

Abstract
Protein interactions with peptides generally have low thermodynamic and mechanical stability. Streptococcus pyogenes fibronectin-binding protein FbaB contains a domain with a spontaneous isopeptide bond between Lys and Asp. By splitting this domain and rational engineering of the fragments, we obtained a peptide (SpyTag) which formed an amide bond to its protein partner (SpyCatcher) in minutes. Reaction occurred in high yield simply upon mixing and amidst diverse conditions of pH, temperature, and buffer. SpyTag could be fused at either terminus or internally and reacted specifically at the mammalian cell surface. Peptide binding was not reversed by boiling or competing peptide. Single-molecule dynamic force spectroscopy showed that SpyTag did not separate from SpyCatcher until the force exceeded 1 nN, where covalent bonds snap. The robust reaction conditions and irreversible linkage of SpyTag shed light on spontaneous isopeptide bond formation and should provide a targetable lock in cells and a stable module for new protein architectures.
Keywords: bacterial attachment, chemical biology, microbiology, protein engineering, single molecule biophysics
Tagging with peptides (e.g., HA, myc, FLAG, His6) is one of the most common ways to detect, purify, or immobilize proteins (1–4). Peptides are very useful minimally disruptive probes (5) but they are also “slippery”- antibodies or other proteins typically bind peptides with low affinity and poor mechanical strength (6–9). We sought to form a rapid covalent bond to a peptide tag without the use of chemical modification, artificial amino acids, or cysteines (disulfide bond formation is reversible and restricted to particular cellular locations).
It has recently been found that Streptococcus pyogenes, like many other Gram-positive bacteria, contains extracellular proteins stabilized by spontaneous intramolecular isopeptide bonds (10). Here we explored the second immunoglobulin-like collagen adhesin domain (CnaB2) from the fibronectin binding protein FbaB, found in invasive strains of S. pyogenes (11,12) and essential for phagocytosis-like uptake of the bacteria by endothelial cells (13). CnaB2 contains a single isopeptide bond conferring exceptional stability: CnaB2 remains folded even at pH 2 or up to 100 °C (12). By splitting CnaB2 into peptide and protein fragments, followed by rational modification of the parts, we developed a peptide tag of 13 amino acids that rapidly formed a covalent bond with its protein partner (138 amino acids, 15 kDa) and characterized the conditions for reaction, cellular specificity of bond formation, and resilience of the reacted product.
Results
Design of SpyTag for Rapid Covalent Bond Formation.
Crystallography and NMR have shown that CnaB2 forms a spontaneous intramolecular isopeptide bond (11, 12); quantum mechanical/molecular mechanical calculations (12) indicate that the unprotonated amine of Lys31 nucleophilically attacks the carbonyl carbon of Asp117 (Fig. 1A), catalyzed by the neighboring Glu77 (Fig. 1B). We hypothesized that splitting the CnaB2 domain into a peptide representing the C-terminal β-strand containing the reactive Asp and a protein partner derived from the rest of the protein would allow the two partners to reconstitute and undergo covalent reaction (Fig. 1C). This reaction occurred initially over hours for each species present at 10 μM, but by rational optimization of the S. pyogenes (Spy) protein partner (termed SpyCatcher) (SI Appendix, Fig. S1) and the peptide tag (termed SpyTag) (SI Appendix, Fig. S2), reaction now occurred in minutes (vide infra). Mixing SpyTag fused to Maltose Binding Protein (SpyTag-MBP) with SpyCatcher led to high yield of a product resistant to boiling in SDS (Fig. 1D). Mutation of the catalytic Glu77 in SpyCatcher (EQ mutant) or the reactive Asp117 in SpyTag (DA mutant) abolished covalent bond formation (Fig. 1D). Isothermal titration calorimetry showed that SpyTag DA-MBP and SpyCatcher formed a noncovalent complex with a Kd of 0.2 μM (SI Appendix, Fig. S3), a high affinity for a peptide-protein interaction (6–8).
Fig. 1.

Spontaneous intermolecular amide bond formation by SpyTag. (A) Amide bond formation between Lys and Asp side chains. (B) Key residues for amide bond formation in CnaB2 shown in stick format, based on PDB 2X5P. (C) Cartoon of SpyTag construction. Streptococcus pyogenes (Spy) CnaB2 was dissected into a large N-terminal fragment (SpyCatcher, left) and a small C-terminal fragment (SpyTag, right). Reactive residues are highlighted in red. (D) SpyTag and SpyCatcher associated covalently. SpyTag-MBP and SpyCatcher were mixed each at 10 μM for 3 h and analyzed after boiling by SDS-PAGE with Coomassie staining, alongside unreactive controls, SpyCatcher E77Q (EQ) and D117A (DA) SpyTag-MBP.
Electrospray ionization mass spectrometry confirmed that reaction of the peptide with SpyCatcher led to covalent bond formation, with the combined mass 18 Da less than the sum of the individual masses, from loss of water (Fig. 2A).
Fig. 2.

Characterization of isopeptide bond formation. (A) Mass spectrometry of reconstitution between Cna peptide and SpyCatcher. The minor peak results from the His6-tag on SpyCatcher leading to some gluconylation (see SI Appendix). (B) Time course of SpyTag-MBP:SpyCatcher covalent complex formation, with each partner at 10 μM at 25 °C, pH 7.0 determined by SDS-PAGE. (C) Rate constant for SpyTag reaction, calculated from triplicate measurements (each point shown) of SpyCatcher depletion under conditions as in (B). The equation for the trend-line and the correlation coefficient are shown.
