular system to radiation: a review. Cancer Invest. 1989;7:287–94. doi: 10.3109/07357908909039849. [DOI] [PubMed] [Google Scholar]
- [8].Hopewell JW, Calvo W, Jaenke R, Reinhold HS, Robbins ME, Whitehouse EM. Microvasculature and radiation damage. Recent Results Cancer Res. 1993;130:1–16. doi: 10.1007/978-3-642-84892-6_1. [DOI] [PubMed] [Google Scholar]
- [9].Jaenke RS, Robbins ME, Bywaters T, Whitehouse E, Rezvani M, Hopewell JW. Capillary endothelium. Target site of renal radiation injury. Lab Invest. 1993;68:396–405. [PubMed] [Google Scholar]
- [10].Lyubimova N, Hopewell JW. Experimental evidence to support the hypothesis that damage to vascular endothelium plays the primary role in the development of late radiation-induced CNS injury. Br J Radiol. 2004;77:488–92. doi: 10.1259/bjr/15169876. [DOI] [PubMed] [Google Scholar]
- [11].Rezvani M, Hopewell JW, Robbins ME. Initiation of non-neoplastic late effects: the role of endothelium and connective tissue. Stem Cells. 1995;13(Suppl 1):248–56. doi: 10.1002/stem.5530130730. [DOI] [PubMed] [Google Scholar]
- [12].Wang J, Zheng H, Ou X, Fink LM, Hauer-Jensen M. Deficiency of microvascular thrombomodulin and up-regulation of protease-activated receptor-1 in irradiated rat intestine: possible link between endothelial dysfunction and chronic radiation fibrosis. Am J Pathol. 2002;160:2063–72. doi: 10.1016/S0002-9440(10)61156-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Alp NJ, Mussa S, Khoo J, et al. Tetrahydrobiopterin-dependent preservation of nitric oxide-mediated endothelial function in diabetes by targeted transgenic GTP-cyclohydrolase I overexpression. J Clin Invest. 2003;112:725–35. doi: 10.1172/JCI17786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pannirselvam M, Simon V, Verma S, Anderson T, Triggle CR. Chronic oral supplementation with sepiapterin prevents endothelial dysfunction and oxidative stress in small mesenteric arteries from diabetic (db/db) mice. Br J Pharmacol. 2003;140:701–6. doi: 10.1038/sj.bjp.0705476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Landmesser U, Dikalov S, Price SR, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–9. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cosentino F, Patton S, d'Uscio LV, et al. Tetrahydrobiopterin alters superoxide and nitric oxide release in prehypertensive rats. J Clin Invest. 1998;101:1530–7. doi: 10.1172/JCI650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cosentino F, Hurlimann D, Delli GC, et al. Chronic treatment with tetrahydrobiopterin reverses endothelial dysfunction and oxidative stress in hypercholesterolaemia. Heart. 2008;94:487–92. doi: 10.1136/hrt.2007.122184. [DOI] [PubMed] [Google Scholar]
- [18].Stroes E, Kastelein J, Cosentino F, et al. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest. 1997;99:41–6. doi: 10.1172/JCI119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dumitrescu C, Biondi R, Xia Y, et al. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc Natl Acad Sci U S A. 2007;104:15081–6. doi: 10.1073/pnas.0702986104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Napoli C, Ignarro LJ. Nitric oxide and pathogenic mechanisms involved in the development of vascular diseases. Arch Pharm Res. 2009;32:1103–8. doi: 10.1007/s12272-009-1801-1. [DOI] [PubMed] [Google Scholar]
