

Abstract
The drying of hydrophobic cavities is believed to play an important role in biophysical phenomena such as the folding of globular proteins, the opening and closing of ligand-gated ion channels, and ligand binding to hydrophobic pockets. We use forward flux sampling, a molecular simulation technique, to compute the rate of capillary evaporation of water confined between two hydrophobic surfaces separated by nanoscopic gaps, as a function of gap, surface size, and temperature. Over the range of conditions investigated (gaps between 9 and 14 Å and surface areas between 1 and 9 nm2), the free energy barrier to evaporation scales linearly with the gap between hydrophobic surfaces, suggesting that line tension makes the predominant contribution to the free energy barrier. The exponential dependence of the evaporation rate on the gap between confining surfaces causes a 10 order-of-magnitude decrease in the rate when the gap increases from 9 to 14 Å. The computed free energy barriers are of the order of 50kT and are predominantly enthalpic. Evaporation rates per unit area are found to be two orders of magnitude faster in confinement by the larger (9 nm2) than by the smaller (1 nm2) surfaces considered here, at otherwise identical conditions. We show that this rate enhancement is a consequence of the dependence of hydrophobic hydration on the size of solvated objects. For sufficiently large surfaces, the critical nucleus for the evaporation process is a gap-spanning vapor tube.
Keywords: hydrophobicity, phase transition, kinetics, rare-event sampling, nanoscale
The behavior of water near hydrophobic surfaces is of interest in a wide range of technological contexts. Examples include the design of self-cleaning materials (1) and antiice coatings (2), and the development of processes for the storage and dissipation of mechanical energy (3). Scientifically, many aspects of hydrophobic hydration are the object of active inquiry (4); examples include the role of density fluctuations in nanoscopic hydrophobic interfaces (5), the entropic or enthalpic character of hydrophobic hydration and its dependence on solute size and thermodynamic conditions (6, 7), and the molecular conformations and solubility of long-chain alkanes in water (8). A fundamental connection between hydrophobicity and biological self-assembly was first pointed out by Kauzmann (9), who showed that the water-mediated tendency for apolar moieties to aggregate is crucial for protein conformational stability. Tanford’s work further contributed to establishing the centrality of water-mediated interactions in biological self-assembly (10). Since these seminal insights, the view has gradually emerged of water as an active participant in life’s processes (11).
Water confined by two impenetrable surfaces is the simplest example of water-mediated interactions between (large) hydrophobic objects. When the distance between such hydrophobic surfaces falls below a critical value, evaporation of water is favored thermodynamically (12). The resulting surface-induced evaporation has been the subject of numerous theoretical and computational studies (e.g., refs. 6, 13–19), with several focusing on biological hydrophobic interfaces (e.g., refs. 20–22).
Previous computational studies of capillary evaporation in hydrophobic confinement have addressed the underlying thermodynamics or have been limited to phenomenological observations of the occurrence or absence of capillary evaporation in finite-time molecular dynamics simulations. Much less attention has been devoted to the equally important matter of evaporation kinetics. Notable exceptions include the important work of Luzar and coworkers (23–25), Bolhuis and Chandler (26), and Xu and Molinero (27). Leung et al. (23) used a combination of umbrella sampling and reactive flux formalism to compute the rate of capillary evaporation of simple point charge (SPC) water (28) in a semiinfinite hydrophobic slit. Subsequently, Luzar (24) used a lattice model to investigate the dependence of the free energy barrier on the separation between the confining surfaces. Bolhuis and Chandler (26) used transition path sampling to study the cavitation of the Lennard–Jones liquid between repulsive surfaces. They focused on the nature of the transition state, and pointed out the relevance of their findings to the hydrophobic effect. Xu and Molinero (27) studied the thermodynamics and kinetics of liquid-vapor oscillations in a coarse-grained model of water in nanoscale hydrophobic confinement.
The drying of hydrophobic cavities is thought to be important in biophysical phenomena such as the folding of globular proteins (4, 6, 9, 10, 20), the opening and closing of ligand-gated ion channels (29), and ligand binding to hydrophobic pockets (30). Thus, knowledge of the rate of capillary evaporation in hydrophobic confinement, and its dependence on temperature, pressure, confinement length scale, size of the confining surfaces, and surface characteristics such as degree of hydrophobicity and curvature, should be useful for a quantitative understanding of several important biophysical phenomena. In this paper, we report on a computational investigation of the effects of surface size, confinement length scale, and temperature on the kinetics of capillary evaporation of water in hydrophobic confinement.