We determined the efficiency of reaction between SpyTag-MBP and SpyCatcher and saw high yielding and rapid reconstitution, with more than 40% forming a covalent bond in the first minute (Fig. 2B). The second-order rate constant was 1.4 × 103 ± 43 M-1 s-1 (SD, n = 3) and the reaction half-time was 74 s (Fig. 2C). Reaction was still rapid with each partner present at twofold or 10-fold lower concentration (SI Appendix, Fig. S4).
SpyTag Reaction was Robust to Diverse Conditions.
We characterized how sensitive the reaction between SpyTag-MBP and SpyCatcher was to the sample conditions. The reaction proceeded similarly at 25 °C as at 37 °C (Fig. 3A). Reaction was also efficient at 4 °C, although significantly slower than at 25 °C (for the 1 min time-point: P < 0.0001, n = 3) (Fig. 3A).
Fig. 3.

Sensitivity of SpyTag reaction to conditions. (A) Temperature dependence of SpyTag-MBP reconstitution with SpyCatcher at 10 μM at pH 7.0 after 1 or 10 min. (B) pH-dependence of reaction as in (A) at 25 °C. (C) SpyTag-MBP and SpyCatcher were incubated each at 10 μM at 25 °C for 1 or 10 min at pH 7.0 in phosphate buffered saline (PBS), phosphate-citrate (PC), Tris, or Hepes buffers. (D) SpyTag-MBP and SpyCatcher were incubated each at 10 μM at 25 °C for 3 h at pH 7.0 in phosphate-citrate buffer containing no detergent (None), 1% Triton X-100 (Tx100), 1% Tween 20, or 0.5% Nonidet P-40 (NP40). All reactions were analyzed by SDS-PAGE and Coomassie staining. (All graphs mean of triplicate ± 1 s.d.; some error bars are too small to be visible.)
We were also interested in the dependence on pH, to see whether SpyTag could be used in low pH compartments of the cell, such as endosomes. Reaction was efficient at all pH values tested from 5 to 8 (Fig. 3B). In fact, the reaction was slightly faster at pH 5 and 6 than at pH 7 (comparing pH 6 with 7 for the 1 min time-point: P < 0.0001, n = 3) (Fig. 3B).
Reaction efficiency was compared in the presence of a range of buffers (PBS, phosphate-citrate, Hepes, Tris), but these had little effect, indicating that the reaction was robust to buffer composition (Fig. 3C). In particular there was no need for Ca2+ or Mg2+, even though these divalent cations are important for the function of many extracellular proteins, such as integrins or sortases. Because there are no cysteines in SpyTag or SpyCatcher, as expected there was no effect of reducing agents on the reaction (SI Appendix, Fig. S5), so that reaction should be efficient in the cytosol/nucleus, secretory pathway, or outside the cell.
We also tested the effect on the reaction of adding detergent, to investigate if SpyTag could be used in cell lysates or in conditions used to stabilize membrane proteins. There was no substantial effect on the reaction in the presence of high concentrations of nonionic detergents (Fig. 3D). We have not come across buffer conditions that impair SpyTag reaction, apart from the presence of sodium dodecyl sulfate from the loading buffer for SDS-PAGE.
SpyTag was Reactive at N-Terminal, Internal, and C-Terminal Sites.
Antibodies often preferentially recognize peptide tags at specific termini (1), while split intein/sortase tags must be at particular sites in the protein (14, 15). To establish the generality of SpyTag function, we tested whether SpyTag could react at different locations in proteins. SpyTag-MBP has the SpyTag internally: SpyTag is preceded by a His6-tag and a thrombin cleavage site at the N terminus and is followed by MBP. N-SpyTag-MBP has SpyTag directly after the initiating formyl-methionine: reaction of N-SpyTag-MBP with SpyCatcher was still efficient (SI Appendix, Fig. S6A). We also generated a construct with two SpyTags: the first SpyTag was between MBP and the three zinc fingers of Zif268, while the second SpyTag was right at the C terminus, following Zif268 (termed MBP-SpyTag-Zif-SpyTag). Both SpyTags were reactive, generating a doubly-branched protein with two SpyCatcher moieties covalently attached (SI Appendix, Fig. S6B).
SpyTag Reaction was Not Reversible over a Day.
Spontaneous isopeptide bond formation is most common between Lys and Asn (10), where there is loss of NH3, which may diffuse away and so help to promote irreversibility. For spontaneous isopeptide bond formation between Lys and Asp, there is loss of H2O but there is approximately 55 M H2O present (at least at the surface of the protein), which could make the SpyTag:SpyCatcher reaction reversible (16) (SI Appendix, Fig. S7A). To test whether the SpyTag reaction would reverse, we allowed SpyCatcher to react with SpyTag-MBP and then, to see if there was any backward reaction, we added a 20-fold excess of Cna peptide to capture any free SpyCatcher (SI Appendix, Fig. S7A). However, overnight incubation with competing free peptide did not lead to a decrease in the SpyTag-MBP:SpyCatcher covalent complex and there was no appearance of the Cna peptide:SpyCatcher complex (SI Appendix, Fig. S7B). Cna peptide gave complete conversion of unreacted SpyCatcher (SI Appendix, Fig. S7B, lane 8). Therefore, the SpyTag reaction did not reverse under these conditions. Note that once the protein is unfolded the isopeptide bond between SpyTag and SpyCatcher should be as chemically stable as a typical amide bond, resisting prolonged boiling.
SpyTag Reacted Efficiently Inside Cells.
Because SpyTag and SpyCatcher are genetically encodable, we tested whether SpyTag-MBP and SpyCatcher could react together inside the cytosol of Escherichia coli. Cells were transformed with a plasmid encoding the proteins individually or bicistronically, induced with IPTG, and subsequently boiled with SDS loading buffer to prevent any ex vivo reaction. Lysates were then analyzed by SDS-PAGE. A covalent complex corresponding to the expected molecular weight of SpyCatcher:SpyTag-MBP could be clearly observed and little unreacted SpyTag-MBP remained (Fig. 4A). This covalent complex was not present when SpyTag-MBP was coexpressed with the control SpyCatcher EQ (Fig. 4A). Mixing lysate or cells from bacteria expressing SpyCatcher or SpyTag-MBP individually did not lead to covalent complex formation (Fig. 4A), indicating that reaction was not occurring ex vivo and consistent with SpyTag being able to react efficiently in the cytosolic environment.