- [21].Harrison DG. Cellular and molecular mechanisms of endothelial cell dysfunction. J Clin Invest. 1997;100:2153–7. doi: 10.1172/JCI119751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Naseem KM. The role of nitric oxide in cardiovascular diseases. Mol Aspects Med. 2005;26:33–65. doi: 10.1016/j.mam.2004.09.003. [DOI] [PubMed] [Google Scholar]
- [23].Li H, Forstermann U. Nitric oxide in the pathogenesis of vascular disease. J Pathol. 2000;190:244–54. doi: 10.1002/(SICI)1096-9896(200002)190:3<244::AID-PATH575>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- [24].Maynard KI, Stewart-Lee AL, Milner P, Burnstock G. X-irradiation attenuates relaxant responses in the rabbit ear artery. Br J Pharmacol. 1992;105:126–8. doi: 10.1111/j.1476-5381.1992.tb14222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Qi F, Sugihara T, Hattori Y, Yamamoto Y, Kanno M, Abe K. Functional and morphological damage of endothelium in rabbit ear artery following irradiation with cobalt60. Br J Pharmacol. 1998;123:653–60. doi: 10.1038/sj.bjp.0701654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sugihara T, Hattori Y, Yamamoto Y, et al. Preferential impairment of nitric oxide-mediated endothelium-dependent relaxation in human cervical arteries after irradiation. Circulation. 1999;100:635–41. doi: 10.1161/01.cir.100.6.635. [DOI] [PubMed] [Google Scholar]
- [27].Zhang XH, Matsuda N, Jesmin S, et al. Normalization by edaravone, a free radical scavenger, of irradiation-reduced endothelial nitric oxide synthase expression. Eur J Pharmacol. 2003;476:131–7. doi: 10.1016/s0014-2999(03)02151-4. [DOI] [PubMed] [Google Scholar]
- [28].Hatoum OA, Otterson MF, Kopelman D, et al. Radiation induces endothelial dysfunction in murine intestinal arterioles via enhanced production of reactive oxygen species. Arterioscler Thromb Vasc Biol. 2006;26:287–94. doi: 10.1161/01.ATV.0000198399.40584.8c. [DOI] [PubMed] [Google Scholar]
- [29].Siegal T, Pfeffer MR, Meltzer A, et al. Cellular and secretory mechanisms related to delayed radiation-induced microvessel dysfunction in the spinal cord of rats. Int J Radiat Oncol Biol Phys. 1996;36:649–59. doi: 10.1016/s0360-3016(96)00357-4. [DOI] [PubMed] [Google Scholar]
- [30].Soloviev AI, Tishkin SM, Parshikov AV, Ivanova IV, Goncharov EV, Gurney AM. Mechanisms of endothelial dysfunction after ionized radiation: selective impairment of the nitric oxide component of endothelium-dependent vasodilation. Br J Pharmacol. 2003;138:837–44. doi: 10.1038/sj.bjp.0705079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Suvorava T, Luksha L, Bulanova KY, Lobanok LM. Dose-rate dependent effects of ionizing radiation on vascular reactivity. Radiat Prot Dosimetry. 2006;122:543–5. doi: 10.1093/rpd/ncl432. [DOI] [PubMed] [Google Scholar]
- [32].Kwon NS, Nathan CF, Stuehr DJ. Reduced biopterin as a cofactor in the generation of nitrogen oxides by murine macrophages. J Biol Chem. 1989;264:20496–501. [PubMed] [Google Scholar]
- [33].Werner ER, Gorren AC, Heller R, Werner-Felmayer G, Mayer B. Tetrahydrobiopterin and nitric oxide: mechanistic and pharmacological aspects. Exp Biol Med (Maywood) 2003;228:1291–302. doi: 10.1177/153537020322801108. [DOI] [PubMed] [Google Scholar]
- [34].Alp NJ, Channon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:413–20. doi: 10.1161/01.ATV.0000110785.96039.f6. [DOI] [PubMed] [Google Scholar]