As will be shown, evaporation requires the formation of a sufficiently large void in the confined region, a rare event. A straightforward molecular dynamics (MD) simulation is therefore incapable of providing quantitative rate information on the basic phenomenon of interest here. Accordingly, we use forward flux sampling (FFS), a technique specifically designed to sample rare events (31–33), in conjunction with MD. Fig. 1 shows schematically the implementation of the calculation. We consider two hydrophobic surfaces separated by a gap d immersed in water at fixed temperature and pressure, and use the FFS technique to calculate the rate at which the confined volume L2d is emptied. We perform the calculation for a range of values of d, L, and temperature. Technical details are provided in the Methods section.
Fig. 1.

Schematic of evaporation rate calculation. Two L × L hydrophobic surfaces (green atoms), separated by a gap d, are immersed in 2,329 (L = 1 nm) or 4,685 (L = 3 nm) water molecules, at atmospheric pressure. Forward flux sampling simulations (31–33) are carried out to compute the rate of capillary evaporation in the confined region of width d, for a range of values of d, L, and temperature.
Results and Discussion
Rates.
Figs. 2 and 3 show the calculated evaporation rate as a function of the gap between hydrophobic surfaces, d, for 1.0 × 0.9 (Fig. 2) and 3.2 × 3 nm2 surfaces (Fig. 3), at 298 K and 1 bar (henceforth, we refer to these as 1 × 1 and 3 × 3 nm2 surfaces, respectively). The characteristic time τ required to nucleate a surface-induced evaporation event is given by [τ ∼ (jA)-1], where A is the surface area and j is the evaporation rate. This time increases by 10 orders of magnitude (from 6.3 × 10-10 to 17.2 s) as the gap between small (1 × 1 nm2) surfaces increases from 9 to 14 Å; similarly, there is a six order-of-magnitude increase in the characteristic evaporation time between large (3 × 3 nm2) surfaces upon increasing the gap from 11 to 14 Å. These numbers suggest constraints on the range of gaps for which capillary evaporation can occur at rates that are dynamically relevant to biophysical phenomena.
Fig. 2.

Calculated evaporation rates. Dependence of the evaporation rate on the gap between 1 × 1 nm2 hydrophobic surfaces, at 298 K. The inset shows, for the same surfaces, Arrhenius plots of the evaporation rate for two values of the gap, corresponding to calculations at T = 298, 348, and 398 K.
Fig. 3.

Calculated evaporation rates. Dependence of the evaporation rate on the gap between 3 × 3 nm2 hydrophobic surfaces, at 298 K.
Free Energy Barrier and Gap Dependence.
In general, the evaporation rate j can be expressed as
| [1] |
where C is a gap-independent preexponential factor, ΔG is the free energy barrier to nucleation, C′ = C exp[ΔS(d)/k], ΔS and ΔH are the entropic and enthalpic contributions to ΔG, and k is Boltzmann’s constant. Eq. 1 implies that, by computing the evaporation rate as a function of the gap d and temperature, one can extract information on ΔG, ΔH, and ΔS. The numerical procedures used to fit the rate data to Eq. 1 and to regress values for ΔG, ΔH, and ΔS are described in SI Text. Briefly, from an Arrhenius plot, ln j vs. 1/T (Fig. 2, Inset), we obtain both ln C′ (intercept) and ΔH (slope = -ΔH/k). The boiling point of the water model used in this work (see Methods) is 397 ± 1 K (34); no boiling occurred during our simulations at 398 K. The intercept, ln C′, was found to be independent of d, implying that the entropic contribution to the free energy is either small or d independent. Using the last expression in Eq. 1, ΔH was found to scale linearly with d, which implies that ΔG is also linear in d. With ΔG = A + Bd, and hence ln j = ln C - A/kT - Bd/kT, we obtain A, B, and ln C by regression of the computed rates, j (T, d). Finally, ΔS is given by the ratio of the intercepts, ΔS/k = ln(C′/C). We find that the free energy barrier is predominantly enthalpic, with TΔS/ΔH ∼ O(10-3) and O(10-1) for the small and large surfaces, respectively. Over the range of conditions investigated in this work, we find that the rate of change of the free energy barrier with respect to the gap, B, is between 4 and 5 kT/Å. Table 1 compares the free energy barriers computed directly from Eq. 1 with the values obtained by rescaling ΔG (at 9.8 Å for 1 × 1 nm2 surfaces; at 12 Å for 3 × 3 nm2 surfaces) assuming linear scaling, ΔG ∼ d. The good agreement shows that, over the range of conditions explored in this work, the free energy barrier scales linearly with the gap between hydrophobic surfaces. As documented in the SI Text, neither a quadratic dependence, ΔG ∼ d2, nor using (d - 2l) instead of d to fit the data, yielded accurate representations of the evaporation rate (here, l is the thickness of the vapor layer adjacent to the hydrophobic surface, which can be clearly seen in Fig. 1; see SI Text for details on the determination of l). It is important to note that, in this work, we use an indirect, kinetic route to calculate ΔG. It would be useful to compute this quantity directly, using free energy sampling techniques.