Fig. 4.

SpyCatcher reacted specifically. (A) SpyTag was reactive inside the E. coli cytosol. SpyTag-MBP and SpyCatcher were expressed in isolation or in the same cells. Cells were lysed and denatured by boiling with SDS buffer, before SDS-PAGE and Coomassie staining. SpyCatcher EQ was a negative control. To show that reconstitution happened within cells, lysates (lane 6) or cells (lane 7) expressing SpyCatcher alone (lane 1) or SpyTag-MBP alone (lane 3) were mixed. (B) SpyTag specificity inside the E. coli cytosol. Proteins were expressed as in (A) and the His6-tags, present on SpyTag-MBP, SpyCatcher (EQ) and anything with which they have reacted, were pulled down with Ni-NTA, before SDS-PAGE and Coomassie staining. (C) Cartoon of targeting SpyTag to the mammalian cell surface, by genetic fusion to ICAM1 linked to GFP, with an HA tag after the signal sequence. (D) HeLa SpyTag-ICAM1-GFP were incubated with dye-labeled SpyCatcher for 15 min and analyzed by fluorescence microscopy. The GFP image (green, left) highlights cells expressing SpyTag-ICAM1-GFP, the 555 image (red, middle) shows the cells to which SpyCatcher bound, and the bright-field image (grayscale, right) shows all the cells. SpyCatcher EQ-555 (lower boxes) is a negative control. (Scale bar, 20 μm).
To address whether SpyCatcher had reacted with other cytosolic proteins, His6-tag containing proteins were pulled down with Ni-NTA. SpyTag-MBP, SpyCatcher, and SpyCatcher EQ all contain an N-terminal His6-tag, and so we would expect to pull-down these proteins and any covalent complexes they formed. As expected SpyTag-MBP and SpyCatcher had efficiently reacted, depleting almost all SpyTag-MBP, but there was minimal pull-down of proteins of other molecular weights as assessed by Coomassie staining, indicating specificity of the SpyTag reaction in the cytosol. Using silver staining and anti-His immunoblotting as more sensitive probes for lower abundance interactions detected a small amount of a second protein pulled down in SpyCatcher-expressing bacteria (SI Appendix, Fig. S8).
SpyTag Reaction was Specific at the Mammalian Cell Surface.
Stable and specific targeting of biophysical probes at the mammalian cell surface is an important challenge in the study of receptor function (1, 15). SpyTag was targeted to the surface of HeLa cells by genetic fusion to green fluorescent protein-labeled Intercellular Adhesion Molecule-1 (ICAM1), to give SpyTag-ICAM1-GFP (Fig. 4C). SpyTag-ICAM1-GFP trafficked efficiently to the plasma membrane (Fig. 4D). Alexa Fluor 555-labeled SpyCatcher added to the medium labeled GFP-positive cells, even those with low expression levels, but did not label neighboring nonexpressing cells (Fig. 4D), showing that there was little nonspecific binding. In addition, cells expressing SpyTag-ICAM1-GFP were not labeled by the negative control dye-labeled SpyCatcher EQ (Fig. 4D). SpyCatcher attachment to cells was resistant to acid wash (SI Appendix, Fig. S9 A and B), suggesting that this cellular labeling was highly stable.
The nature of the labeling was further explored by immunoblotting against the HA tag on SpyTag-ICAM1-GFP, showing an adduct of SpyTag-ICAM1-GFP consistent with interaction with SpyCatcher and stable to boiling in SDS, indicating that the reaction was covalent (SI Appendix, Fig. S9C). To see if SpyCatcher had reacted with other cellular proteins, we blotted against the His6-tag on SpyCatcher. The anti-His blot showed a new band, corresponding to the molecular weight of SpyCatcher:SpyTag-ICAM1-GFP, while there was no detectable reaction on cells not expressing SpyTag-ICAM1-GFP (SI Appendix, Fig. S9C). These blots indicate that SpyCatcher showed high specificity to detect SpyTag amidst the diverse other surface proteins (17) and that the reaction was efficient at cellular expression levels.
SpyTag Formed Mechanically Stable Bonds with SpyCatcher.
Because the environment of the SpyTag:SpyCatcher reactive residues lowers the activation energy for amide bond formation, it is possible that the activation energy for amide bond hydrolysis could also be decreased. We showed that under mild conditions, in the presence of excess competitor, there was no sign of breakage of the bond to SpyTag (SI Appendix, Fig. S7). However, it was possible that pulling on SpyTag would distort the energy profile and lead to efficient bond breakage (18), making the SpyTag system mechanically labile. We assessed the mechanical stability of SpyTag at the single-molecule level, using dynamic force spectroscopy with an atomic force microscope (AFM). We fused SpyCatcher to two I27 domains, which provide a fingerprint from their characteristic unfolding force, to validate that one is observing specific cantilever bending from pulling on a single molecule (19) (Fig. 5A).
Fig. 5.

Dynamic force spectroscopy of the SpyTag:SpyCatcher interaction. (A) Cartoon of the constructs for testing the SpyTag:SpyCatcher interaction by AFM. (B) Representative force extension curve for Cys-I272-SpyCatcher forming a strong interaction with SpyTag-MBP-Cys, showing regions corresponding to PEG/agarose stretching, unfolding of two I27 domains (enlarged in the inset), and covalent bond breakage. (C) Histogram of different bond breakage forces for Cys-I272-SpyCatcher interactions with SpyTag-MBP-Cys. (D) Table of statistics of breakage forces. SpyTag:SpyCatcher was compared with the control SpyTag:SpyCatcher EQ. SpyTag was also tested against a cantilever only coated with PEG (SpyTag:PEG), or SpyTag was preblocked with free SpyCatcher before testing with a SpyCatcher-coated cantilever.