- [35].Gorren AC, Mayer B. Tetrahydrobiopterin in nitric oxide synthesis: a novel biological role for pteridines. Curr Drug Metab. 2002;3:133–57. doi: 10.2174/1389200024605154. [DOI] [PubMed] [Google Scholar]
- [36].Andrew PJ, Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc Res. 1999;43:521–31. doi: 10.1016/s0008-6363(99)00115-7. [DOI] [PubMed] [Google Scholar]
- [37].Gorren AC, List BM, Schrammel A, et al. Tetrahydrobiopterin-free neuronal nitric oxide synthase: evidence for two identical highly anticooperative pteridine binding sites. Biochemistry. 1996;35:16735–45. doi: 10.1021/bi961931j. [DOI] [PubMed] [Google Scholar]
- [38].Rodriguez-Crespo I, Moenne-Loccoz P, Loehr TM. Ortiz de Montellano PR. Endothelial nitric oxide synthase: modulations of the distal heme site produced by progressive N-terminal deletions. Biochemistry. 1997;36:8530–8. doi: 10.1021/bi970192j. [DOI] [PubMed] [Google Scholar]
- [39].Klatt P, Schmid M, Leopold E, Schmidt K, Werner ER, Mayer B. The pteridine binding site of brain nitric oxide synthase. Tetrahydrobiopterin binding kinetics, specificity, and allosteric interaction with the substrate domain. J Biol Chem. 1994;269:13861–6. [PubMed] [Google Scholar]
- [40].Ghosh DK, Wu C, Pitters E, et al. Characterization of the inducible nitric oxide synthase oxygenase domain identifies a 49 amino acid segment required for subunit dimerization and tetrahydrobiopterin interaction. Biochemistry. 1997;36:10609–19. doi: 10.1021/bi9702290. [DOI] [PubMed] [Google Scholar]
- [41].Gorren AC, Kungl AJ, Schmidt K, Werner ER, Mayer B. Electrochemistry of pterin cofactors and inhibitors of nitric oxide synthase. Nitric Oxide. 2001;5:176–86. doi: 10.1006/niox.2001.0332. [DOI] [PubMed] [Google Scholar]
- [42].Reif A, Frohlich LG, Kotsonis P, et al. Tetrahydrobiopterin inhibits monomerization and is consumed during catalysis in neuronal NO synthase. J Biol Chem. 1999;274:24921–9. doi: 10.1074/jbc.274.35.24921. [DOI] [PubMed] [Google Scholar]
- [43].Kotsonis P, Frohlich LG, Shutenko ZV, Horejsi R, Pfleiderer W, Schmidt HH. Allosteric regulation of neuronal nitric oxide synthase by tetrahydrobiopterin and suppression of auto-damaging superoxide. Biochem J. 2000;346(Pt 3):767–76. [PMC free article] [PubMed] [Google Scholar]
- [44].Thony B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347(Pt 1):1–16. [PMC free article] [PubMed] [Google Scholar]
- [45].Hattori Y, Nakanishi N, Kasai K, Murakami Y, Shimoda S. Tetrahydrobiopterin and GTP cyclohydrolase I in a rat model of endotoxic shock: relation to nitric oxide synthesis. Exp Physiol. 1996;81:665–71. doi: 10.1113/expphysiol.1996.sp003967. [DOI] [PubMed] [Google Scholar]
- [46].Widder JD, Chen W, Li L, et al. Regulation of tetrahydrobiopterin biosynthesis by shear stress. Circ Res. 2007;101:830–8. doi: 10.1161/CIRCRESAHA.107.153809. [DOI] [PubMed] [Google Scholar]
- [47].De Bono JP, Channon KM. Endothelial cell tetrahydrobiopterin: going with the flow. Circ Res. 2007;101:752–4. doi: 10.1161/CIRCRESAHA.107.162503. [DOI] [PubMed] [Google Scholar]
- [48].Maita N, Hatakeyama K, Okada K, Hakoshima T. Structural basis of biopterin-induced inhibition of GTP cyclohydrolase I by GFRP, its feedback regulatory protein. J Biol Chem. 2004;279:51534–40. doi: 10.1074/jbc.M409440200. [DOI] [PubMed] [Google Scholar]