Table 1.
Comparison of free energy barriers for evaporation between small (1 × 1 nm2) and large (3 × 3 nm2) surfaces, calculated directly from computed evaporation rates at 298 K, and by assuming linear dependence of the barrier on the gap size
| Gap, Å | ΔG/kT, small surface (Eq. 1)* | ΔG/kT, small surface (linear scaling)† | ΔG/kT, large surface (Eq. 1)* | ΔG/kT, large surface (linear scaling)† |
| 9.0 | 42.5 | 42.0 | — | — |
| 9.8 | 45.7 | 45.7 | — | — |
| 11 | 50.4 | 51.3 | 57.8 | 55.8 |
| 12 | 55.5 | 55.9 | 60.9 | 60.9 |
| 13 | 59.5 | 60.6 | 64.5 | 65.9 |
| 14 | 66.5 | 65.2 | 71.7 | 71.0 |
*Free energy barriers obtained from evaporation rate calculations (ΔG/kT = ln C - ln j), with preexponential factor obtained from as explained in the SI Text.
†Free energy barriers calculated assuming linear dependence of ΔG on d, ΔG(d) = ΔG(9.8 Å) × d/9.8 for small surfaces, and ΔG(d) = ΔG(12 Å) × d/12 for large surfaces.
The linear scaling of ΔG with d suggests that the predominant contribution to ΔG, over the range of conditions, size of the hydrophobic surfaces, and gaps investigated here, comes from line tension (35, 36). To rationalize this observation, we consider the formation of a cylindrical vapor tube of radius r between two solid surfaces separated by a distance d, the gap being otherwise filled with liquid (SI Text). As will be shown below, the critical nucleus for sufficiently large surfaces is indeed a cylindrical tube. The equilibrium state of a macroscopic system corresponds to a condition of minimum free energy (e.g., minimum Gibbs free energy for a closed system at fixed temperature and pressure; minimum Helmholtz free energy for a closed system at fixed temperature and volume). For an open system possessing both an interface (e.g., vapor–liquid) and a line along which three phases are in contact (e.g., solid–liquid–vapor), this free energy is given by Ω = -PV + γF + λL and is called the grand potential. Here, P denotes pressure; V, volume; F, interfacial area; L, the linear dimension along which three phases are in contact; γ, the vapor–liquid interfacial tension; and λ is the line tension associated with three-phase contact along the circumference of the cylinder’s base. The free energy cost of forming a gap-spanning vapor tube is given by
| [2] |
The above expression assumes that the surface is perfectly nonwetting (contact angle 180°). The derivation of Eq. 2 is provided in the SI Text.
The free energy maximum occurs for a tube radius r∗, given by
 |
[3] |
in correspondence to which the free energy barrier is
 |
[4] |
In the absence of a line tension contribution, the free energy barrier scales quadratically with the gap, a well-known result (14). Using typical values [γ ∼ 0.07 N/m (ref. 12), d ∼ 1 nm, λ ∼ 10-5 dyn (refs. 35 and 37)], the relative magnitude of the three terms on the right-hand side of Eq. 4 is approximately 1∶11∶33, indicating that line tension makes the predominant contribution to the free energy barrier. The literature includes reports of positive as well as negative line tensions (37). Our observations are consistent with positive line tensions of magnitudes such as are reported in the literature (35, 37).
Surface Size Dependence.
As shown in Figs. 2 and 3, evaporation rate calculations were performed at 11, 12, 13, and 14 Å gaps for both the small (1 × 1 nm2) and large (3 × 3 nm2) surfaces. For a given gap, evaporation is much faster for the larger surfaces: the rate for the 3 × 3 nm2 hydrophobic surfaces is 40 times larger than for the 1 × 1 nm2 surfaces when the gap is 11 Å, and 358 times larger when the gap is 14 Å. Table 2 lists the average water density and compressibility in the confined region for the different gap sizes, and for small (1 × 1 nm2) and large (3 × 3 nm2) surfaces. Effective compressibilities were obtained from the fluctuation equation  , where KT is the isothermal compressibility, V is the confined volume, and angle brackets denote thermal average.
, where KT is the isothermal compressibility, V is the confined volume, and angle brackets denote thermal average.
Table 2.
Comparison of mean density and compressibility of water at 298 K and 1 bar confined between small (1 × 1 nm2) and large (3 × 3 nm2) surfaces
| Gap, Å |
Density,* g/mL |
105 x compressibility,* 1/ bar |
||
| small surface | large surface | small surface | large surface | |
| 11 | 1.477 (0.595) | 1.384 (0.558) | 13.5 (33.6) | 18.3 (45.3) |
| 12 | 1.369 (0.620) | 1.268 (0.574) | 13.6 (30.2) | 16.5 (36.4) |
| 13 | 1.332 (0.659) | 1.235 (0.611) | 14.4(29.2) | 15.3(30.9) |
| 14 | 1.302 (0.691) | 1.209 (0.642) | 13.8(26.0) | 14.2(26.7) |
*The width of the confined region was calculated as d - 2σO-W (numbers without parenthesis) or as d (numbers in parenthesis).