For the interaction between SpyTag and SpyCatcher we observed traces with a breakage force > 1 nN (Fig. 5 B and C), consistent with mechanochemistry taking place as the cantilever breaks a covalent bond when it retracts (18). The median breakage force for the covalent interaction was 1.9 nN, which is > 20 times stronger than streptavidin-biotin or antibody-antigen interactions (20, 21). C-N and C-C bonds are not predicted to break until 5 nN (18), so it is likely that was not the isopeptide bond, nor another bond in the protein which broke, but rather a weaker link in the chain, such as a C-S bond linking the proteins to the cantilever/bead. The nonreactive SpyCatcher EQ, after 1,940 tests with SpyTag, formed zero interactions above 500 pN (Fig. 5D, comparing SpyCatcher and SpyCatcher EQ: Fisher’s exact test P < 0.0001). As further negative controls, with no SpyCatcher on the cantilever or with SpyTag on the bead preblocked with free SpyCatcher, there were also zero force traces above 500 pN (Fig. 5D), consistent with the specificity of the > 500 pN interactions between SpyTag and SpyCatcher seen by AFM.
The cantilever was lowered for 20 s, in which time we would expect only a fraction of covalent bonds to form (Fig. 2B), so that we could probe the early stages of the SpyTag:SpyCatcher interaction. The unusually long contact times of this experiment made it challenging to avoid nonspecific interaction between the cantilever and the bead surface (22). Hence there was a low background of tests where the polyethylene glycol (PEG) and agarose linkers were extended for the negative controls, using no SpyCatcher or using preblocked SpyTag (Fig. 5D), giving force traces < 500 pN. The frequency of PEG extensions for SpyCatcher EQ interacting with SpyTag was comparable to these two controls (Fig. 5D). Therefore there was no detectable interaction between SpyCatcher EQ and SpyTag in this experiment. However, the frequency giving force traces < 500 pN for SpyCatcher interacting with SpyTag was significantly higher than for the other three conditions (chi-squared P < 0.0001) (Fig. 5D), supporting that the 100–200 pN peak for SpyTag and SpyCatcher (Fig. 5C) was a specific interaction. This peak was likely the force from a noncovalent interaction between SpyTag and SpyCatcher, as SpyTag docks and forms β-sheet hydrogen bonds. 100–200 pN is strong for a protein-protein interaction (23,24) and, in concert with the isothermal titration calorimetry data (SI Appendix, Fig. S3), the AFM results suggest that the SpyTag:SpyCatcher interaction has unusual mechanical strength even before the covalent bond forms.
Discussion
Formation of an amide bond from reaction of a carboxylate with an amine is thermodynamically unfavorable under standard biochemical conditions (16); the equilibrium constant for two amino acids reacting to form a dipeptide plus water is approximately 10-3 (16). Hence proteases reach equilibrium with almost complete hydrolysis of their peptide substrates, while protein synthesis depends on ATP-dependent amino acid activation by amino-acyl tRNA synthetases. Studies attempting to use proteases to catalyze amide bond formation have established that factors moving the equilibrium towards bond formation are a low dielectric constant to facilitate deprotonation (removing the energetic cost from dehydration of the charges) and trapping the product, either by transfer to an organic phase, precipitation, or an exergonic binding interaction (16). Therefore, for intermolecular amide bond formation by SpyTag, the orientation of the reactive groups, the hydrophobic environment, and proton-shuffling by Glu77 all accelerate the reaction kinetics, but the environment created in the SpyTag:SpyCatcher complex is also likely to determine that the position of equilibrium lies firmly on the side of bond formation; we found that SpyTag formed the covalent complex with high yield both in vitro (Figs. 1 and 2) and inside E. coli (Fig. 4).
Isopeptide bond formation is a spontaneous posttranslational modification found in several extracellular proteins from Gram-positive bacteria, including human pathogens such as Staphylococcus aureus, Corynebacterium diphtheriae, and Streptococcus pneumoniae (10). There has been little indication of how fast isopeptide bond formation occurs, because the protein recovered from recombinant expression had reacted to completion (12, 25). With this split system we provide the clearest evidence that reaction can occur in minutes, consistent with the biological time scale for export of these proteins (13, 26). Regarding the evolution of this posttranslational modification, our search for a fast reacting peptide tag showed that every change we tried to the residues in the C-terminal β-strand, even conservative changes pointing away from the protein partner, greatly reduced the speed of reaction (SI Appendix, Fig. S2B), suggesting that the reactive aspartic acid must be precisely aligned for reaction and that there is strong evolutionary selection for the stability the bond provides to the protein. Because residues in the C-terminal β-strand could not be changed, we managed to increase the efficiency of reaction by including an extra 5 C-terminal amino acids (SI Appendix, Fig. S2), which were not seen in the crystal structure (11) but may have an interaction with the main domain, facilitating docking of the peptide with SpyCatcher.