- [49].Gesierich A, Niroomand F, Tiefenbacher CP. Role of human GTP cyclohydrolase I and its regulatory protein in tetrahydrobiopterin metabolism. Basic Res Cardiol. 2003;98:69–75. doi: 10.1007/s00395-003-0394-y. [DOI] [PubMed] [Google Scholar]
- [50].Ishii M, Shimizu S, Wajima T, et al. Reduction of GTP cyclohydrolase I feedback regulating protein expression by hydrogen peroxide in vascular endothelial cells. J Pharmacol Sci. 2005;97:299–302. doi: 10.1254/jphs.sc0040146. [DOI] [PubMed] [Google Scholar]
- [51].Chalupsky K, Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2005;102:9056–61. doi: 10.1073/pnas.0409594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gao L, Chalupsky K, Stefani E, Cai H. Mechanistic insights into folic acid-dependent vascular protection: Dihydrofolate reductase (DHFR)-mediated reduction in oxidant stress in endothelial cells and angiotensin II-infused mice: A novel HPLC-based fluorescent assay for DHFR activity. J Mol Cell Cardiol. 2009;47(6):752–60. doi: 10.1016/j.yjmcc.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang J, Boerma M, Fu Q, Kulkarni A, Fink LM, HauerJensen M. Simvastatin ameliorates radiation enteropathy development after localized, fractionated irradiation by a protein C-independent mechanism. Int J Radiat Oncol Biol Phys. 2007;68:1483–90. doi: 10.1016/j.ijrobp.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Haydont V, Bourgier C, Pocard M, et al. Pravastatin Inhibits the Rho/CCN2/extracellular matrix cascade in human fibrosis explants and improves radiation-induced intestinal fibrosis in rats. Clin Cancer Res. 2007;13:5331–40. doi: 10.1158/1078-0432.CCR-07-0625. [DOI] [PubMed] [Google Scholar]
- [55].Tamura Y, Naemura A, Inoue A, et al. Impaired endothelial function may be due to decreased aortic tetrahydrobiopterin, assessed by a new flow-mediated vasodilation in vivo in hypercholesterolemic/atherogenic mice. Blood Coagul Fibrinolysis. 2009 doi: 10.1097/MBC.0b013e328331fd18. [DOI] [PubMed] [Google Scholar]
- [56].Shinozaki K, Nishio Y, Okamura T, et al. Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in the aortas of insulin-resistant rats. Circ Res. 2000;87:566–73. doi: 10.1161/01.res.87.7.566. [DOI] [PubMed] [Google Scholar]
- [57].Pieper GM. Acute amelioration of diabetic endothelial dysfunction with a derivative of the nitric oxide synthase cofactor, tetrahydrobiopterin. J Cardiovasc Pharmacol. 1997;29:8–15. doi: 10.1097/00005344-199701000-00002. [DOI] [PubMed] [Google Scholar]
- [58].Heitzer T, Brockhoff C, Mayer B, et al. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers : evidence for a dysfunctional nitric oxide synthase. Circ Res. 2000;86:E36–41. doi: 10.1161/01.res.86.2.e36. [DOI] [PubMed] [Google Scholar]
- [59].Heitzer T, Krohn K, Albers S, Meinertz T. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia. 2000;43:1435–8. doi: 10.1007/s001250051551. [DOI] [PubMed] [Google Scholar]
- [60].Higashi Y, Sasaki S, Nakagawa K, et al. Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am J Hypertens. 2002;15:326–32. doi: 10.1016/s0895-7061(01)02317-2. [DOI] [PubMed] [Google Scholar]
- [61].Settergren M, Bohm F, Malmstrom RE, Channon KM, Pernow J. L-arginine and tetrahydrobiopterin protects against ischemia/reperfusion-induced endothelial dysfunction in patients with type 2 diabetes mellitus and coronary artery disease. Atherosclerosis. 2009;204:73–8. doi: 10.1016/j.atherosclerosis.2008.08.034. [DOI] [PubMed] [Google Scholar]