It can be seen that, for a given value of the gap, the density of confined water decreases and its compressibility increases, as the size of the confining surface increases. This observation is consistent with Stillinger’s important insight regarding the structure of aqueous interfaces near large nonpolar objects (38), with the theoretical description of the manner in which soft interfaces arise on nanoscopic scales (6), and with subsequent results from simulations of capillary evaporation using lattice models (25). Thus, the marked increase in evaporation rate with the size of the confining surfaces is a manifestation of the length-scale dependence of hydrophobicity, whereby the interface between water and a hydrophobic object evolves from hard and liquid-like to soft and vapor-like as the size of the solvated object increases (6, 7). Accordingly, penetration into the metastable region is accomplished both by bringing a given pair of hydrophobic surfaces closer together or by enlarging the hydrophobic surface area while keeping the gap unchanged.
A complementary theoretical perspective on the size-dependence of evaporation for a given gap follows from considering the critical gap dc, between hydrophobic surfaces below which confined liquid water becomes metastable with respect to the vapor. This quantity is given by (6, 12, 13, 24)
 |
[5] |
where Δp is the difference between the imposed pressure and the saturation pressure at the given temperature, and the immersed surfaces are assumed to be L × L squares. For L ∼ 1 nm, the second term in brackets in the right-hand side denominator of Eq. 5 is of order 103, whereupon the following simplified result follows (12):
| [6] |
This result implies that, if nanoscale pairs of hydrophobic surfaces of different size are immersed in water, the supersaturation will increase with the size of immersed surface, even if the gap between pairs of surfaces is fixed. Hence we expect that the evaporation rate will increase with the characteristic size of the hydrophobic surfaces. Generalization of Eq. 5 and relation 6 to include line tension is discussed in the SI Text.
For the 1 × 1 nm2 surfaces, [6] also suggests that the vapor may be metastable with respect to the confined liquid because d > L/2. It should be emphasized, however, that the continuum picture on which [6] is predicated breaks down at molecular length scales (12). Thus, it is the scaling dc ∼ L, not the precise coefficient, that is sufficient to rationalize the L dependence of the computed evaporation rates.
Transition State.
In order to investigate the nature of the transition state leading to evaporation, calculations were performed at gaps of 9.8 Å (small surfaces) and 12 Å (large surfaces), at 298 K and 1 bar. Configurations that upon randomizing the molecular velocities have equal probability of reaching the vapor state (empty gap space) or returning to the liquid state constitute the transition state ensemble (26, 39–41). Members of this ensemble were harvested by a three-step computational procedure described in Methods.
The fraction of trajectories that, starting from a given configuration, reach the vapor state without first returning to the liquid state constitutes the committor probability for that configuration (41). Figs. 4 and 5 show the committor probabilities for the various configurations. Each curve corresponds to a fixed number of water molecules in the confined region, N (small surfaces) or to a range of N values (large surfaces). The horizontal line corresponding to a committor value of one-half identifies the members of the transition state ensemble. For the small surfaces (Fig. 4), the transition state is mostly composed of configurations with a single molecule remaining in the confined region. It can be seen that, even when as few as three or four molecules remain in the confined space, the majority of trajectories initiated from such configurations return to the liquid state. Fig. 5 (large surfaces) shows a different picture. The curve corresponding to 176 ≤ N ≤ 180, for which the majority of configurations lead to evaporation, crosses the 50% committor value almost orthogonally (compare with the behavior of the N = 1 curve in Fig. 4). This observation indicates that N by itself is not a good order parameter for identifying transition states, a conclusion substantiated in Fig. 6. Shown there are three configurations corresponding to N = 179, 180, and 190 (Fig. 6 A–C, respectively). The committor probability of the configuration shown in Fig. 6A is only 4%, even though the number of water molecules in the confined region, 179, is the smallest of the three cases considered. The committor probabilities for the configurations shown in Fig. 6 B and C are 52% and 83%, respectively. It is clear that the pathway to evaporation involves the formation of a vapor tube of critical diameter (14, 23, 26). The configuration depicted in Fig. 6A, though “farther along” the route toward the vapor phase as measured by N, is in reality very far from vaporizing, because it lacks a sufficiently large cavity.
Fig. 4.