Our results also provide insight into the mechanical stability of spontaneous isopeptide bonds. The pilin Spy0128, has been analyzed by dynamic force spectroscopy and shown to be resistant to stretching because of its isopeptide bond, but forces > 1,000 pN (sufficient to break covalent bonds) were not explored (27). Also, in Spy0128 the reaction was between Lys and Asn (25, 28), rather than Lys and Asp for CnaB2 (16). Like Spy0128, in CnaB2 the isopeptide is between the N-terminal and C-terminal β-strands, so that, if the isopeptide does not break, then the force on the protein will not cause any extension of the protein chain. Intermolecular reconstitution can be achieved with split Spy0128 but the partners react orders of magnitude slower, undergo side reactions, and the protein is undesirably large (29). The stability of the SpyTag interaction before the isopeptide bond forms may relate to the known mechanical strength of long parallel β-strands, where multiple hydrogen bonds have to break simultaneously (23). The resilience of SpyTag:SpyCatcher to pulling forces after isopeptide bond formation suggests that the CnaB2 domain may be exposed to high force in bridging S. pyogenes to fibronectin during cell invasion (13). Precise attachment of proteins to AFM tips, to understand protein folding and resistance to extension, is usually achieved by introducing cysteines. However, carbon-sulfur or sulfur-sulfur bonds break at lower forces than amide bonds (18) and introducing free cysteines often disrupts folding of proteins containing disulfides; instead fusing a protein of interest to SpyTag may prove a useful handle to allow AFM tips to pull on specific proteins, even on the cell surface.
Peptide tags are a central tool in molecular biology (1). However, the reversibility of peptide binding often compromises sensitivity of immunoassays, stability of arrays/nano-assemblies, and purity of protein purification (1, 30, 31). SpyTag is a short peptide with fundamentally different binding characteristics to peptide tags in current use, forming a covalent isopeptide bond to its protein partner. Reaction requires only mixing and is rapid, high yielding, robust to experimental conditions, and shows good specificity. The reactivity of SpyTag at 37 °C means that SpyTag can be used in live cell imaging of mammalian cells and also potentially in transgenic mammalian systems. The reactivity at 4 °C, combined with the resistance to commonly used detergents, should allow the use of SpyTag for pull-downs in cell lysate, where the low temperature minimizes proteolysis. The range of pH values over which SpyTag can react and the absence of Cys in SpyTag or SpyCatcher should facilitate its use in the cytosol, nucleus, outside the cell, but also in the low pH endosomes and lysosomes. This robustness of spontaneous isopeptide formation to various conditions may be because docking of the peptide and protein partner could create a microenvironment buried from the surface. Note that derivatizing SpyCatcher with an N-hydroxy succinimide (NHS) activated dye did not block reaction with SpyTag (Fig. 4D), suggesting a reduced accessibility of Lys31 to exogenous labels, which will facilitate surface attachment of SpyCatcher or conjugation with other biophysical probes.
There are a number of ways to engineer peptides to react with small molecules, most commonly SNAP-tag (an engineered protein tag for multiprotein labeling in living cells) and HaloTag (2, 4, 32, 33), but unlike SpyTag, these methods are not suitable for directing interactions with other proteins. Coiled coils can drive specific protein association in living cells but have limited stability (34). Native chemical ligation is an elegant way to link proteins (35, 36) but specificity and yield appear to be limited in cells (36). Sortase can be used to link together two proteins in vitro or to link a peptide with a small molecule on the surface of living cells, but requires high [Ca2+] (15), which is damaging in the cytosol or nucleus. Photoreactive amino acids are powerful for identifying unknown binding partners (37,38,39) but (i) require UV which is often damaging in living systems, (ii) do not react with a defined partner, and (iii) typically give low yields (38). A protein containing an artificial alkyne amino acid can react with another protein containing an artificial azido amino acid (40) but the reaction must be catalyzed by toxic CuI. Furthermore, the reaction would not just be between the tagged proteins but also between the free amino acids. Split inteins can covalently splice proteins together and have the great advantage of leaving no trace after reaction (14, 41, 42), but must be located at defined termini and may undergo side reactions (43). Thus SpyTag has distinctive features in terms of flexibility of reaction conditions, specificity in cells, and high yield of reaction, in comparison to existing chemical biology approaches for irreversible protein targeting.
Limitations of SpyTag are that it is not traceless, in contrast to split intein-mediated ligation, and that the reaction rate is still far from the diffusion limit, although future work with phage or ribosome display (44) should be able to improve the on-rate. The rapid on-rate makes the femtomolar affinity streptavidin-biotin interaction so useful for isolation of biotin-conjugates at trace concentrations (31, 45). However, the strongest noncovalent interactions, such as streptavidin-biotin, can be broken in seconds by molecular motors (46, 47) or in milliseconds by shear forces (48), so it has been very difficult to provide barriers or locks in cellular systems using current tools. Molecular motors will not be able to break the covalent bond formed by SpyTag, which should be targetable to specific proteins, compartments or cell types, expressed from inducible promoters. Overall, SpyTag should enable new possibilities for cell biologists to probe the effect of force in cells (49) and for biochemists to create new protein architectures (34).
Materials and Methods
In Vitro Reconstitution Reactions.
Cloning, protein expression, statistical analysis, isothermal titration calorimetry, AFM, microscopy, in vivo reconstitution, mass spectrometry, and immunoblotting are described fully in the SI Appendix. Briefly, SpyTag-MBP, SpyCatcher, and variants of these proteins were expressed in E. coli and purified using Ni-NTA resin.
Amide bond formation between the protein and peptide binding partners was monitored by SDS-PAGE. To demonstrate covalent reconstitution (Fig. 1D), proteins were mixed at 10 μM in PBS pH 7.4 at 25 °C for 3 h. All quantified reactions were performed in triplicate. To stop reactions, samples were heated in SDS loading buffer on a Bio-Rad C1000 thermal cycler at 95 °C for 7 min. SDS-PAGE was performed on 14% polyacrylamide gels, using an X-cel SureLock (Life Technologies) at 200 V for approximately 1 h. Gels were stained with Instant Blue Coomassie stain (Triple Red Ltd.) and band intensities were quantified using a Gel Doc XR imager and Image Lab 3.0 software (Bio-Rad).