- [62].Fiege B, Ballhausen D, Kierat L, et al. Plasma tetrahydrobiopterin and its pharmacokinetic following oral administration. Mol Genet Metab. 2004;81:45–51. doi: 10.1016/j.ymgme.2003.09.014. [DOI] [PubMed] [Google Scholar]
- [63].Sawabe K, Wakasugi KO, Hasegawa H. Tetrahydrobiopterin uptake in supplemental administration: elevation of tissue tetrahydrobiopterin in mice following uptake of the exogenously oxidized product 7,8-dihydrobiopterin and subsequent reduction by an anti-folate-sensitive process. J Pharmacol Sci. 2004;96:124–33. doi: 10.1254/jphs.fp0040280. [DOI] [PubMed] [Google Scholar]
- [64].Hasegawa H, Sawabe K, Nakanishi N, Wakasugi OK. Delivery of exogenous tetrahydrobiopterin (BH4) to cells of target organs: role of salvage pathway and uptake of its precursor in effective elevation of tissue BH4. Mol Genet Metab. 2005;86(Suppl 1):S2–10. doi: 10.1016/j.ymgme.2005.09.002. [DOI] [PubMed] [Google Scholar]
- [65].Tiefenbacher CP, Bleeke T, Vahl C, Amann K, Vogt A, Kubler W. Endothelial dysfunction of coronary resistance arteries is improved by tetrahydrobiopterin in atherosclerosis. Circulation. 2000;102:2172–9. doi: 10.1161/01.cir.102.18.2172. [DOI] [PubMed] [Google Scholar]
- [66].Tiefenbacher CP, Lee CH, Kapitza J, Dietz V, Niroomand F. Sepiapterin reduces postischemic injury in the rat heart. Pflugers Arch. 2003;447:1–7. doi: 10.1007/s00424-003-1131-y. [DOI] [PubMed] [Google Scholar]
- [67].Vasquez-Vivar J, Martasek P, Whitsett J, Joseph J, Kalyanaraman B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: an EPR spin trapping study. Biochem J. 2002;362:733–9. doi: 10.1042/0264-6021:3620733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tarpey MM. Sepiapterin treatment in atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22:1519–21. doi: 10.1161/01.atv.0000038144.37823.bf. [DOI] [PubMed] [Google Scholar]
- [69].Hattori Y, Nakanishi N, Akimoto K, Yoshida M, Kasai K. HMG-CoA reductase inhibitor increases GTP cyclohydrolase I mRNA and tetrahydrobiopterin in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:176–82. doi: 10.1161/01.atv.0000054659.72231.a1. [DOI] [PubMed] [Google Scholar]
- [70].Wenzel P, Daiber A, Oelze M, et al. Mechanisms underlying recoupling of eNOS by HMG-CoA reductase inhibition in a rat model of streptozotocin-induced diabetes mellitus. Atherosclerosis. 2008;198:65–76. doi: 10.1016/j.atherosclerosis.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Williams JP, Hernady E, Johnston CJ, et al. Effect of administration of lovastatin on the development of late pulmonary effects after whole-lung irradiation in a murine model. Radiat Res. 2004;161:560–7. doi: 10.1667/rr3168. [DOI] [PubMed] [Google Scholar]
- [72].Haydont V, Gilliot O, Rivera S, et al. Successful mitigation of delayed intestinal radiation injury using pravastatin is not associated with acute injury improvement or tumor protection. Int J Radiat Oncol Biol Phys. 2007;68:1471–82. doi: 10.1016/j.ijrobp.2007.03.044. [DOI] [PubMed] [Google Scholar]