Identification of the transition state ensemble. Each curve gives the probability, computed over 100 runs launched from a given configuration after randomizing the velocities, that such runs will reach the vapor state (no water molecules in the confined region) without first returning to the liquid state. This probability is plotted as a function of configuration number, with configurations ranked in order of increasing committor probability. All of the configurations along a given line have the same number of confined water molecules (N = 0, 1, 2, 3, or 4). Conditions are d = 9.8 Å, T = 298 K, P = 1 bar, L = 1 nm. The transition state ensemble corresponds to those configurations with equal probability of reaching the vapor state or of returning to the liquid state (dashed line). Along each line, the number of configurations has been normalized so as to lie between 1 and 100. For example, if there are m ≠ 100 configurations with N = 1, their number has been scaled by 100/m.
Fig. 5.

Identification of the transition state ensemble. Same as Fig. 4, but for d = 12 Å, T = 298 K, P = 1 bar, L = 3 nm. Because of the larger number of confined water molecules compared to the L = 1 case (Fig. 4), these have been combined into groups for ease of representation. Thus, each curve corresponds to a range of N values.
Fig. 6.

Selected configurations intermediate between the confined liquid and vapor states, for d = 12 Å, T = 298 K, P = 1 bar, and L = 3 nm. The hydrophobic surfaces, whose boundary is traced by the yellow line, have been removed for ease of visualization. The view is along the direction perpendicular to the surfaces. The number of confined water molecules and committor probability for these configurations are (179, 0.04), (180, 0.52), and (190, 0.83) for A–C, respectively. Gap-spanning vapor tubes are clearly visible in B and C.
Conclusions
The present calculations suggest that there is a narrow range of gaps (ca. 5–15 Å) between hydrophobic surfaces within which capillary evaporation occurs at rates that may be relevant to biological assembly phenomena. Over the range of gaps (9.0–14 Å), surface areas (1–9 nm2), and temperatures investigated here (298 ≤ T ≤ 398 K), the predominant contribution to the free energy barrier to evaporation comes from line tension. We find that free energy barriers are predominantly enthalpic and increase in proportion to the gap between surfaces at a rate of 4–5 kT/Å. We observe a marked increase in the rate of capillary evaporation (on a per unit area basis) upon increasing the size of the hydrophobic surface. Recent simulations have shown that capillary drying is involved in the closing of the pentameric pore in a ligand-gated ion channel (29). The possible relevance of capillary drying to other biophysical phenomena deserves investigation.
FFS is a powerful technique that enables rate calculations spanning more than 10 orders of magnitude (e.g., characteristic evaporation times ranging from 6 × 10-10 to 17 s for the 1 × 1 nm2 surfaces; see Fig. 2 and text). Numerical analysis of the transition state ensemble shows that, for sufficiently large surfaces, the critical nucleus is a gap-spanning vapor tube (14, 23, 26). On smaller surfaces, the transition state ensemble consists largely of configurations containing as little as a single water molecule in the confined space.
Methods
Forward Flux Sampling.
Consider a system with two locally stable states designated by A (e.g., confined liquid) and B (e.g., confined vapor), which are separated by a free energy barrier much larger than the thermal energy. The goal is to find the thermally averaged rate at which the system evolves from A to B. Consider a property that can distinguish state A from state B. For the present problem, it is clear that the number of water molecules in the confined region, N, is such a property. For convenience, we consider the corresponding intensive property, ρ, the average value of which is ρA in state A and ρB in state B(ρA > ρB). The evolution from A to B can be described by “interfaces” λi (i = 0,1,2,3,…,n) which are collections of configurations with the same value of ρ, say ρi (31–33). Let ρi > ρi+1. State A (liquid) is uniquely defined as comprising all configurations with ρ > ρ0 and state B (vapor) comprises configurations with ρ < ρn. We chose ρ0 to be one standard deviation away from the mean liquid density in the confined region. FFS comprises of two steps. In the first step, we calculated the flux from state A to the first interface λ1 (42, 43). An O (50 ns) MD simulation was conducted at liquid conditions, and each time the simulation reached λ1, the configuration at λ1 was stored. Because A is the locally stable state, on most occasions, the trajectory reaching λ1 returned back to A. If on a rare occasion it reached B, then the simulation was stopped and restarted from a random initial condition in A. The flux to reach λ1 from A was calculated by dividing the number of λ1 crossings that originate from λ0 by the total time spent by the MD trajectory within the liquid basin (ρ > ρ0).The location of λ1 was chosen so as to ensure that 500–700 independent trajectories from λ0 cross λ1. Uncorrelated configurations were ensured by storing configurations separated by at least 2 ps. In the second step, the conditional probability of a trajectory starting from λi and reaching λi+1 before reaching λ0 [denoted by P(λi+1|λi)] is determined. In order to find P(λ2|λ1), a number of MD trajectories are started from the configurations stored at λ1 after velocity randomization and are propagated until they reach either λ2 or λ0. P(λ2|λ1) is simply the fraction of trajectories that reach λ2 out of all the trajectories started from λ1. The configurations at λ2 are stored for further propagation to λ3, and steps are repeated until the system reaches λn. The rate of the transition from A to B is then given by (31–33, 42, 43)
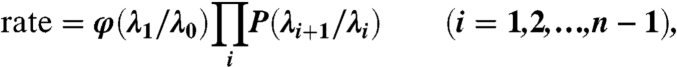 |
[7] |
where φ(λ1|λ0) is the flux of trajectories that leave λ0(ρ > ρ0) and reach λ1. Interfaces λi were chosen to ensure that similar statistics of trajectory crossings are obtained at each interface. From each configuration at λi, 100 trajectories are shot (each with randomized velocities), and the location of λi+1 is selected such that P(λi+1|λi) ∼ 0.01. Numerical checks were conducted for both the small (1 × 1 nm2) and large (3 × 3 nm2) walls. In the former case, for d = 9 Å at 298 K, evaporation occurred fast enough that it could be computed directly by MD. Comparison of FFS and direct MD rates yielded excellent agreement (1.67 × 109 vs. 1.69 × 109 nm-2 s-1, respectively, the latter averaged over 127 evaporation transitions). For the large walls case, the number of interfaces for the case d = 14 Å at 334 K was changed from three (N = 260, 240, 226) to four (N = 260, 240, 226, 200). The calculated evaporation rates were 1.21 × 105 and 1.22 × 105 nm-2s-1.