Reactions for analyzing speed, pH-dependence, temperature-dependence, and the reversibility of amide bond formation were performed by mixing 10 μM of each protein in 40 mM Na2HPO4 with 20 mM citric acid pH 7.0 (phosphate-citrate) for the indicated time at 25 °C, or at the indicated temperature and pH. (PBS alone would not enable proper buffering over the pH range explored.) For determining temperature dependence all reactions were incubated in a Bio-Rad C1000 thermal cycler at 4, 25, and 37 °C with a heated lid to prevent evaporation.
To calculate the rate constant, SpyTag-MBP and SpyCatcher at 10 μM were mixed in triplicate in phosphate-citrate and incubated at 25 °C for 1, 3, or 5 min, in the linear part of the reaction. Samples were then heated to 95 °C for 7 min in SDS loading buffer and analyzed on 14% SDS-PAGE with Coomassie staining. Unreacted SpyCatcher concentration was quantified from band intensity as above. 1/[unreacted Spy Catcher] was plotted against time and a straight line, whose gradient corresponds to the second order rate constant, was fitted using the “LINEST” linear least squares curve-fitting routine in Excel. The units were converted from μM-1 min-1 to M-1 s-1. The correlation coefficient and error bars to the gradient were calculated using the LINEST function in Excel. The reaction half-time was calculated from 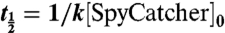 .
.
To test reversibility, 10 μM SpyTag-MBP and 10 μM SpyCatcher in phosphate-citrate were incubated for 3 h at 25 °C to allow reaction to take place. 100 μM Cna peptide (RSGAHIVMVDAGSR, made by solid-phase synthesis at 98% purity by Insight Biotechnology Ltd.) was then added and incubated at 25 °C for 16 h, before SDS-PAGE. In control experiments, 10 μM SpyCatcher or SpyCatcher EQ was incubated with 100 μM Cna peptide for 3 h at 25 °C in the same buffer. Also, 10 μM SpyCatcher or SpyCatcher EQ was incubated with 10 μM SpyTag-MBP for 3 h at 25 °C in the same buffer.
The survey of SpyCatcher reactivity against the series of peptide-MBP variants was performed by mixing each protein at 10 μM in PBS pH 7.4 and incubating at 25 °C for 30 min.
For analyzing the effect of various buffers on the covalent reconstitution between SpyCatcher and SpyTag-MBP, each protein was mixed at 10 μM in either phosphate buffered saline (PBS) pH 7.4, phosphate-citrate, 50 mM Tris (tris-hydroxymethyl aminomethane) pH 7.0, or 50 mM Hepes [4-(2-hydroxyethyl)-1-piperazine ethanesulfonate] pH 7.0 and incubated at 25 °C for 1 or 10 min.
For analyzing the effect of detergent on amide bond formation, SpyCatcher and SpyTag-MBP were mixed at 10 μM for 3 h at 25 °C in either PBS pH 7.4, or PBS pH 7.4 containing 1% Triton X-100, 1% Tween 20, or 0.5% Nonidet P-40.
Concentration-dependence of amide bond formation was analyzed by incubating SpyCatcher and SpyTag-MBP both at 1, 5, or 10 μM in phosphate-citrate pH 7.0 at 25 °C for the indicated times.
For analyzing the effect of reducing agent on amide bond formation, SpyCatcher and SpyTag-MBP were mixed at 10 μM in phosphate-citrate pH 7.0 with or without 10 mM final concentration of freshly prepared dithiothreitol (DTT) for 10 min at 25 °C.
To determine how SpyTag reacted at terminal or internal locations, N-SpyTag-MBP or MBP-SpyTag-Zif-SpyTag was incubated with SpyCatcher or SpyCatcher EQ for 30 min at pH 7.0 in phosphate-citrate buffer at 25 °C and analyzed by SDS-PAGE and Coomassie staining; proteins were present at 10 μM, except for those marked 3× which were present at 30 μM.
Calculation of percent reconstitution was performed by dividing the intensity of the band for the covalent complex by the intensity of all the bands in the lane, then multiplying by 100. To analyze the speed of SpyCatcher:SpyTag-MBP covalent complex formation, the reduction in intensity of the band for SpyCatcher was monitored relative to a control not incubated with SpyTag-MBP.
Cell Culture and Labeling.
HeLa cells were grown in DMEM with 10% Fetal Calf Serum, 50 U/mL penicillin and 50 μg/mL streptomycin. HeLa cells were transfected with a plasmid encoding SpyTag-ICAM1-GFP using TurboFect (Fermentas) following manufacturer’s instructions. After 48 h, transfectants were selected and maintained with 5 μg/mL puromycin. HeLa cells stably expressing SpyTag-ICAM1-GFP were washed with PBS containing 5 mM MgCl2 (PBS-Mg) and incubated in PBS-Mg containing 1% BSA and 5 μM SpyCatcher-Alexa Fluor 555 or 5 μM SpyCatcher EQ-Alexa Fluor 555 for 15 min at 25 °C. Cells were then washed twice in PBS-Mg at 4 °C and imaged live.
Supplementary Material
Acknowledgments.
Funding was provided by the Clarendon Fund (B.Z., J.O.F.), the National Institutes of Health (V.T.M., E.C.), the National Science Foundation (V.T.M.), the James and Esther King Biomedical Research Program (V.T.M), the School of Biology of the University of St. Andrews (U.S-L.), Oxford University Department of Biochemistry (B.Z., E.C.C. and M.H.), St. Peter’s College (B.Z.), New College (J.O.F.), and Worcester College (E.C.C. and M.H.).
Footnotes
Conflict of interest statement: M.H. and B.Z. are authors on a patent application regarding peptide targeting via spontaneous amide bond formation (United Kingdom Patent Application No. 1002362.0).