- [73].Fu Q, Wang J, Boerma M, et al. Involvement of heat shock factor 1 in statin-induced transcriptional upregulation of endothelial thrombomodulin. Circ Res. 2008;103:369–77. doi: 10.1161/CIRCRESAHA.108.174607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Zhou Q, Zhao Y, Li P, Bai X, Ruan C. Thrombomodulin as a marker of radiation-induced endothelial cell injury. Radiat Res. 1992;131:285–9. [PubMed] [Google Scholar]
- [75].Holler V, Buard V, Gaugler MH, et al. Pravastatin limits radiationinduced vascular dysfunction in the skin. J Invest Dermatol. 2009;129:1280–91. doi: 10.1038/jid.2008.360. [DOI] [PubMed] [Google Scholar]
- [76].Huang A, Vita JA, Venema RC, Keaney JF., Jr. Ascorbic acid enhances endothelial nitric-oxide synthase activity by increasing intracellular tetrahydrobiopterin. J Biol Chem. 2000;275:17399–406. doi: 10.1074/jbc.M002248200. [DOI] [PubMed] [Google Scholar]
- [77].Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem. 2003;278:22546–54. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- [78].d'Uscio LV, Milstien S, Richardson D, Smith L, Katusic ZS. Long-term vitamin C treatment increases vascular tetrahydrobiopterin levels and nitric oxide synthase activity. Circ Res. 2003;92:88–95. doi: 10.1161/01.res.0000049166.33035.62. [DOI] [PubMed] [Google Scholar]
- [79].Kennedy M, Bruninga K, Mutlu EA, Losurdo J, Choudhary S, Keshavarzian A. Successful and sustained treatment of chronic radiation proctitis with antioxidant vitamins E and C. Am J Gastroenterol. 2001;96:1080–4. doi: 10.1111/j.1572-0241.2001.03742.x. [DOI] [PubMed] [Google Scholar]
- [80].Halperin EC, Gaspar L, George S, Darr D, Pinnell S. A double-blind, randomized, prospective trial to evaluate topical vitamin C solution for the prevention of radiation dermatitis. CNS Cancer Consortium. Int J Radiat Oncol Biol Phys. 1993;26:413–6. doi: 10.1016/0360-3016(93)90958-x. [DOI] [PubMed] [Google Scholar]
- [81].Wagdi P, Fluri M, Aeschbacher B, Fikrle A, Meier B. Cardioprotection in patients undergoing chemo- and/or radiotherapy for neoplastic disease. A pilot study. Jpn Heart J. 1996;37:353–9. doi: 10.1536/ihj.37.353. [DOI] [PubMed] [Google Scholar]
- [82].van Etten RW, de Koning EJ, Verhaar MC, Gaillard CA, Rabelink TJ. Impaired NO-dependent vasodilation in patients with Type II (non-insulin-dependent) diabetes mellitus is restored by acute administration of folate. Diabetologia. 2002;45:1004–10. doi: 10.1007/s00125-002-0862-1. [DOI] [PubMed] [Google Scholar]
- [83].Verhaar MC, Wever RM, Kastelein JJ, van Dam T, Koomans HA, Rabelink TJ. 5-methyltetrahydrofolate, the active form of folic acid, restores endothelial function in familial hypercholesterolemia. Circulation. 1998;97:237–41. doi: 10.1161/01.cir.97.3.237. [DOI] [PubMed] [Google Scholar]
- [84].Woo KS, Chook P, Lolin YI, Sanderson JE, Metreweli C, Celermajer DS. Folic acid improves arterial endothelial function in adults with hyperhomocystinemia. J Am Coll Cardiol. 1999;34:2002–6. doi: 10.1016/s0735-1097(99)00469-6. [DOI] [PubMed] [Google Scholar]
- [85].Moens AL, Vrints CJ, Claeys MJ, Timmermans JP, Champion HC, Kass DA. Mechanisms and potential therapeutic targets for folic acid in cardiovascular disease. Am J Physiol Heart Circ Physiol. 2008;294:H1971–H1977. doi: 10.1152/ajpheart.91503.2007. [DOI] [PubMed] [Google Scholar]