Transition State Ensemble.
The three-step procedure for harvesting the transition state ensemble is as follows. In the first step, an appropriate value was determined for the number of confined water molecules characterizing configurations from which subsequent trajectory “launches” were performed. This appropriate number was determined by starting molecular dynamics runs from states along the various interfaces λi used in the evaporation rate calculations and identifying an interface from which the probability of reaching the vapor state is significantly less than one, but nonvanishing. The number of confined water molecules so selected was 7 and 198 for the small and large walls, respectively. At 298 K and 1 bar, the corresponding probabilities of reaching the vapor phase were 0.026 and 0.015, respectively. In the second step, molecular dynamics runs were launched from these starting configurations (i.e., from configurations with 7 and 198 water molecules confined between the small and large walls, respectively); a subset of these reached successive interfaces on the way to the vapor phase, and these configurations were saved. For the small wall simulations, O(104) runs were launched from N = 7, 150 of which reached N = 1. Another O(104) runs were launched from N = 7, of which 400 reached N = 5. This procedure yielded O(102) configurations in each of the milestones N = 1, 2, 3, 4, and 5. Similar calculations for the large walls case yielded O(102) configurations in each of the three milestone ranges 176 ≤ N ≤ 180, 181 ≤ N ≤ 185, and 186 ≤ N ≤ 190, the grouping being necessary because of the much larger number of molecules. In the third step, 100 trajectories were launched starting from each of the candidate configurations (i.e., 100 trajectories starting from each of the O(102) N = 1 configurations, 100 from each of the O(102) N = 2, etc.).
Molecular Dynamics.
Mimicking the arrangement of carbon atoms in graphene sheets, the hydrophobic walls were represented by a rigid, hexagonal lattice of Lennard–Jones (LJ) atoms with a lattice constant of 1.4 Å. The walls were kept fixed, parallel to each other, separated by a distance d, and symmetrically located with respect to the center of the simulation box. The extended simple point charge water model was used throughout (44). The LJ parameters for water–wall interaction were taken as εO-W = 0.0289 kcal/mol and σO-W = 3.283 Å (18). MD simulations were conducted in the isothermal-isobaric (NPT) ensemble at 298 K and 1 bar in a periodic simulation box, using a Nosé–Hoover thermostat and barostat (45, 46). All simulations were performed using the Large-Scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) MD package (47). The number of simulated water molecules was 2,329 for the small wall system and 4,685 for the large wall system. The Particle-Particle Particle-Mesh (PPPM) Ewald method was used to compute long-range corrections to electrostatic interactions (48, 49). The k-space vector was taken to be 0.295 Å-1, and calculations were performed on a 25 × 36 × 36 grid, with rms precision of 6 × 10-5, the standard PPPM Ewald parameters in LAMMPS.
Supplementary Material
Acknowledgments.
We are grateful to Alenka Luzar and Amish Patel for insightful comments on this work. The financial support of Unilever UK Central Resources is gratefully acknowledged. Computations were performed at the Terascale Infrastructure for Groundbreaking Research in Engineering and Science (TIGRESS) facility at Princeton University.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1116167109/-/DCSupplemental.