This article is a PNAS Direct Submission.
See Author Summary on page 4347 (volume 109, number 12).
Data deposition: The sequence reported in this paper has been deposited in the GenBank database, www.ncbi.nlm.nih.gov/genbank (accession no. JQ478411).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1115485109/-/DCSupplemental.
References
- 1.Jarvik JW, Telmer CA. Epitope tagging. Annu Rev Genet. 1998;32:601–618. doi: 10.1146/annurev.genet.32.1.601. [DOI] [PubMed] [Google Scholar]
- 2.Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science. 1998;281:269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 3.Allen KN, Imperiali B. Lanthanide-tagged proteins--an illuminating partnership. Curr Opin Chem Biol. 2010;14:247–254. doi: 10.1016/j.cbpa.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Halo TL, Appelbaum J, Hobert EM, Balkin DM, Schepartz A. Selective recognition of protein tetraserine motifs with a cell-permeable, pro-fluorescent bis-boronic acid. J Am Chem Soc. 2009;131:438–439. doi: 10.1021/ja807872s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huh WK, et al. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 6.Stanfield RL, Wilson IA. Protein-peptide interactions. Curr Opin Struct Biol. 1995;5:103–113. doi: 10.1016/0959-440x(95)80015-s. [DOI] [PubMed] [Google Scholar]
- 7.Korndorfer IP, Skerra A. Improved affinity of engineered streptavidin for the Strep-tag II peptide is due to a fixed open conformation of the lid-like loop at the binding site. Protein Sci. 2002;11:883–893. doi: 10.1110/ps.4150102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houk KN, Leach AG, Kim SP, Zhang X. Binding affinities of host-guest, protein-ligand, and protein-transition-state complexes. Angew Chem Int Ed Engl. 2003;42:4872–4897. doi: 10.1002/anie.200200565. [DOI] [PubMed] [Google Scholar]
- 9.Horne WS, Gellman SH. Foldamers with heterogeneous backbones. Acc Chem Res. 2008;41:1399–1408. doi: 10.1021/ar800009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang HJ, Baker EN. Intramolecular isopeptide bonds: protein crosslinks built for stress? Trends Biochem Sci. 2010;36:229–237. doi: 10.1016/j.tibs.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 11.Oke M, et al. The scottish structural proteomics facility: targets, methods and outputs. Journal of Structural and Functional Genomics. 2010;11:167–180. doi: 10.1007/s10969-010-9090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagan RM, et al. NMR spectroscopic and theoretical analysis of a spontaneously formed Lys-Asp isopeptide bond. Angew Chem Int Ed Engl. 2010;49:8421–8425. doi: 10.1002/anie.201004340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amelung S, et al. The FbaB-type fibronectin-binding protein of Streptococcus pyogenes promotes specific invasion into endothelial cells. Cell Microbiol. 2011;13:1200–1211. doi: 10.1111/j.1462-5822.2011.01610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zettler J, Schutz V, Mootz HD. The naturally split Npu DnaE intein exhibits an extraordinarily high rate in the protein trans-splicing reaction. FEBS Lett. 2009;583:909–914. doi: 10.1016/j.febslet.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Popp MW, Antos JM, Grotenbreg GM, Spooner E, Ploegh HL. Sortagging: a versatile method for protein labeling. Nat Chem Biol. 2007;3:707–708. doi: 10.1038/nchembio.2007.31. [DOI] [PubMed] [Google Scholar]
- 16.Kullmann W. Proteases as catalytic agents in peptide synthetic chemistry—shifting the extent of peptide-bond synthesis from a quantite negligible to a quantite considerable. J Protein Chem. 1985;4:1–22. [Google Scholar]
- 17.McDonald CA, Yang JY, Marathe V, Yen TY, Macher BA. Combining results from lectin affinity chromatography and glycocapture approaches substantially improves the coverage of the glycoproteome. Mol Cell Proteomics. 2009;8:287–301. doi: 10.1074/mcp.M800272-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beyer MK, Clausen-Schaumann H. Mechanochemistry: the mechanical activation of covalent bonds. Chem Rev. 2005;105:2921–2948. doi: 10.1021/cr030697h. [DOI] [PubMed] [Google Scholar]
- 19.Marszalek PE, et al. Mechanical unfolding intermediates in titin modules. Nature. 1999;402:100–103. doi: 10.1038/47083. [DOI] [PubMed] [Google Scholar]
- 20.Moy VT, Florin EL, Gaub HE. Intermolecular forces and energies between ligands and receptors. Science. 1994;266:257–259. doi: 10.1126/science.7939660. [DOI] [PubMed] [Google Scholar]
- 21.Neuert G, Albrecht C, Pamir E, Gaub HE. Dynamic force spectroscopy of the digoxigenin-antibody complex. FEBS Lett. 2006;580:505–509. doi: 10.1016/j.febslet.2005.12.052. [DOI] [PubMed] [Google Scholar]
- 22.Celik E, Moy VT. Nonspecific interactions in AFM force spectroscopy measurements. J Mol Recognit. 2012;25:53–56. doi: 10.1002/jmr.2152. [DOI] [PubMed] [Google Scholar]
- 23.Crampton N, Brockwell DJ. Unravelling the design principles for single protein mechanical strength. Curr Opin Struct Biol. 2010;20:508–517. doi: 10.1016/j.sbi.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 24.Huang J, Nagy SS, Koide A, Rock RS, Koide S. A peptide tag system for facile purification and single-molecule immobilization. Biochemistry. 2009;48:11834–11836. doi: 10.1021/bi901756n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang HJ, Coulibaly F, Clow F, Proft T, Baker EN. Stabilizing isopeptide bonds revealed in gram-positive bacterial pilus structure. Science. 2007;318:1625–1628. doi: 10.1126/science.1145806. [DOI] [PubMed] [Google Scholar]