- [86].Gao L, Chalupsky K, Stefani E, Cai H. Mechanistic insights into folic acid-dependent vascular protection: Dihydrofolate reductase (DHFR)-mediated reduction in oxidant stress in endothelial cells and angiotensin II-infused mice: A novel HPLC-based fluorescent assay for DHFR activity. J Mol Cell Cardiol. 2009;47:752–60. doi: 10.1016/j.yjmcc.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Moulder JE, Fish BL, Cohen EP, Bonsib SM. Angiotensin II receptor antagonists in the prevention of radiation nephropathy. Radiat Res. 1996;146:106–10. [PubMed] [Google Scholar]
- [88].Moulder JE, Fish BL, Cohen EP. Radiation nephropathy is treatable with an angiotensin converting enzyme inhibitor or an angiotensin II type-1 (AT1) receptor antagonist. Radiother Oncol. 1998;46:307–15. doi: 10.1016/s0167-8140(97)00175-8. [DOI] [PubMed] [Google Scholar]
- [89].Molteni A, Moulder JE, Cohen EP, et al. Prevention of radiationinduced nephropathy and fibrosis in a model of bone marrow transplant by an angiotensin II receptor blocker. Exp Biol Med (Maywood) 2001;226:1016–23. doi: 10.1177/153537020122601108. [DOI] [PubMed] [Google Scholar]
- [90].Molteni A, Moulder JE, Cohen EF, et al. Control of radiationinduced pneumopathy and lung fibrosis by angiotensin-converting enzyme inhibitors and an angiotensin II type 1 receptor blocker. Int J Radiat Biol. 2000;76:523–32. doi: 10.1080/095530000138538. [DOI] [PubMed] [Google Scholar]
- [91].Molteni A, Wolfe LF, Ward WF, et al. Effect of an angiotensin II receptor blocker and two angiotensin converting enzyme inhibitors on transforming growth factor-beta (TGF-beta) and alpha-actomyosin (alpha SMA), important mediators of radiation-induced pneumopathy and lung fibrosis. Curr Pharm Des. 2007;13:1307–16. doi: 10.2174/138161207780618777. [DOI] [PubMed] [Google Scholar]
- [92].Robbins ME, Payne V, Tommasi E, et al. The AT1 receptor antagonist, L-158,809, prevents or ameliorates fractionated whole-brain irradiation-induced cognitive impairment. Int J Radiat Oncol Biol Phys. 2009;73:499–505. doi: 10.1016/j.ijrobp.2008.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Satoh M, Fujimoto S, Arakawa S, et al. Angiotensin II type 1 receptor blocker ameliorates uncoupled endothelial nitric oxide synthase in rats with experimental diabetic nephropathy. Nephrol Dial Transplant. 2008;23:3806–13. doi: 10.1093/ndt/gfn357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Berbee M, Fu Q, Boerma M, Wang J, Kumar KS, Hauer-Jensen M. gamma-Tocotrienol ameliorates intestinal radiation injury and reduces vascular oxidative stress after total-body irradiation by an HMG-CoA reductase-dependent mechanism. Radiat Res. 2009;171:596–605. doi: 10.1667/RR1632.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Ghosh SP, Kulkarni S, Hieber K, et al. Gamma-tocotrienol, a tocol antioxidant as a potent radioprotector. Int J Radiat Biol. 2009;85:598–606. doi: 10.1080/09553000902985128. [DOI] [PubMed] [Google Scholar]
- [96].Parker RA, Pearce BC, Clark RW, Gordon DA, Wright JJ. Tocotrienols regulate cholesterol production in mammalian cells by post-transcriptional suppression of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 1993;268:11230–8. [PubMed] [Google Scholar]
- [97].Song BL, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of 3-hydroxy-3-methylglutaryl coenzyme a reductase stimulated by delta- and gamma-tocotrienols. J Biol Chem. 2006;281:25054–61. doi: 10.1074/jbc.M605575200. [DOI] [PubMed] [Google Scholar]