References
- 1.Blossey R. Self-cleaning surfaces—virtual realities. Nat Mater. 2003;2:301–306. doi: 10.1038/nmat856. [DOI] [PubMed] [Google Scholar]
- 2.Cao L, Jones AK, Sikka VK, Wu J, Gao D. Anti-icing superhydrophobic coatings. Langmuir. 2009;25:12444–12448. doi: 10.1021/la902882b. [DOI] [PubMed] [Google Scholar]
- 3.Eroshenko V, Robert-Charles R, Soulard M, Patarin J. Energetics: A new field of applications for hydrophobic zeolites. J Am Chem Soc. 2001;123:8129–8130. doi: 10.1021/ja011011a. [DOI] [PubMed] [Google Scholar]
- 4.Chandler D. Interfaces and the driving force for hydrophobic assembly. Nature. 2005;437:640–647. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- 5.Acharya H, Vembanur S, Jamadagni SN, Garde S. Mapping hydrophobicity at the nanoscale: Applications to heterogeneous surfaces and proteins. Faraday Discuss. 2010;146:353–365. doi: 10.1039/b927019a. [DOI] [PubMed] [Google Scholar]
- 6.Lum K, Chandler D, Weeks JD. Hydrophobicity at small and large length scales. J Phys Chem B. 1999;103:4570–4577. [Google Scholar]
- 7.Ashbaugh HS, Pratt LR. Colloquium: Scaled particle theory ad the length scales of hydrophobicity. Rev Mod Phys. 2006;78:159–178. [Google Scholar]
- 8.Ferguson AL, Debenedetti PG, Panagiotopoulos AZ. Solubility and molecular conformations of n-alkane chains in water. J Phys Chem B. 2009;113:6405–6414. doi: 10.1021/jp811229q. [DOI] [PubMed] [Google Scholar]
- 9.Kauzmann W. Some forces in the interpretation of protein denaturation. Adv Protein Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 10.Tanford C. Contribution of hydrophobic interactions to the stability of the globular conformation of proteins. J Am Chem Soc. 1962;84:4240–4247. [Google Scholar]
- 11.Ball PJ. Water as an active constituent in cell biology. Chem Rev. 2008;108:74–108. doi: 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]
- 12.Cerdeiriña CA, Debenedetti PG, Rossky PJ, Giovambattista N. Evaporation length scales of confined water and some common organic liquids. J Phys Chem Lett. 2011;2:1000–1003. [Google Scholar]
- 13.Lum K, Luzar A. Pathway to surface-induced phase transition of a confined fluid. Phys Rev E. 1997;56:R6283–R6286. [Google Scholar]
- 14.Lum K, Chandler D. Phase diagram and free energies of vapor films and tubes for a confined fluid. Int J Thermophys. 1998;19:845–855. [Google Scholar]
- 15.Luzar A, Bratko D, Blum L. Monte Carlo simulation of hydrophobic interaction. J Chem Phys. 1987;86:2955–2959. [Google Scholar]
- 16.Bérard DR, Attard P, Patey GN. Cavitation of a Lennard-Jones fluid between hard walls, and the possible relevance to the attraction measured between hydrophobic surfaces. J Chem Phys. 1993;98:7236–7244. [Google Scholar]
- 17.Giovambattista N, Rossky PJ, Debenedetti PG. Effect of pressure on the phase behavior and structure of water confined between nanoscale hydrophobic and hydrophilic plates. Phys Rev E. 2006;73:041604. doi: 10.1103/PhysRevE.73.041604. [DOI] [PubMed] [Google Scholar]
- 18.Choudhury N, Pettitt BM. The dewetting transition and the hydrophobic effect. J Am Chem Soc. 2007;129:4847–4852. doi: 10.1021/ja069242a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eun C, Berkowitz ML. Fluctuations in number of water molecules confined between nanoparticles. J Phys Chem B. 2010;114:13410–13414. doi: 10.1021/jp1072654. [DOI] [PubMed] [Google Scholar]
- 20.Zhou R, Huang X, Margulis CJ, Berne BJ. Hydrophobic collapse in multidomain protein folding. Science. 2004;305:1605–1609. doi: 10.1126/science.1101176. [DOI] [PubMed] [Google Scholar]
- 21.Berne BJ, Weeks JD, Zhou R. Dewetting and hydrophobic interaction in physical and biological systems. Annu Rev Phys Chem. 2009;60:85–103. doi: 10.1146/annurev.physchem.58.032806.104445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giovambattista N, Lopez CF, Rossky PJ, Debenedetti PG. Hydrophobicity of protein surfaces: Separating geometry from chemistry. Proc Natl Acad Sci USA. 2008;105:2274–2279. doi: 10.1073/pnas.0708088105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leung K, Luzar A, Bratko D. Dynamics of capillary drying in water. Phys Rev Lett. 2003;90:065502. doi: 10.1103/PhysRevLett.90.065502. [DOI] [PubMed] [Google Scholar]