- 26.Ton-That H, Marraffini LA, Schneewind O. Protein sorting to the cell wall envelope of Gram-positive bacteria. Biochim Biophys Acta. 2004;1694:269–278. doi: 10.1016/j.bbamcr.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 27.Alegre-Cebollada J, Badilla CL, Fernandez JM. Isopeptide bonds block the mechanical extension of pili in pathogenic Streptococcus pyogenes. J Biol Chem. 2010;285:11235–11242. doi: 10.1074/jbc.M110.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu XQ, et al. Autocatalytic intramolecular isopeptide bond formation in gram-positive bacterial pili: a QM/MM simulation. J Am Chem Soc. 2011;133:478–485. doi: 10.1021/ja107513t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zakeri B, Howarth M. Spontaneous intermolecular amide bond formation between side chains for irreversible peptide targeting. J Am Chem Soc. 2010;132:4526–4527. doi: 10.1021/ja910795a. [DOI] [PubMed] [Google Scholar]
- 30.Huang J, Makabe K, Biancalana M, Koide A, Koide S. Structural basis for exquisite specificity of affinity clamps, synthetic binding proteins generated through directed domain-interface evolution. J Mol Biol. 2009;392:1221–1231. doi: 10.1016/j.jmb.2009.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez P, et al. Isolation of transcription factor complexes by in vivo biotinylation tagging and direct binding to streptavidin beads. Methods Mol Biol. 2006;338:305–323. doi: 10.1385/1-59745-097-9:305. [DOI] [PubMed] [Google Scholar]
- 32.Nonaka H, Tsukiji S, Ojida A, Hamachi I. Non-enzymatic covalent protein labeling using a reactive tag. J Am Chem Soc. 2007;129:15777–15779. doi: 10.1021/ja074176d. [DOI] [PubMed] [Google Scholar]
- 33.Uttamapinant C, et al. A fluorophore ligase for site-specific protein labeling inside living cells. Proc Natl Acad Sci USA. 2010;107:10914–10919. doi: 10.1073/pnas.0914067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woolfson DN. The design of coiled-coil structures and assemblies. Adv Protein Chem. 2005;70:79–112. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- 35.Kent SB. Total chemical synthesis of proteins. Chem Soc Rev. 2009;38:338–351. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- 36.Chattopadhaya S, Abu Bakar FB, Srinivasan R, Yao SQ. In vivo imaging of a bacterial cell division protein using a protease-assisted small-molecule labeling approach. Chembiochem. 2008;9:677–680. doi: 10.1002/cbic.200700647. [DOI] [PubMed] [Google Scholar]
- 37.Chin JW, et al. Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli. J Am Chem Soc. 2002;124:9026–9027. doi: 10.1021/ja027007w. [DOI] [PubMed] [Google Scholar]
- 38.Lee HS, Dimla RD, Schultz PG. Protein-DNA photo-crosslinking with a genetically encoded benzophenone-containing amino acid. Bioorg Med Chem Lett. 2009;19:5222–5224. doi: 10.1016/j.bmcl.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suchanek M, Radzikowska A, Thiele C. Photo-leucine and photo-methionine allow identification of protein-protein interactions in living cells. Nat Methods. 2005;2:261–267. doi: 10.1038/nmeth752. [DOI] [PubMed] [Google Scholar]
- 40.Hancock SM, Uprety R, Deiters A, Chin JW. Expanding the genetic code of yeast for incorporation of diverse unnatural amino acids via a pyrrolysyl-tRNA synthetase/tRNA pair. J Am Chem Soc. 2010;132:14819–14824. doi: 10.1021/ja104609m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi J, Muir TW. Development of a tandem protein trans-splicing system based on native and engineered split inteins. J Am Chem Soc. 2005;127:6198–6206. doi: 10.1021/ja042287w. [DOI] [PubMed] [Google Scholar]
- 42.Tavassoli A, Benkovic SJ. Split-intein mediated circular ligation used in the synthesis of cyclic peptide libraries in E. coli. Nat Protoc. 2007;2:1126–1133. doi: 10.1038/nprot.2007.152. [DOI] [PubMed] [Google Scholar]
- 43.Siebold C, Erni B. Intein-mediated cyclization of a soluble and a membrane protein in vivo: function and stability. Biophys Chem. 2002;96:163–171. doi: 10.1016/s0301-4622(02)00012-1. [DOI] [PubMed] [Google Scholar]
- 44.Griffiths AD, Duncan AR. Strategies for selection of antibodies by phage display. Curr Opin Biotechnol. 1998;9:102–108. doi: 10.1016/s0958-1669(98)80092-x. [DOI] [PubMed] [Google Scholar]
- 45.Laitinen OH, Hytonen VP, Nordlund HR, Kulomaa MS. Genetically engineered avidins and streptavidins. Cell Mol Life Sci. 2006;63:2992–3017. doi: 10.1007/s00018-006-6288-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chivers CE, et al. A streptavidin variant with slower biotin dissociation and increased mechanostability. Nat Methods. 2010;7:391–393. doi: 10.1038/nmeth.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perumal SK, Raney KD, Benkovic SJ. Analysis of the DNA translocation and unwinding activities of T4 phage helicases. Methods. 2010;51:277–288. doi: 10.1016/j.ymeth.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pierres A, Touchard D, Benoliel AM, Bongrand P. Dissecting streptavidin-biotin interaction with a laminar flow chamber. Biophys J. 2002;82:3214–3223. doi: 10.1016/S0006-3495(02)75664-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vogel V, Sheetz MP. Cell fate regulation by coupling mechanical cycles to biochemical signaling pathways. Curr Opin Cell Biol. 2009;21:38–46. doi: 10.1016/j.ceb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