- 24.Luzar A. Activation barrier scaling for spontaneous evaporation of confined water. J Phys Chem B. 2004;108:19859–19866. [Google Scholar]
- 25.Leung K, Luzar A. Dynamics of capillary evaporation. II. Free energy barriers. J Chem Phys. 2000;113:5845–5852. [Google Scholar]
- 26.Bolhuis PG, Chandler D. Transition path sampling of cavitation between molecular scale solvophobic surfaces. J Chem Phys. 2000;113:8154–8160. [Google Scholar]
- 27.Xu L, Molinero V. Liquid-vapor oscillations of water nanoconfined between hydrophobic disks: Thermodynamics and kinetics. J Phys Chem B. 2010;114:7320–7328. doi: 10.1021/jp102443m. [DOI] [PubMed] [Google Scholar]
- 28.Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. In: Intermolecular Forces. Pullman B, editor. Dordrecht, The Netherlands: Reidel; 1981. pp. 331–342. [Google Scholar]
- 29.Zhu F, Hummer G. Pore opening and closing of a pentameric ligand-gated ion channel. Proc Natl Acad Sci USA. 2010;107:19814–19819. doi: 10.1073/pnas.1009313107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Modis Y, Ogata S, Clements D, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci USA. 2003;100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allen RJ, Warren PB, ten Wolde PR. Sampling rare switching events in biochemical networks. Phys Rev Lett. 2005;94:018104. doi: 10.1103/PhysRevLett.94.018104. [DOI] [PubMed] [Google Scholar]
- 32.Allen RJ, Frenkel D, ten Wolde PR. Simulating rare events in equilibrium or nonequilibrium stochastic systems. J Chem Phys. 2006;124:024102. doi: 10.1063/1.2140273. [DOI] [PubMed] [Google Scholar]
- 33.Allen RJ, Valeriani C, ten Wolde PR. Forward flux sampling for rare event simulations. J Phys Condens Matter. 2009;21:463102. doi: 10.1088/0953-8984/21/46/463102. [DOI] [PubMed] [Google Scholar]
- 34.Chen B, Xing J, Siepmann JI. Development of polarizable water force fields for phase equilibrium calculations. J Phys Chem B. 2000;104:2391–2401. [Google Scholar]
- 35.Widom B. Line tension and the shape of a sessile drop. J Phys Chem. 1995;99:2803–2806. [Google Scholar]
- 36.Lefevre B, et al. Intrusion and extrusion of water in hydrophobic mesopores. J Chem Phys. 2004;120:4927–4938. doi: 10.1063/1.1643728. [DOI] [PubMed] [Google Scholar]
- 37.Toshev BV, Platikanov D, Scheludko A. Line tension in three-phase equilibrium systems. Langmuir. 1988;4:489–499. [Google Scholar]
- 38.Stillinger FH. Structure in aqueous solutions of nonpolar solutes from the standpoint of scaled-particle theory. J Solution Chem. 1973;2:141–158. [Google Scholar]
- 39.Bolhuis PG, Dellago C, Chandler D. Sampling ensembles of deterministic transition pathways. Faraday Discuss. 1998;110:421–436. [Google Scholar]
- 40.Dellago C, Bolhuis PG, Geissler PL. Transition path sampling. Adv Chem Phys. 2002;123:1–84. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- 41.Bolhuis PG, Chandler D, Dellago C, Geissler PL. Transition path sampling: Throwing ropes over rough mountain passes, in the dark. Annu Rev Phys Chem. 2002;53:291–318. doi: 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- 42.Borrero EE, Escobedo FA. Optimizing the sampling and staging for simulations of rare events via forward flux sampling schemes. J Chem Phys. 2008;129:024115. doi: 10.1063/1.2953325. [DOI] [PubMed] [Google Scholar]
- 43.Velez-Vega C, Borrero EE, Escobedo FA. Kinetics and reaction coordinate for the isomerization of alanine dipeptide by a forward flux sampling protocol. J Chem Phys. 2009;130:225101. doi: 10.1063/1.3147465. [DOI] [PubMed] [Google Scholar]
- 44.Berendsen HJC, Grigera JR, Straatsma TP. The missing term in effective pair potentials. J Phys Chem. 1987;91:6269–6271. [Google Scholar]
- 45.Hoover WG. Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 46.Hoover WG. Constant pressure equations of motion. Phys Rev A. 1986;34:2499–2500. doi: 10.1103/physreva.34.2499. [DOI] [PubMed] [Google Scholar]
- 47.Plimpton SJ. Fast parallel algorithms for short-range molecular dynamics. J Comp Phys. 1995;117:1–19. [Google Scholar]
- 48.Hockney RW, Eastwood JW. Computer Simulations Using Particles. New York: McGraw-Hill; 1981. pp. 267–304. [Google Scholar]
- 49.Frenkel D, Smit B. Understanding Molecular Simulation from Algorithms to Applications. 2nd Ed. San Diego: Academic; 2002. pp. 310–316. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.